Design of Anti-HIV Ligands by Means of Minimal Topological Difference (MTD) Method

Abstract
:1. Introduction
2. The MTD (Minimal Topological Difference) Method
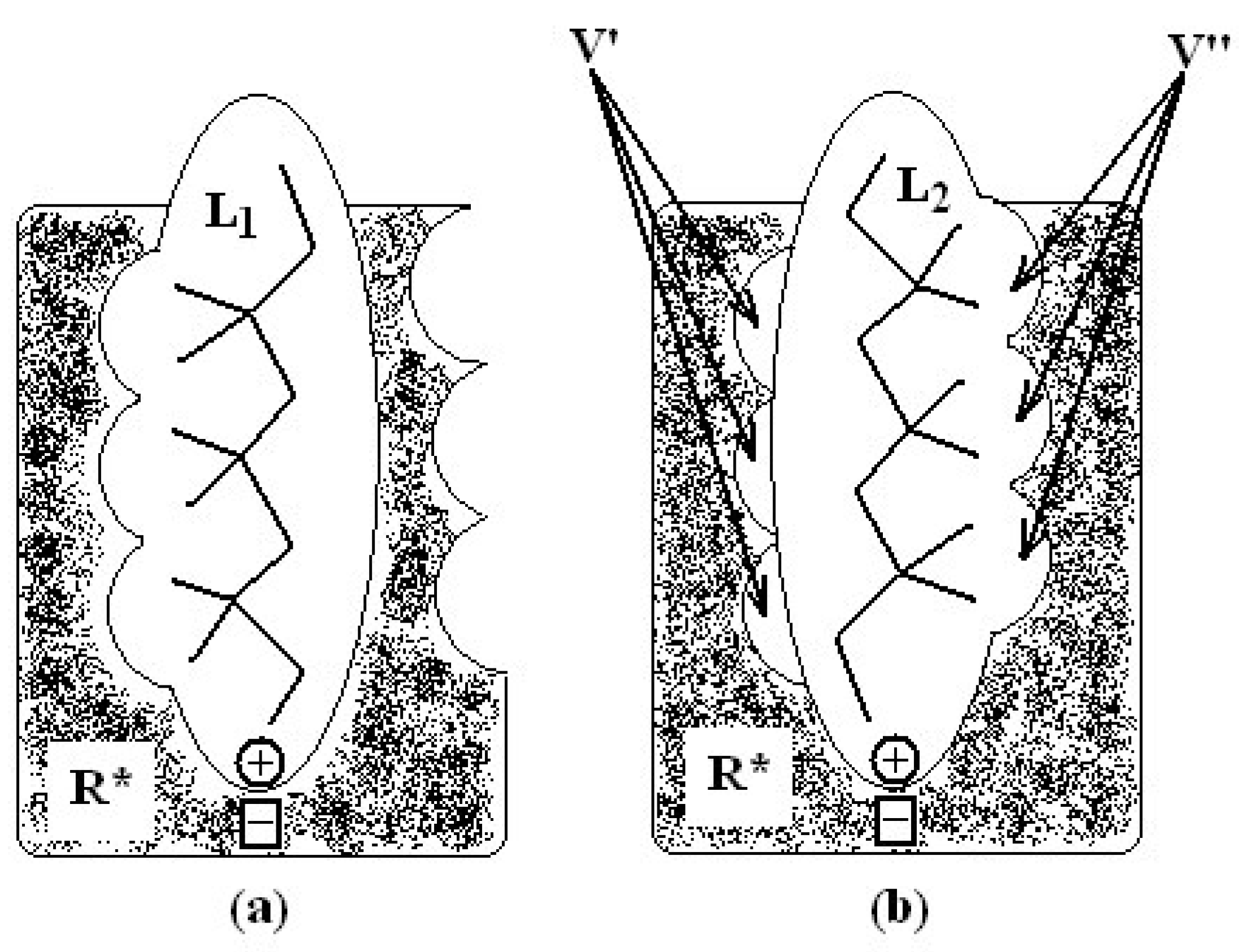
- molecular superposition, based on certain common molecular elements or on a “pharmacophoral” constellation” of the atoms, and
- defining equivalent positions.

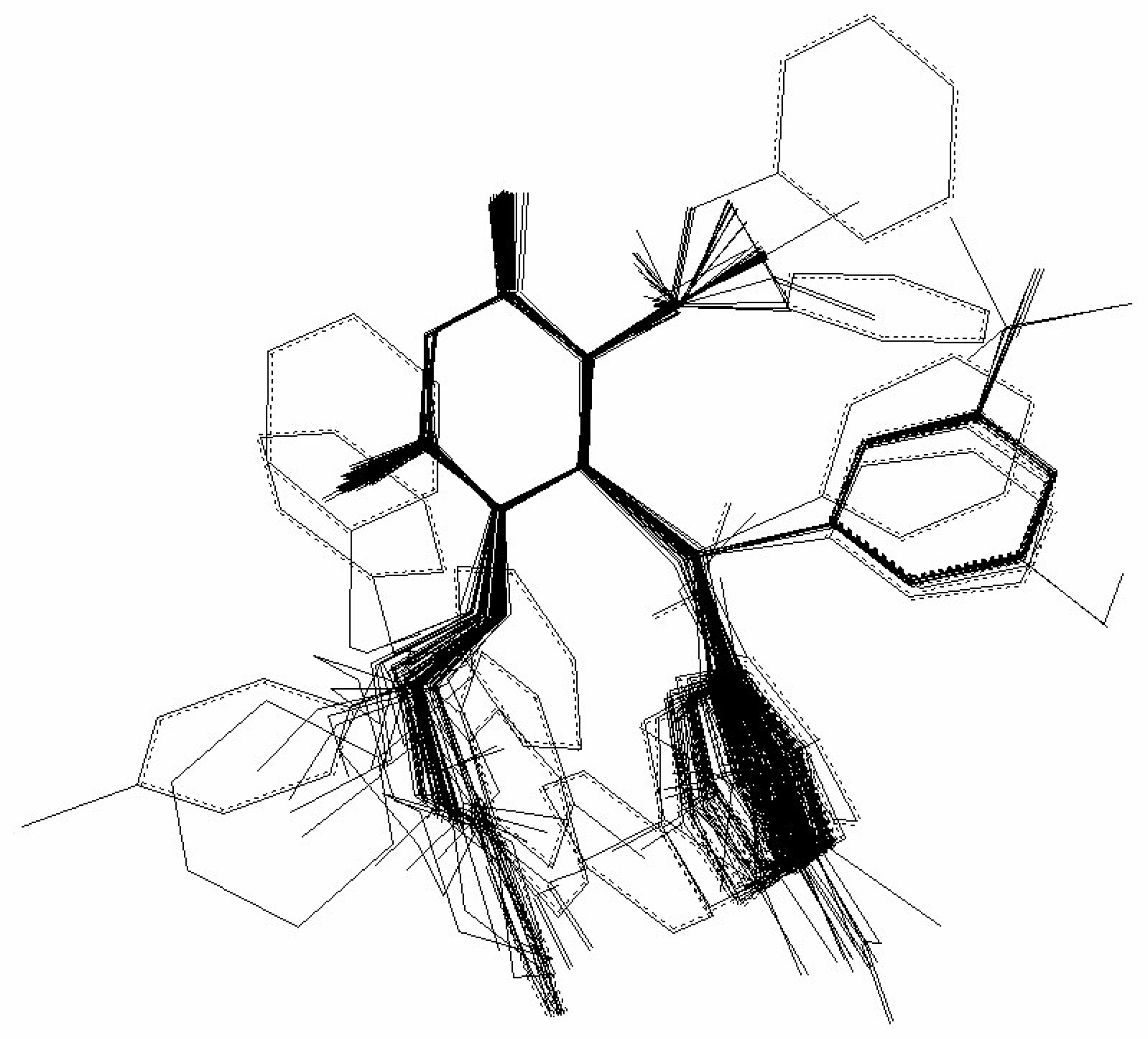
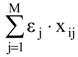 in (2) represents the summation over both the vertices of cavity of R that are not occupied by the ligand Li as well as of those from the wall of R being occupied by the atoms of Li, unless the hydrogen atoms, in equation (1), respectively. As a consequence, the key relation (2) gives the analytical quantification of the steric relation between each ligand „i” of the total tested N respecting the available M vertices of receptor R in the course of bonding. Thus, the degree of steric misfit for the molecule Li with respect to the receptor, i.e. MTDi, is defined as the sum of the number of unoccupied cavity vertices of H and the number of occupied wall vertices of H. If Li shows more low energy conformations, k = 1,…,Ci (Ci is the number of conformations of molecule I), the conformation that fits best in the receptor site will be retained through the modified version of equation (2):
in (2) represents the summation over both the vertices of cavity of R that are not occupied by the ligand Li as well as of those from the wall of R being occupied by the atoms of Li, unless the hydrogen atoms, in equation (1), respectively. As a consequence, the key relation (2) gives the analytical quantification of the steric relation between each ligand „i” of the total tested N respecting the available M vertices of receptor R in the course of bonding. Thus, the degree of steric misfit for the molecule Li with respect to the receptor, i.e. MTDi, is defined as the sum of the number of unoccupied cavity vertices of H and the number of occupied wall vertices of H. If Li shows more low energy conformations, k = 1,…,Ci (Ci is the number of conformations of molecule I), the conformation that fits best in the receptor site will be retained through the modified version of equation (2):
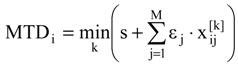
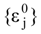 attribution set, the beginning chart S0. Then, one has to calculate the
attribution set, the beginning chart S0. Then, one has to calculate the 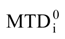 values and, by employing the multi-linear regression analysis, the regression coefficients in the above equation (4) and the associate correlation coefficient are computed. The algorithm continues with one by one modification of εj attributions resulting in a number of 2M mono-substituted charts, with the respective MTDi values together with 2M correlation equations of type (4) and their correlation coefficients. The mono-substituted chart with the highest correlation coefficient r from all 2M outputs is taken as starting point for the next cycle. The process continues until the regression coefficient r doesn’t increase anymore. The final increase anymore. The final {εj} set of values, j = 1,M, represents the optimized receptor chart, S*, and provides through their employment in equation (2) the optimized predictor variables MTD* for the QSAR problem [18].
values and, by employing the multi-linear regression analysis, the regression coefficients in the above equation (4) and the associate correlation coefficient are computed. The algorithm continues with one by one modification of εj attributions resulting in a number of 2M mono-substituted charts, with the respective MTDi values together with 2M correlation equations of type (4) and their correlation coefficients. The mono-substituted chart with the highest correlation coefficient r from all 2M outputs is taken as starting point for the next cycle. The process continues until the regression coefficient r doesn’t increase anymore. The final increase anymore. The final {εj} set of values, j = 1,M, represents the optimized receptor chart, S*, and provides through their employment in equation (2) the optimized predictor variables MTD* for the QSAR problem [18]. 3. Computational Details

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
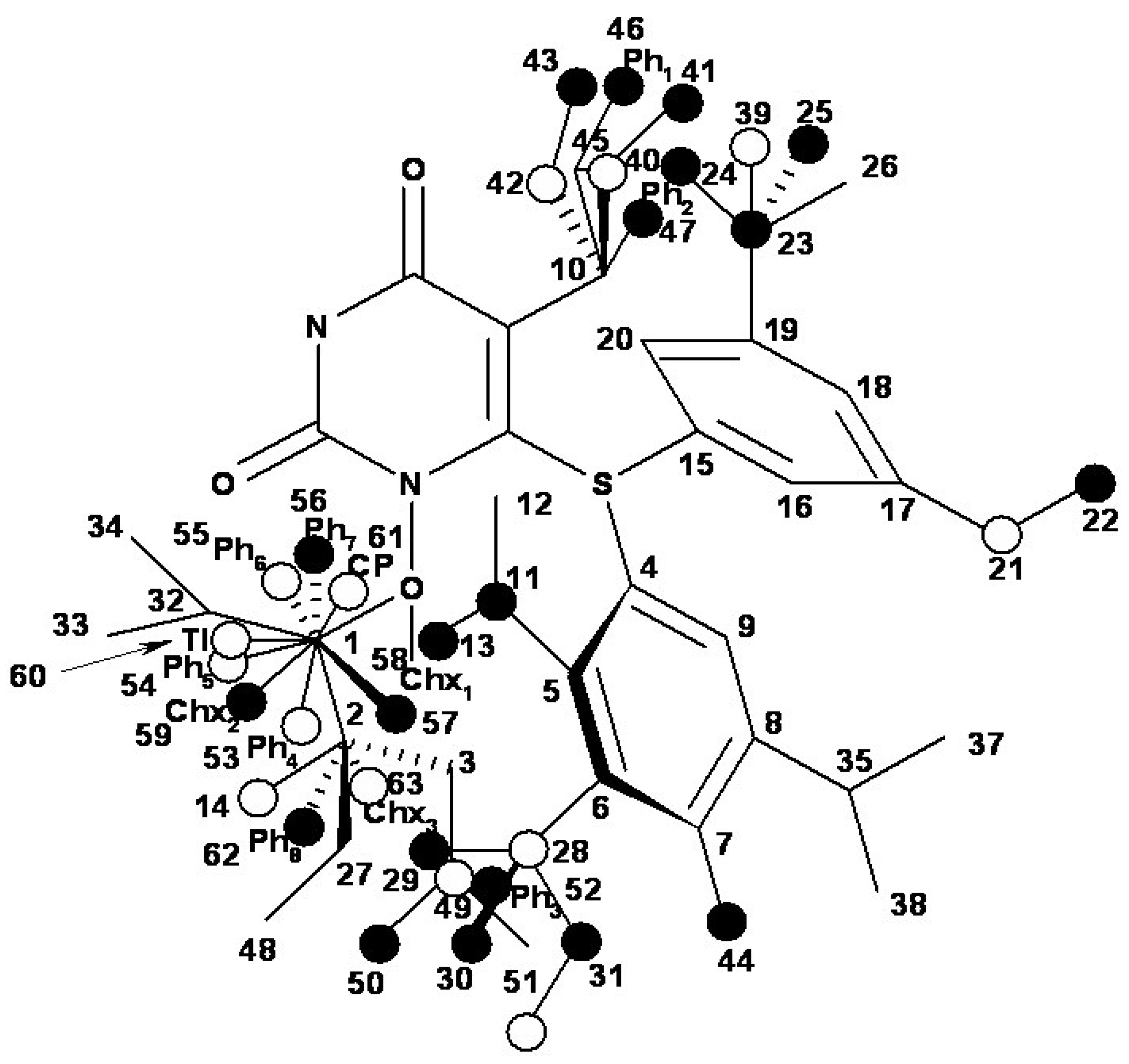{kind=link}
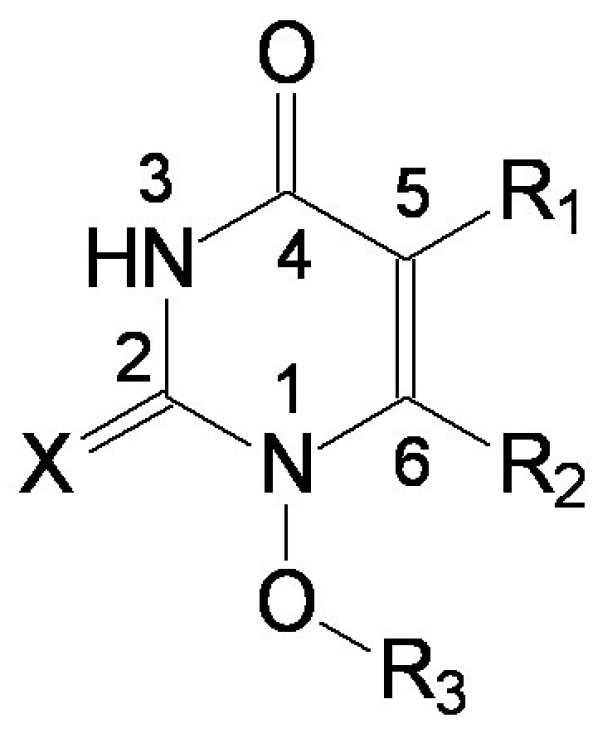
| i/Li | R1 | R2 | R3 | X |
|---|---|---|---|---|
| 1 | methyl | 2-methylphenylthio | 2-hidroxyethyl | O |
| 2 | methyl | 2-nitrophenhylthio | 2-hidroxyethyl | O |
| 3 | methyl | 2-methoxyphenylthio | 2-hidroxyethyl | O |
| 4 | methyl | 3-metilfeniltio | 2-hidroxietil | O |
| 5 | methyl | 3-ethylphenylthio | 2-hidroxyethyl | O |
| 6 | methyl | 3- tertbuthylphenylthio | 2-hidroxyethyl | O |
| 7 | methyl | 3-trifluoromethylphenylthio | 2-hidroxyethyl | O |
| 8 | methyl | 3-fluorophenylthio | 2-hidroxyethyl | O |
| 9 | methyl | 3-clorophenylthio | 2-hidroxyethyl | O |
| 10 | methyl | 3-bromophenylthio | 2-hidroxyethyl | O |
| 11 | methyl | 3-iodophenylthio | 2-hidroxyethyl | O |
| 12 | methyl | 3-nitrophenylthio | 2-hidroxyethyl | O |
| 13 | methyl | 3-hidroxyphenylthio | 2-hidroxyethyl | O |
| 14 | methyl | 3-metoxyphenylthio | 2-hidroxyethyl | O |
| 15 | methyl | 3,5-dimetilphenylthio | 2-hidroxyethyl | O |
| 16 | methyl | 3,5-diclorophenylthio | 2-hidroxyethyl | O |
| 17 | methyl | 3,5-dimethylphenylthio | 2-hidroxyethyl | S |
| 18 | methyl | 3-metoxycarbonylphenylthio | 2-hidroxyethyl | O |
| 19 | methyl | 3-acetylphenylthio | 2-hidroxyethyl | O |
| 20 | methyl | 3-cyanophenylthio | 2-hidroxyethyl | O |
| 21 | allyl | phenylthio | 2-hidroxyethyl | O |
| 22 | ethyl | phenylthio | 2-hidroxyethyl | S |
| 23 | propyl | phenylthio | 2-hidroxyethyl | S |
| 24 | isopropyl | phenylthio | 2-hidroxyethyl | S |
| 25 | ethyl | 3,5-dimethylphenylthio | 2-hidroxyethyl | S |
| 26 | isopropyl | 3,5-dimethylphenylthio | 2-hidroxyethyl | S |
| 27 | ethyl | 3,5-diclorophenylthio | 2-hidroxyethyl | S |
| 28 | ethyl | phenylthio | 2-hidroxyethyl | O |
| 29 | propyl | phenylthio | 2-hidroxyethyl | O |
| 30 | isopropyl | phenylthio | 2-hidroxyethyl | O |
| 31 | ethyl | 3,5-dimetylphenylthio | 2-hidroxyethyl | O |
| 32 | isopropyl | 3,5-dimetylphenylthio | 2-hidroxyethyl | O |
| 33 | ethyl | 3,5-diclorophenylthio | 2-hidroxyethyl | O |
| 34 | methyl | 4-metylphenylthio | 2-hidroxyethyl | O |
| 35 | methyl | phenylthio | 2-hidroxyethyl | O |
| 36 | methyl | phenylthio | 2-hidroxyethyl | S |
| 37 | iodo | phenylthio | 2-hidroxyethyl | O |
| 38 | ethenyl | phenylthio | 2-hidroxyethyl | O |
| 39 | 2-phenylethenyl | phenylthio | 2-hidroxyethyl | O |
| 40 | benzyl | phenylthio | 2-hidroxyethyl | O |
| 41 | methyl | phenylthio | 2-metoxyethyl | O |
| 42 | methyl | phenylthio | 2-acetyloxyethyl | O |
| 43 | methyl | phenylthio | 2-benzoyloxyethyl | O |
| 44 | methyl | phenylthio | ethyl | O |
| 45 | methyl | phenylthio | 2-cloroethyl | O |
| 46 | methyl | phenylthio | 2-azidoethyl | O |
| 47 | methyl | phenylthio | 2-fluoroethyl | O |
| 48 | methyl | phenylthio | propyl | O |
| 49 | methyl | phenylthio | benzyl | O |
| 50 | ethyl | phenylthio | ethyl | O |
| 51 | ethyl | phenylthio | ethyl | S |
| 52 | ethyl | 3,5-dimethylphenylthio | ethyl | O |
| 53 | ethyl | 3,5-dimethylphenhylthio | ethyl | S |
| 54 | ethyl | phenylthio | benzyl | O |
| 55 | ethyl | 3,5-dimethylphenylthio | benzyl | O |
| 56 | ethyl | phenylthio | benzyl | S |
| 57 | ethyl | 3,5-dimethylphenylthio | benzyl | S |
| 58 | isopropyl | phenylthio | ethyl | O |
| 59 | isopropyl | phenylthio | benzyl | O |
| 60 | isopropyl | phenylthio | ethyl | S |
| 61 | isopropyl | phenylthio | benzyl | S |
| 62 | methyl | phenylthio | methyl | O |
| 63 | methyl | phenylthio | butyl | O |
| 64 | methyl | phenylthio | methyl | S |
| 65 | methyl | phenylthio | propyl | S |
| 66 | ethyl | 3,5-diclorophenylthio | ethyl | S |
| 67 | ethyl | phenylthio | isopropyl | S |
| 68 | ethyl | phenylthio | cyclohexyl | S |
| 69 | ethyl | phenylthio | cyclohexylmethyl | S |
| 70 | ethyl | phenylthio | 4-methylbenzyl | S |
| 71 | ethyl | phenylthio | 4-clorobenzyl | S |
| 72 | ethyl | phenylthio | 2-phenylethyl | S |
| 73 | ethyl | 3,5-diclorophenylthio | ethyl | O |
| 74 | ethyl | phenylthio | isopropyl | O |
| 75 | ethyl | phenylthio | cyclohexyl | O |
| 76 | ethyl | phenylthio | cyclohexylmethyl | O |
| 77 | ethyl | phenylthio | 2-ciclohexylethyl | O |
| 78 | cyclopropyl | phenylthio | ethyl | S |
| 79 | cyclopropyl | phenylthio | ethyl | O |
- flexible torsion angles variation: ±60º - ±180º;
- energetic criteria to accept a conformation: 4 kcal/mol above the minimum;
- conformation cancellation: for atoms with distances less than 0.5 Å and torsion angles differences less than 15º;
- duplicate conformations: energy differences less than 0.05 kcal/mol;
- optimization program: MM+;
- optimization algorithm: Polak-Ribiere;
- optimization RMS (root mean square value) gradient: 0.05 kcal/mol;
- maximum iterations: 250;
- maximum optimizations: 250;
- maximum retained conformations: 20.
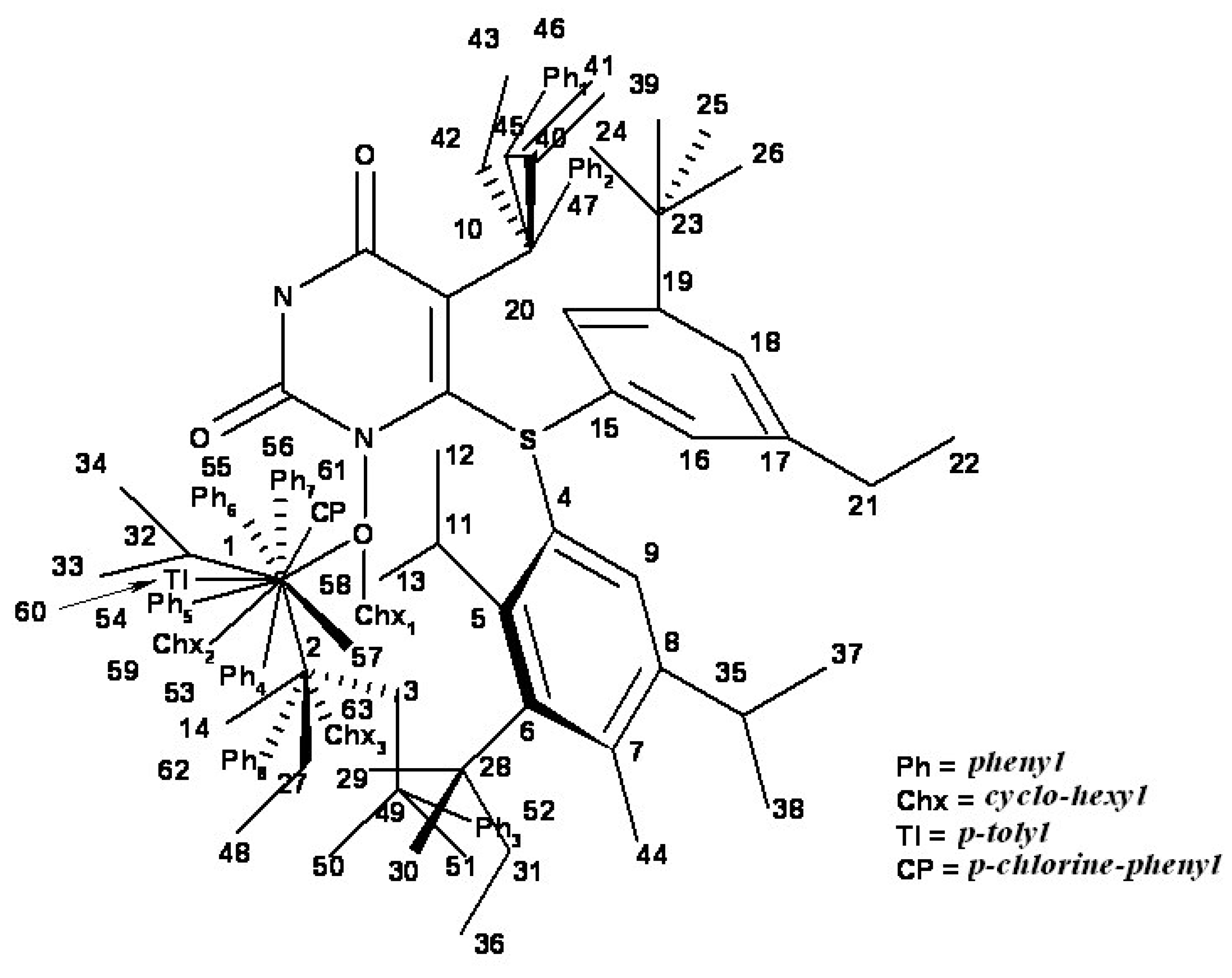
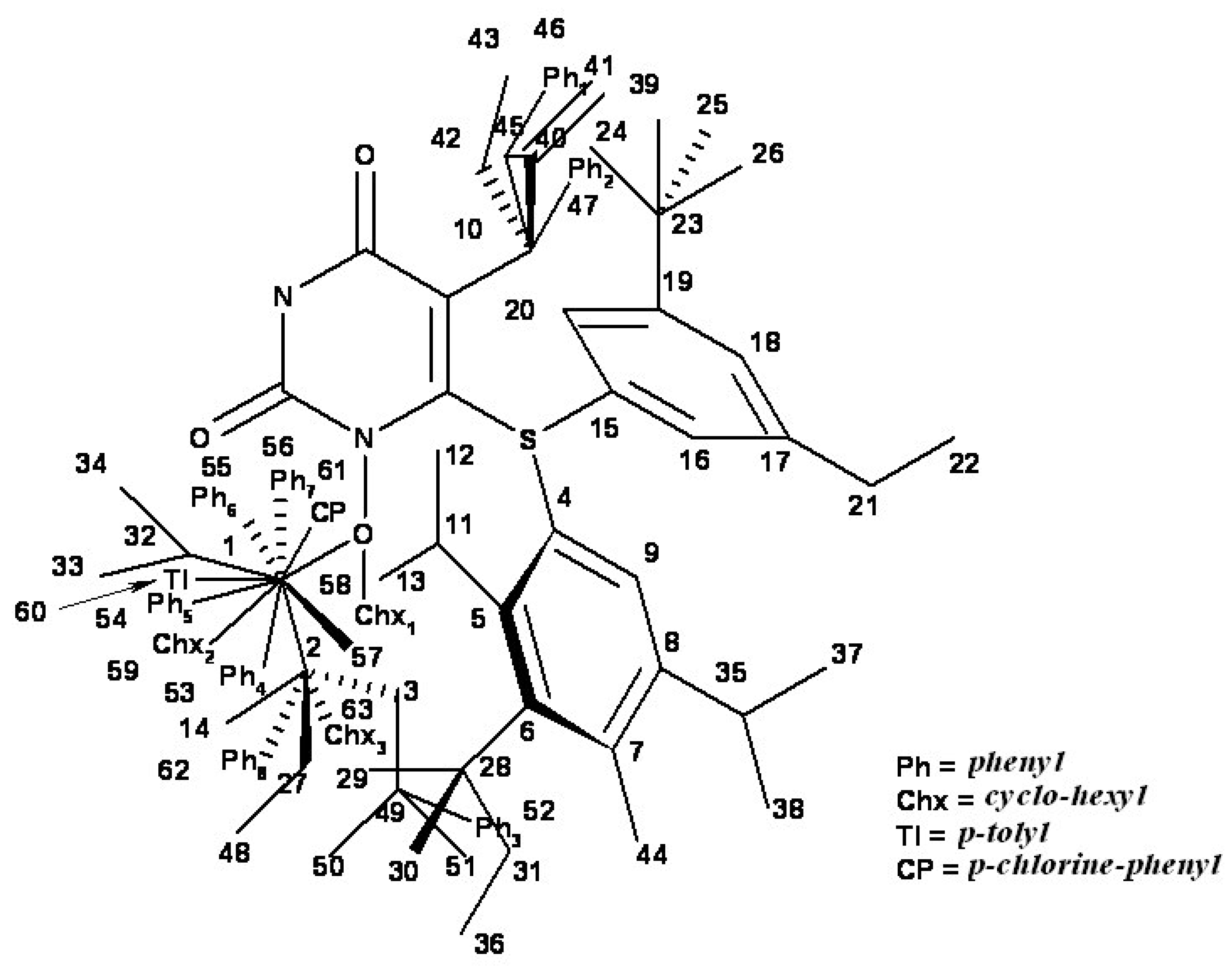
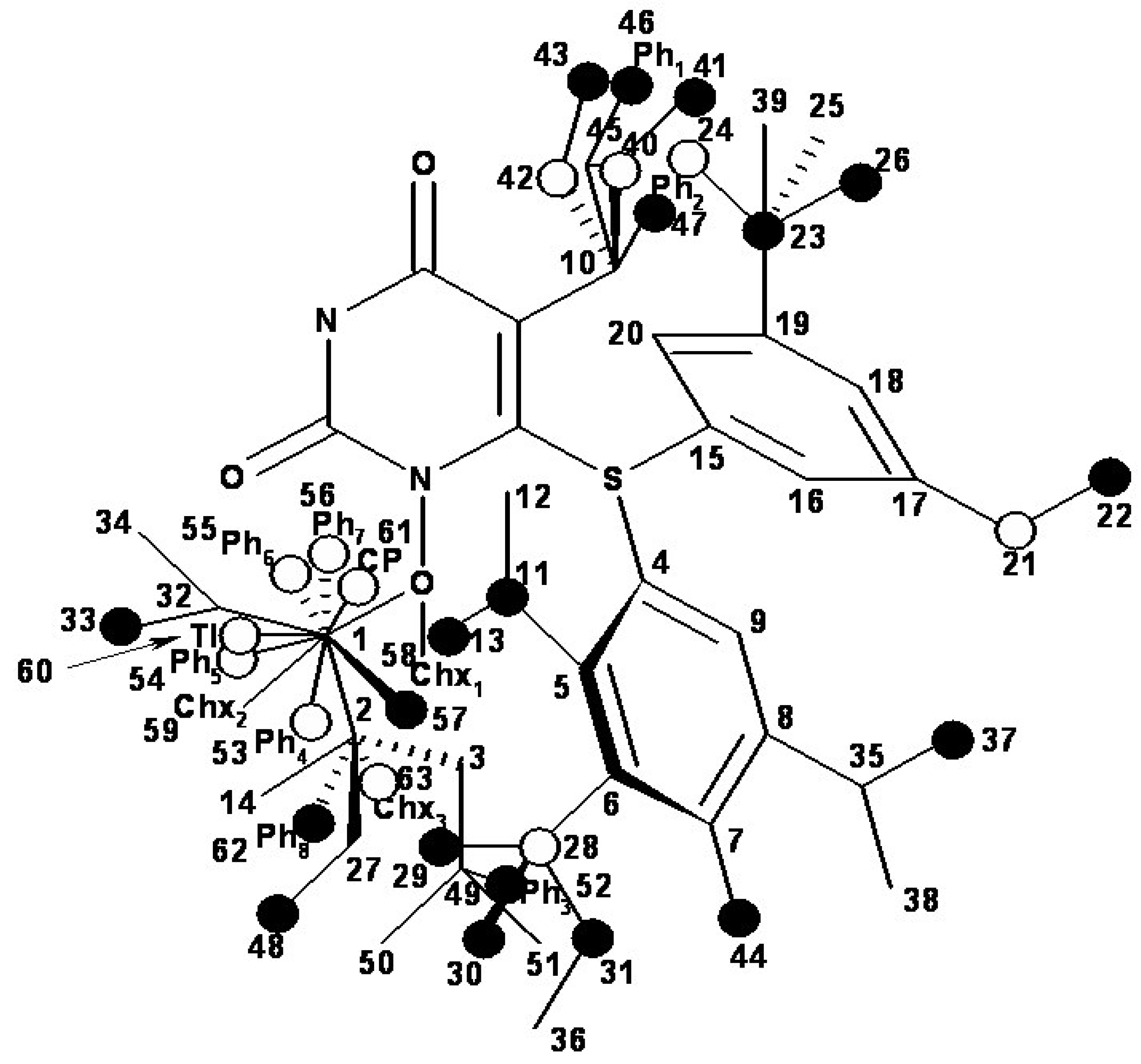
| i/Li | Vertices occupancy in hypermolecule | i/Li | Vertices occupancy in hypermolecule |
|---|---|---|---|
| 1 | 1,2,3,4,5,6,7,8,9,10,11 | 41 | 1,2,4,5,6,7,8,9,10,27,48 |
| 2 | 1,2,3,4,5,6,7,8,9,10,11,12,13 | 42 | 1,2,3,4,5,6,7,8,9,10,49,50,51 |
| 3 | 1,2,4,5,6,7,8,9,10,11,12,14 | 43 | 1,2,3,4,5,6,7,8,9,10,49,50,51,52 |
| 4 | 1,2,3,4,5,6,7,8,9,10,11 | 44 | 1,2,4,5,6,7,8,9,10 |
| 5 | 1,2,3,10,15,16,17,18,19,20,21,22 | 45 | 1,2,3,4,5,6,7,8,9,10 |
| 6 | 1,2,10,14,15,16,17,18,19,20,23,24,25,26 | 46 | 1,2,3,4,5,6,7,8,9,10,49,50 |
| 7 | 1,2,4,5,6,7,8,9,10,27,28,29,30,31 | 47 | 1,2,3,4,5,6,7,8,9,10 |
| 8 | 1,4,5,6,7,8,9,10,28,32,33 | 48 | 1,2,3,4,5,6,7,8,9,10 |
| 9 | 1,2,3,10,15,16,17,18,19,20,23 | 49 | 1,4,5,6,7,8,9,10,53 |
| 10 | 1,2,3,10,15,16,17,18,19,20,23 | 50 | 1,2,4,5,6,7,8,9,10,40 |
| 11 | 1,2,3,10,15,16,17,18,19,20,23 | 51 | 1,2,3,4,5,6,7,8,9,10,42 |
| 12 | 1,2,3,4,5,6,7,8,9,10,28,29,31 | 52 | 1,2,3,4,5,6,7,8,9,10,28,35,40 |
| 13 | 1,2,3,10,15,16,17,18,19,20,23 | 53 | 1,2,3,4,5,6,7,8,9,10,28,35,42 |
| 14 | 1,10,15,16,17,18,19,20,23,26,32,34 | 54 | 1,4,5,6,7,8,9,10,40,53 |
| 15 | 1,2,3,10,15,16,17,18,19,20,21,23 | 55 | 1,4,5,6,7,8,9,10,28,35,40,53 |
| 16 | 1,2,3,4,5,6,7,8,9,10,28,35 | 56 | 1,4,5,6,7,8,9,10,42,54 |
| 17 | 1,2,3,4,5,6,7,8,9,10,28,35 | 57 | 1,4,5,6,7,8,9,10,28,35,42,55 |
| 18 | 1,2,3,4,5,6,7,8,9,10,28,29,31,36 | 58 | 1,2,3,4,5,6,7,8,9,10,40,42 |
| 19 | 1,2,3,4,5,6,7,8,9,10,35,37,38 | 59 | 1,4,5,6,7,8,9,10,40,42,53 |
| 20 | 1,2,10,15,16,17,18,19,20,23,27,39 | 60 | 1,2,3,4,5,6,7,8,9,10,40,42 |
| 21 | 1,2,3,4,5,6,7,8,9,10,40,41 | 61 | 1,4,5,6,7,8,9,10,40,42,56 |
| 22 | 1,4,5,6,7,8,9,10,32,34,42 | 62 | 1,4,5,6,7,8,9,10 |
| 23 | 1,4,5,6,7,8,9,10,32,34,42,43 | 63 | 1,2,3,4,5,6,7,8,9,10,49 |
| 24 | 1,10,15,16,17,18,19,20,32,34,40,42 | 64 | 1,4,5,6,7,8,9,10 |
| 25 | 1,4,5,6,7,8,9,10,28,32,34,35,40 | 65 | 1,2,3,4,5,6,7,8,9,10 |
| 26 | 1,4,5,6,7,8,9,10,28,32,34,35,40,42 | 66 | 1,2,4,5,6,7,8,9,10,28,35,40 |
| 27 | 1,4,5,6,7,8,9,10,28,32,34,35,40 | 67 | 1,2,4,5,6,7,8,9,10,40,57 |
| 28 | 1,2,3,4,5,6,7,8,9,10,40 | 68 | 4,5,6,7,8,9,10,40,58 |
| 29 | 1,2,3,4,5,6,7,8,9,10,40,41 | 69 | 1,4,5,6,7,8,9,10,40,59 |
| 30 | 1,2,3,4,5,6,7,8,9,10,40,42 | 70 | 1,4,5,6,7,8,9,10,40,60 |
| 31 | 1,2,3,4,5,6,7,8,9,10,28,35,40 | 71 | 1,4,5,6,7,8,9,10,41,61 |
| 32 | 1,2,3,4,5,6,7,8,9,10,28,35,40,42 | 72 | 1,2,4,5,6,7,8,9,10,40,62 |
| 33 | 1,2,4,5,6,7,8,9,10,27,28,35,40 | 73 | 1,4,5,6,7,8,9,10,28,35,42,57 |
| 34 | 1,4,5,6,7,8,9,10,32,34,44 | 74 | 1,2,4,5,6,7,8,9,10,40 |
| 35 | 1,4,5,6,7,8,9,10,32,33 | 75 | 4,5,6,7,8,9,10,40,58 |
| 36 | 1,4,5,6,7,8,9,10,32,34 | 76 | 1,4,5,6,7,8,9,10,42,59 |
| 37 | 1,2,10,14,15,16,17,18,19,20 | 77 | 1,2,4,5,6,7,8,9,10,40,63 |
| 38 | 1,2,3,4,5,6,7,8,9,10,45 | 78 | 1,2,3,4,5,6,7,8,9,10,40,45 |
| 39 | 1,2,3,4,5,6,7,8,9,10,45,46 | 79 | 1,2,4,5,6,7,8,9,10,40,45 |
| 40 | 1,2,4,5,6,7,8,9,10,27,47 |

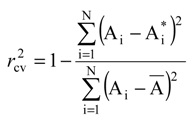
 value, approximate the percent from the (experimental) biological activity variance around the average A that can be predicted with the help of the correlation equation (4) and of the optimized chart.
value, approximate the percent from the (experimental) biological activity variance around the average A that can be predicted with the help of the correlation equation (4) and of the optimized chart. 4. Results and Discussions
- the mono-parametric model of hidrophobicity:
- the case of linear mono-parametric model where MTD is the steric parameter:
- and the combined correlation
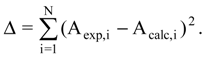
| i/Li | logP | MTD* | MTD*logP | AlogP | AMTD | AMTD+LogP | Aexp |
|---|---|---|---|---|---|---|---|
| 1 | 2.14 | 13 | 15 | 5.529 | 5.052 | 4.778 | 4.15 |
| 2 | 1.62 | 14 | 16 | 5.118 | 4.218 | 3.851 | 3.85 |
| 3 | 1.42 | 13 | 14 | 4.960 | 5.052 | 5.101 | 4.72 |
| 4 | 2.14 | 13 | 15 | 5.529 | 5.052 | 4.778 | 5.59 |
| 5 | 2.53 | 12 | 14 | 5.837 | 5.886 | 5.643 | 5.57 |
| 6 | 3.3 | 13 | 16 | 6.445 | 5.052 | 4.670 | 4.92 |
| 7 | 2.55 | 14 | 16 | 5.853 | 4.218 | 4.304 | 4.35 |
| 8 | 1.81 | 12 | 13 | 5.268 | 5.886 | 5.965 | 5.48 |
| 9 | 2.19 | 13 | 15 | 5.568 | 5.052 | 4.803 | 4.89 |
| 10 | 2.46 | 13 | 15 | 5.781 | 5.052 | 4.934 | 5.24 |
| 11 | 2.93 | 13 | 15 | 6.153 | 5.052 | 5.164 | 5,00 |
| 12 | 1.62 | 13 | 15 | 5.118 | 5.052 | 4.525 | 4.47 |
| 13 | 1.39 | 13 | 15 | 4.936 | 5.052 | 4.412 | 4.09 |
| 14 | 1.42 | 14 | 15 | 4.960 | 4.218 | 4.427 | 4.66 |
| 15 | 2.61 | 12 | 14 | 5.900 | 5.886 | 5.682 | 6.59 |
| 16 | 2.71 | 11 | 13 | 5.979 | 6.720 | 6.404 | 5.89 |
| 17 | 3.25 | 11 | 13 | 6.406 | 6.720 | 6.668 | 6.66 |
| 18 | 1.4 | 13 | 14 | 4.944 | 5.052 | 5.091 | 5.10 |
| 19 | 0.98 | 13 | 14 | 4.612 | 5.052 | 4.886 | 5.14 |
| 20 | 1.71 | 13 | 14 | 5.189 | 5.052 | 5.242 | 5,00 |
| 21 | 2.25 | 12 | 14 | 5.616 | 5.886 | 5.506 | 5.60 |
| 22 | 2.72 | 11 | 13 | 5.987 | 6.720 | 6.409 | 6.96 |
| 23 | 3.11 | 12 | 14 | 6.295 | 5.886 | 5.926 | 5,00 |
| 24 | 3.05 | 10 | 12 | 6.248 | 7.554 | 7.244 | 7.23 |
| 25 | 3.65 | 10 | 12 | 6.722 | 7.554 | 7.537 | 8.11 |
| 26 | 3.98 | 9 | 11 | 6.982 | 8.388 | 8.372 | 8.30 |
| 27 | 3.75 | 10 | 12 | 6.801 | 7.554 | 7.586 | 7.37 |
| 28 | 2.07 | 11 | 13 | 5.473 | 6.720 | 6.092 | 6.92 |
| 29 | 2.46 | 12 | 14 | 5.781 | 5.886 | 5.608 | 5.47 |
| 30 | 2.4 | 10 | 12 | 5.734 | 7.554 | 6.927 | 7.20 |
| 31 | 3 | 10 | 12 | 6.208 | 7.554 | 7.220 | 7.89 |
| 32 | 3.33 | 9 | 11 | 6.469 | 8.388 | 8.055 | 8.57 |
| 33 | 3.1 | 10 | 12 | 6.287 | 7.554 | 7.269 | 7.85 |
| 34 | 2.14 | 13 | 15 | 5.529 | 5.052 | 4.778 | 3.66 |
| 35 | 1.67 | 13 | 14 | 5.157 | 5.052 | 5.223 | 5.15 |
| 36 | 2.32 | 12 | 14 | 5.671 | 5.886 | 5.540 | 6.01 |
| 37 | 2.03 | 12 | 13 | 5.442 | 5.886 | 6.073 | 5.44 |
| 38 | 1.85 | 12 | 14 | 5.300 | 5.886 | 5.311 | 5.69 |
| 39 | 3.42 | 13 | 15 | 6.540 | 5.052 | 5.403 | 5.22 |
| 40 | 3.28 | 13 | 15 | 6.429 | 5.052 | 5.335 | 4.37 |
| 41 | 1.95 | 13 | 14 | 5.379 | 5.052 | 5.360 | 5.06 |
| 42 | 1.8 | 12 | 14 | 5.260 | 5.886 | 5.286 | 5.17 |
| 43 | 3.71 | 13 | 15 | 6.769 | 5.052 | 5.544 | 5.12 |
| 44 | 2.46 | 12 | 14 | 5.781 | 5.886 | 5.608 | 6.48 |
| 45 | 2.82 | 12 | 14 | 6.066 | 5.886 | 5.784 | 5.82 |
| 46 | 3.15 | 12 | 14 | 6.327 | 5.886 | 5.945 | 5.24 |
| 47 | 2.27 | 12 | 14 | 5.631 | 5.886 | 5.516 | 5.96 |
| 48 | 2.93 | 12 | 14 | 6.153 | 5.886 | 5.838 | 5.48 |
| 49 | 3.89 | 11 | 14 | 6.911 | 6.720 | 6.306 | 7.06 |
| 50 | 2.85 | 9 | 11 | 6.090 | 8.388 | 7.821 | 7.72 |
| 51 | 3.5 | 9 | 11 | 6.603 | 8.388 | 8.138 | 7.58 |
| 52 | 3.79 | 10 | 12 | 6.832 | 7.554 | 7.606 | 8.24 |
| 53 | 4.44 | 9 | 11 | 7.346 | 8.388 | 8.597 | 8.30 |
| 54 | 4.29 | 10 | 12 | 7.227 | 7.554 | 7.850 | 8.23 |
| 55 | 5.22 | 9 | 11 | 7.962 | 8.388 | 8.977 | 8.55 |
| 56 | 4.94 | 10 | 12 | 7.741 | 7.554 | 8.167 | 8.09 |
| 57 | 5.87 | 9 | 13 | 8.475 | 8.388 | 7.947 | 8.14 |
| 58 | 3.18 | 12 | 14 | 6.350 | 5.886 | 5.960 | 7.99 |
| 59 | 4.62 | 12 | 13 | 7.488 | 5.886 | 7.337 | 8.51 |
| 60 | 3.83 | 12 | 14 | 6.864 | 5.886 | 6.277 | 7.89 |
| 61 | 5.27 | 12 | 14 | 8.001 | 5.886 | 6.980 | 8.14 |
| 62 | 2.11 | 10 | 12 | 5.505 | 7.554 | 6.786 | 5.68 |
| 63 | 3.32 | 12 | 14 | 6.461 | 5.886 | 6.028 | 5.33 |
| 64 | 2.11 | 11 | 13 | 5.505 | 6.720 | 6.112 | 5.66 |
| 65 | 2.93 | 11 | 14 | 6.153 | 6.720 | 5.838 | 5.92 |
| 66 | 4.54 | 10 | 12 | 7.425 | 7.554 | 7.972 | 7.89 |
| 67 | 3.91 | 12 | 14 | 6.927 | 5.886 | 6.316 | 6.66 |
| 68 | 4.74 | 12 | 14 | 7.583 | 5.886 | 6.721 | 5.79 |
| 69 | 5.06 | 11 | 13 | 7.835 | 6.720 | 7.551 | 6.45 |
| 70 | 5.4 | 11 | 13 | 8.104 | 6.720 | 7.717 | 7.11 |
| 71 | 5.45 | 11 | 13 | 8.144 | 6.720 | 7.742 | 7.92 |
| 72 | 5.19 | 11 | 14 | 7.938 | 6.720 | 6.941 | 7.04 |
| 73 | 3.89 | 10 | 12 | 6.911 | 7.554 | 7.654 | 8.13 |
| 74 | 3.27 | 11 | 13 | 6.421 | 6.720 | 6.678 | 6.47 |
| 75 | 4.1 | 11 | 13 | 7.077 | 6.720 | 7.083 | 5.40 |
| 76 | 4.41 | 11 | 13 | 7.322 | 6.720 | 7.234 | 6.35 |
| 77 | 4.17 | 11 | 13 | 7.132 | 6.720 | 7.117 | 7.02 |
| 78 | 3.33 | 11 | 13 | 6.469 | 6.720 | 6.707 | 7.02 |
| 79 | 2.68 | 11 | 13 | 5.955 | 6.720 | 6.390 | 7.00 |
(N = 79; r = 0.673; F = 63.85)
(N = 79; r = 0.830; F = 169.9; r2cv = 0.685)

(N = 79; r = 0.882; F = 133.2; r2cv = 0.774)

5. Conclusions
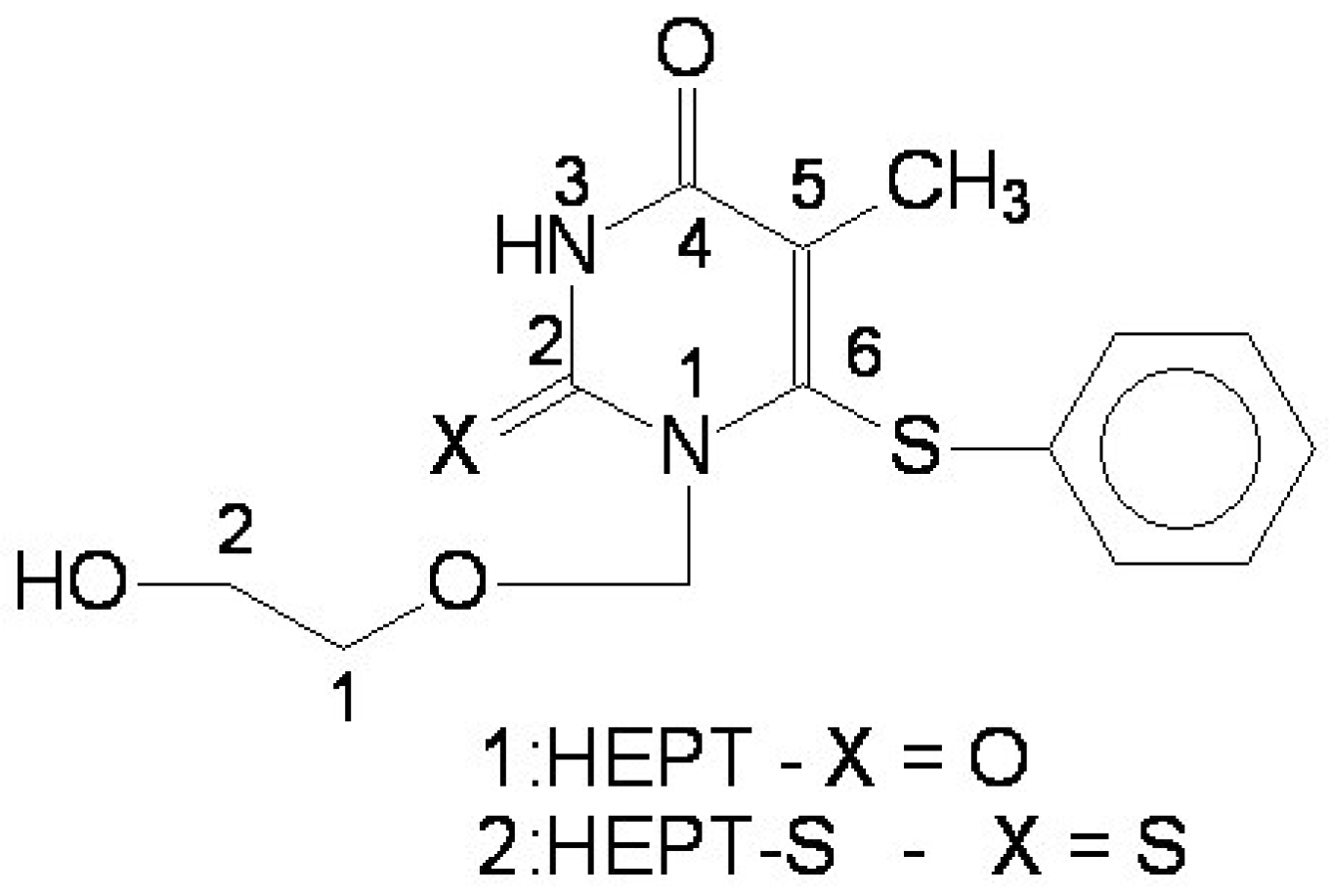
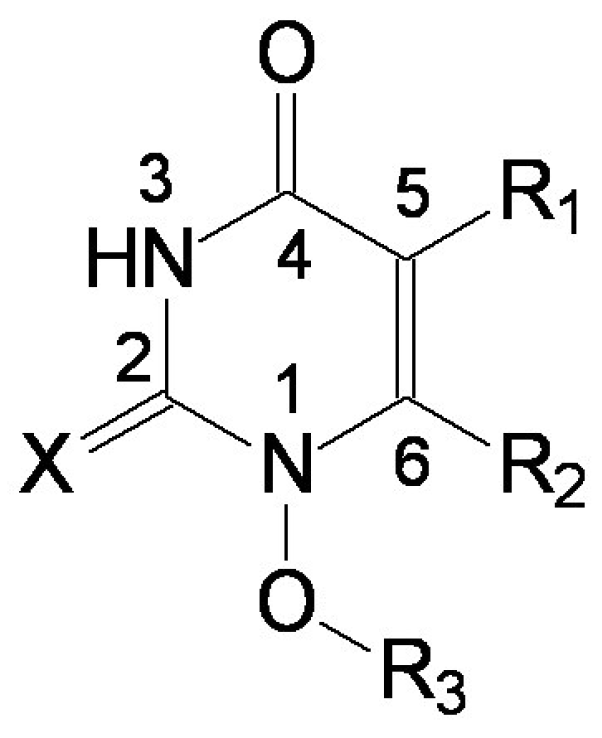
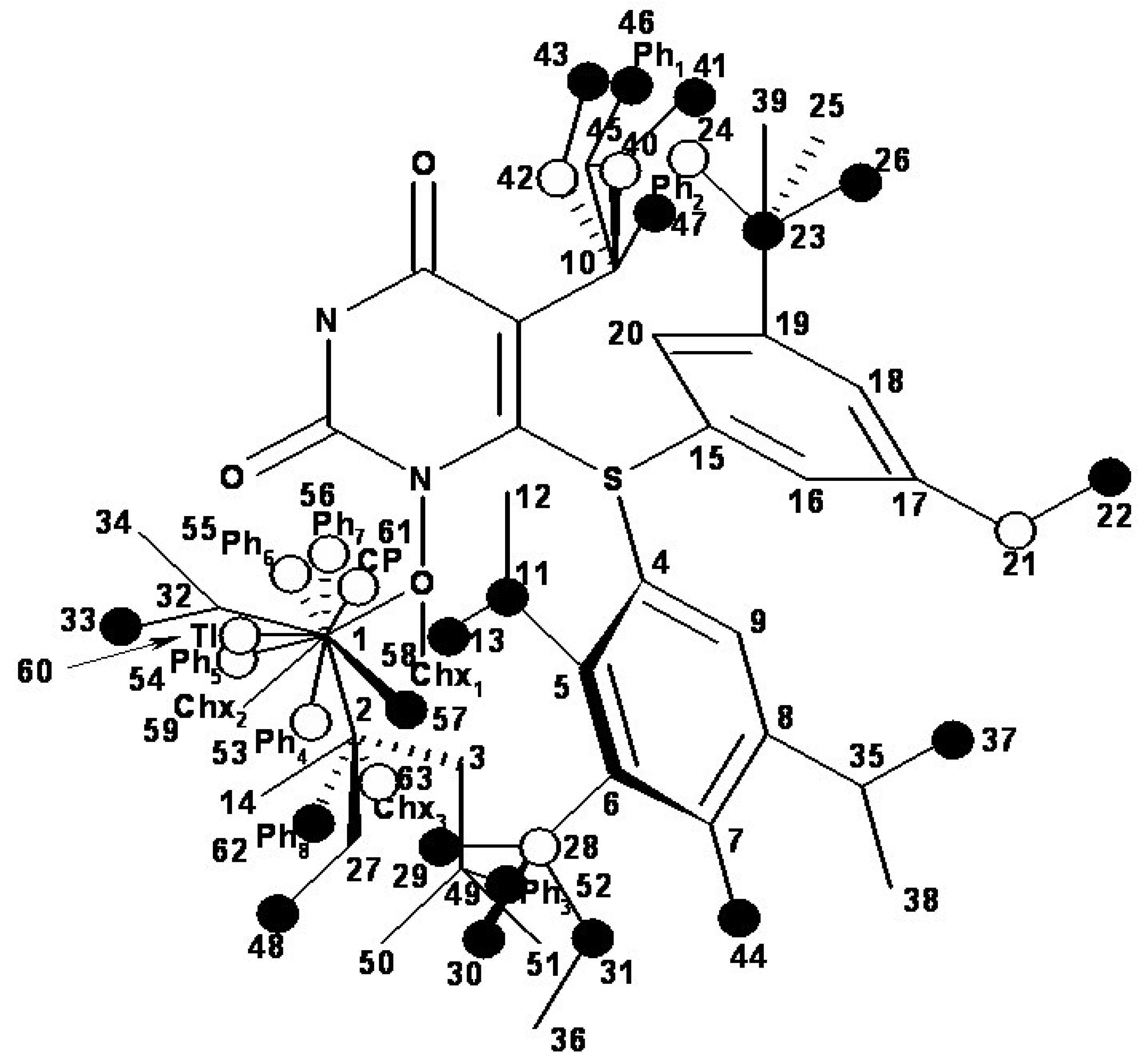
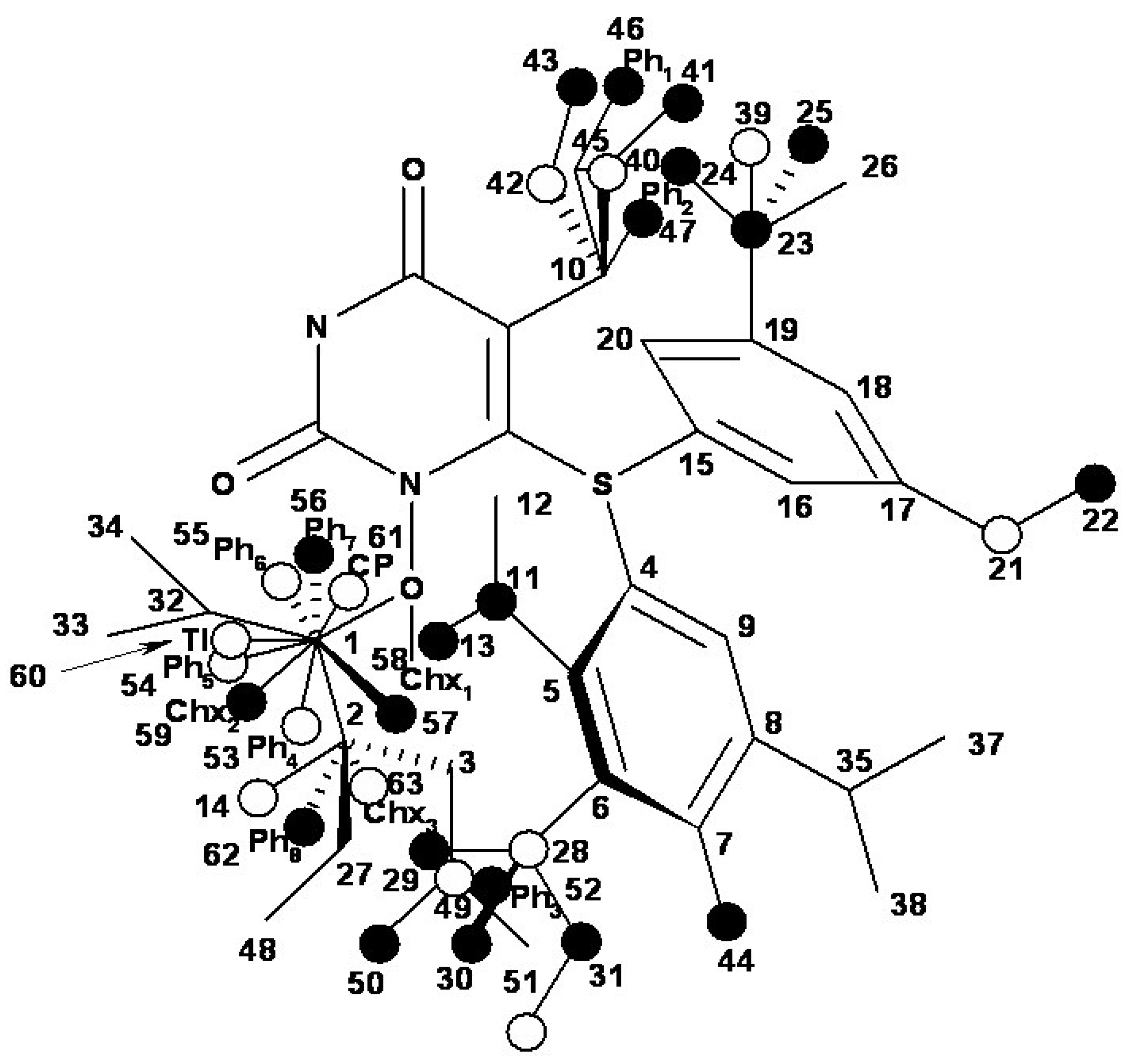
- the region of R1 substitute (see Table I) in the common pirymidin-2,4(1H,3H)-dione structure (corresponding to position 5 on this structure) with generally substitutes of small volume (alkyl, arylalkyl);
- the region of R2 substitute in the common structure (position 6) with phenyl-thyo rests un-substituted or substituted;
- the region of R3 substitute in the common structure (position N-1 in the primary structure of Figure 3) with the highest structural variation.
Acknowledgements
References
- Clavel, F.; Guyader, M.; Guetard, M.; Salle, M.; Montagnier, L.; Alizon, M. Molecular cloning and polymorphism of the human immune deficiency virus type 2. Nature 1986, 324, 691–695. [Google Scholar] [CrossRef]
- Guyader, M.; Emerman, M.; Sonigo, P.; Clavel, F.; Montagnier, M.; Alizon, M. Genome organization and transactivation of the human immunodeficiency virus type 2. Nature 1987, 326, 662–669. [Google Scholar] [CrossRef]
- Fauci, A.S. The human immunodeficiency virus: infectivity and mechanisms of pathogenesis. Science 1988, 239, 617–622. [Google Scholar]
- Barre-Sinoussi, F.; Chermann, J.-C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; Rozenbaum, W.; Montagnier, L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar]
- Popovic, M.; Sarngadharan, M.G.; Read, E.; Gallo, R.C. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 1984, 224, 497–500. [Google Scholar]Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; White, G.; Foster, P.; Markham, P.D. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar]
- Fauci, A.S.; Lane, H.C. Human Immunodeficiency virus disease: AIDS and related disorders. In Harrison’s Principles of Internal Medicine, 16th ed.; Kasper, D. L., Braunwald, E., Fauci, A. S., Hauser, S. L., Longo, D. L., Jameson, J. L., Eds.; McGraw-Hill Companies Inc., 2005; pp. 1076–1139. [Google Scholar]
- De Clercq, E. HIV-1-specific RT inhibitors: highly selective inhibitors of human immunodeficiency virus type 1 that are specifically targeted at the viral reverse transcriptase. Med. Res. Rev. 1993, 13, 229–258. [Google Scholar] [CrossRef]
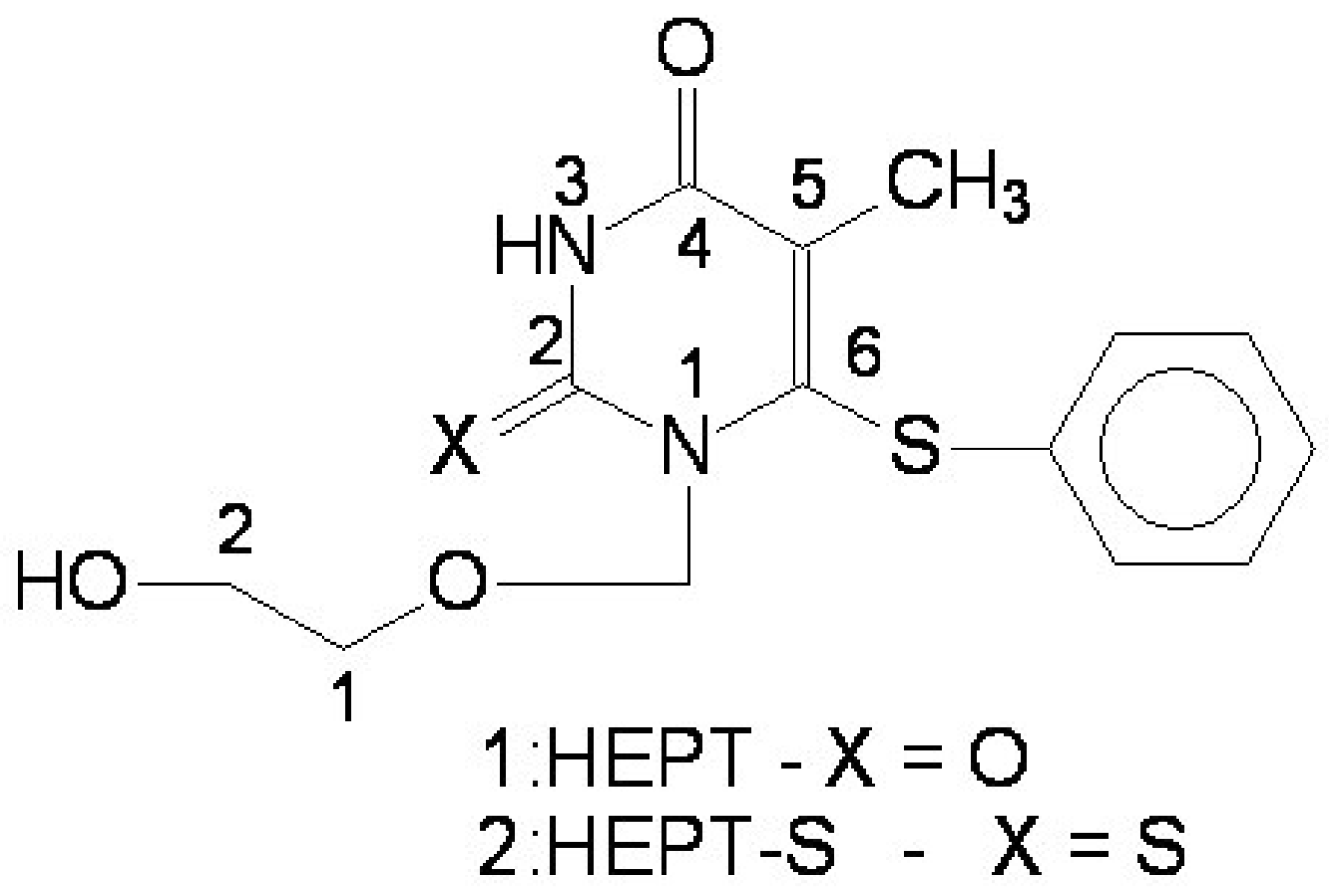
- Miyasaka, T.; Tanaka, H.; Baba, M.; Hayakawa, H.; Walker, R.T.; Balzarini, J.; De Clercq, E. A novel lead for specific anti-HIV-1 agents: 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine. J. Med. Chem. 1989, 32, 2507–2509. [Google Scholar]Baba, M.; Tanaka, H.; De Clercq, E.; Pauwels, R.; Balzarini, J.; Schols, D.; Nakashima, H.; Perno, C.F.; Walker, R.T.; Miyasaka, T. Highly specific inhibition of human immunodeficiency virus type 1 by a novel 6-substituted acyclouridine derivative. Biochem. Biophys. Res. Commun. 1989, 165, 1375–1381. [Google Scholar]
- Tanaka, H.; Hirayama, M.; Suzuki, M.; Miyasaka, T.; Matsuda, A.; Ueda, T. A lithiation route to c-5 substitution of an imidazole nucleoside and its application to the synthesis of 3-deazaguanosine. Tetrahedron 1986, 42, 1971–1980. [Google Scholar] [CrossRef]
- Tanaka, H.; Takashima, H.; Ubasawa, M.; Sekiya, K.; Nitta, I.; Baba, M.; Shigeta, S.; Walker, R.T.; De Clerq, E. Structure-activity relationships of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine analogues: effect of substitutions at the C-6 phenyl ring and at the C-5 position on anti-HIV-1 activity. J. Med. Chem. 1992, 35, 337–345. [Google Scholar]
- Balaban, A. T.; Chiriac, A.; Motoc, I; Simon, Z. Steric Fit in QSAR; Springer: Berlin, (Lecture Notes in Chemistry Series); 1980. [Google Scholar]
- Simon, Z; Chiriac, A.; Holban, S.; Ciubotariu, D.; Mihalas, G. I. Minimum Steric Difference. The MTD Method for QSAR Studies; Res. Studies Press (Wiley): Letchworth, 1984. [Google Scholar]
- Ciubotariu, D.; Deretey, E.; Oprea, T. I.; Sulea, T.; Simon, Z.; Kurunczi, L.; Chiriac, A. Multiconformational Minimal Steric Difference. Structure-Acetylcholinesterase Hydrolysis Rates Relations for Acetic Acid Esters. Quant. Struct. Act. Relat. 1993, 12, 367–372. [Google Scholar] [CrossRef]
- Simon, Z.; Chiriac, A.; Ciubotariu, D.; Mureşan, S.; Bologa, C.; Sulea, T.; Kurunczi, L. Metoda MTD (The MTD method). In Relaţii cantitative structură chimică – activitate biologică (QSAR). Metoda MTD (Quantitative chemical structure – biological activity relationships studies (QSAR). The MTD method); Chiriac, A., Ciubotariu, D., Simon, Z., Eds.; Mirton Publishing House: Timişoara, Romania, 1996; Chapter 5. [Google Scholar]
- Ciubotariu, D.; Gogonea, V.; Medeleanu, M. QSAR Studies by Molecular Descriptions; Diudea, M. V., Ed.; Nova Science Publ. Inc.: Huntington New York, 2001; Chapter 10. [Google Scholar]
- Ciubotariu, D.; Medeleanu, M.; Vlaia, V.; Olariu, T.; Ciubotariu, C.; Dragoş, D.; Seiman, C. Molecular van der Waals Space and Topological Indices from the Distance Matrix. Molecules 2004, 9, 1053–1078. [Google Scholar]
- Duda-Seiman, C. Doctoral Thesis: QSAR Studies of Pyrimidinic Compounds with Anti-HIV Activity; West University of Timişoara, 2005. [Google Scholar]
- Chiriac, A; Ciubotariu, D.; Funar-Timoftei, S.; Kurunczi, L.; Mracec, M.; Mracec, M.; Szabadai, Z.; Seclaman, E.; Simon, Z. QSAR and 3D-QSAR in Timişoara. 1972-2005. Rev. Roum. Chim. 2006, 51, 79–99. [Google Scholar]
- Duda-Seiman, C.; Duda-Seiman, D.; Hegheş, A.; Nuţiu, R.; Ciubotariu, D.; Suceveanu, N. Modelarea compuSilor pirimidinici cu activitate anti-HIV (Molecular modeling of pyrimidinic compounds with anti-HIV activity). Revista de Medicină şi Farmacie (Journal of Medicine and Pharmacy ) 2004, 50 (Supl. II), 144–149, ISSN 1221-2229. [Google Scholar]
- Tanaka, H.; Baba, M.; Ubasawa, M.; Takashima, H.; Sekiya, K.; Nitta, I.; Shigeta, S.; Walker, R.T.; De Clercq, E.; Miyasaka, T. Synthesis and anti-HIV activity of 2-, 3-, and 4-substituted analogues of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT). J. Med. Chem. 1991, 34, 1394–1399. [Google Scholar] [CrossRef]
- Tanaka, H.; Baba, M.; Saito, S.; Miyasaka, T.; Takashima, H.; Sekiya, K.; Ubasawa, M.; Nitta, I.; Walker, R.T.; Nakashima, H.; De Clercq, E. Specific anti-HIV-1 "acyclonucleosides" which cannot be phosphorylated: synthesis of some deoxy analogues of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine. J. Med. Chem. 1991, 34, 1508–1511. [Google Scholar] [CrossRef]
- HyperChem 7.01, Program package. Hypercube Inc., 2002.
- Olah, M.; Ciubotariu, D.; Ciubotariu, C.; Seiman, C.; Dragoş, D.; Pasere, M.; Medeleanu, M. Quantitative treatment of steric effects with the aid of molecular van der Waals descriptors for size and shape of substituents. In International Course & Conference on the Interface among Mathematics, Chemistry and Computer Sciences; Book of Abstracts; Ante, Graovac, Biserka, Pokrič, Vilko, Smrečky, Eds.; Inter University Centre: Dubrovnik, Croatia, June 24–29th; 2002; p. 55, ISBN 953-6690-22-5. [Google Scholar]
- Dean, P.M. Molecular Foundations of Drug-Receptor Interactions, 1st ed.; Vol. 1, Cambridge University Press: Cambridge, UK, 1987. [Google Scholar]
© 2006 by MDPI, (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Duda-Seiman, C.; Duda-Seiman, D.; Dragos, D.; Medeleanu, M.; Careja, V.; Putz, M.V.; Lacrama, A.-M.; Chiriac, A.; Nutiu, R.; Ciubotariu, D. Design of Anti-HIV Ligands by Means of Minimal Topological Difference (MTD) Method. Int. J. Mol. Sci. 2006, 7, 537-555. https://doi.org/10.3390/i7110537
Duda-Seiman C, Duda-Seiman D, Dragos D, Medeleanu M, Careja V, Putz MV, Lacrama A-M, Chiriac A, Nutiu R, Ciubotariu D. Design of Anti-HIV Ligands by Means of Minimal Topological Difference (MTD) Method. International Journal of Molecular Sciences. 2006; 7(11):537-555. https://doi.org/10.3390/i7110537
Chicago/Turabian StyleDuda-Seiman, Corina, Daniel Duda-Seiman, Dan Dragos, Mihai Medeleanu, Valentin Careja, Mihai V. Putz, Ana-Maria Lacrama, Adrian Chiriac, Remus Nutiu, and Dan Ciubotariu. 2006. "Design of Anti-HIV Ligands by Means of Minimal Topological Difference (MTD) Method" International Journal of Molecular Sciences 7, no. 11: 537-555. https://doi.org/10.3390/i7110537
APA StyleDuda-Seiman, C., Duda-Seiman, D., Dragos, D., Medeleanu, M., Careja, V., Putz, M. V., Lacrama, A.-M., Chiriac, A., Nutiu, R., & Ciubotariu, D. (2006). Design of Anti-HIV Ligands by Means of Minimal Topological Difference (MTD) Method. International Journal of Molecular Sciences, 7(11), 537-555. https://doi.org/10.3390/i7110537