1J(CH) Couplings
In
figure 1, the change ΔJ in
1J(CH) as a function of the H...O distance for system I is presented. It corresponds to the difference between the
1J(CH) value for the complex at the optimized geometry and that for the isolated NCH molecule. This change will be identified by ΔJ
C. The electric field effect is shown in the same figure. It corresponds to the difference between the
1J(CH) value calculated in the presence of the H
2O electric field for each H...O distance considering the optimized geometry of the NCH molecule in the complex and the
1J(CH) value of the isolated NCH molecule. This change will be identified as ΔJ
E. Results in
figure 1 were obtained at both RPA and SOPPA levels for the Fermi contact (FC) term of the coupling, as it has been shown previously that other terms are unimportant to define the trend of
1J(CH) couplings in C-H...O interactions [
13]. In
figure 2 the same ΔJ
C and ΔJ
E changes are shown for the CH
4 molecule in system II.
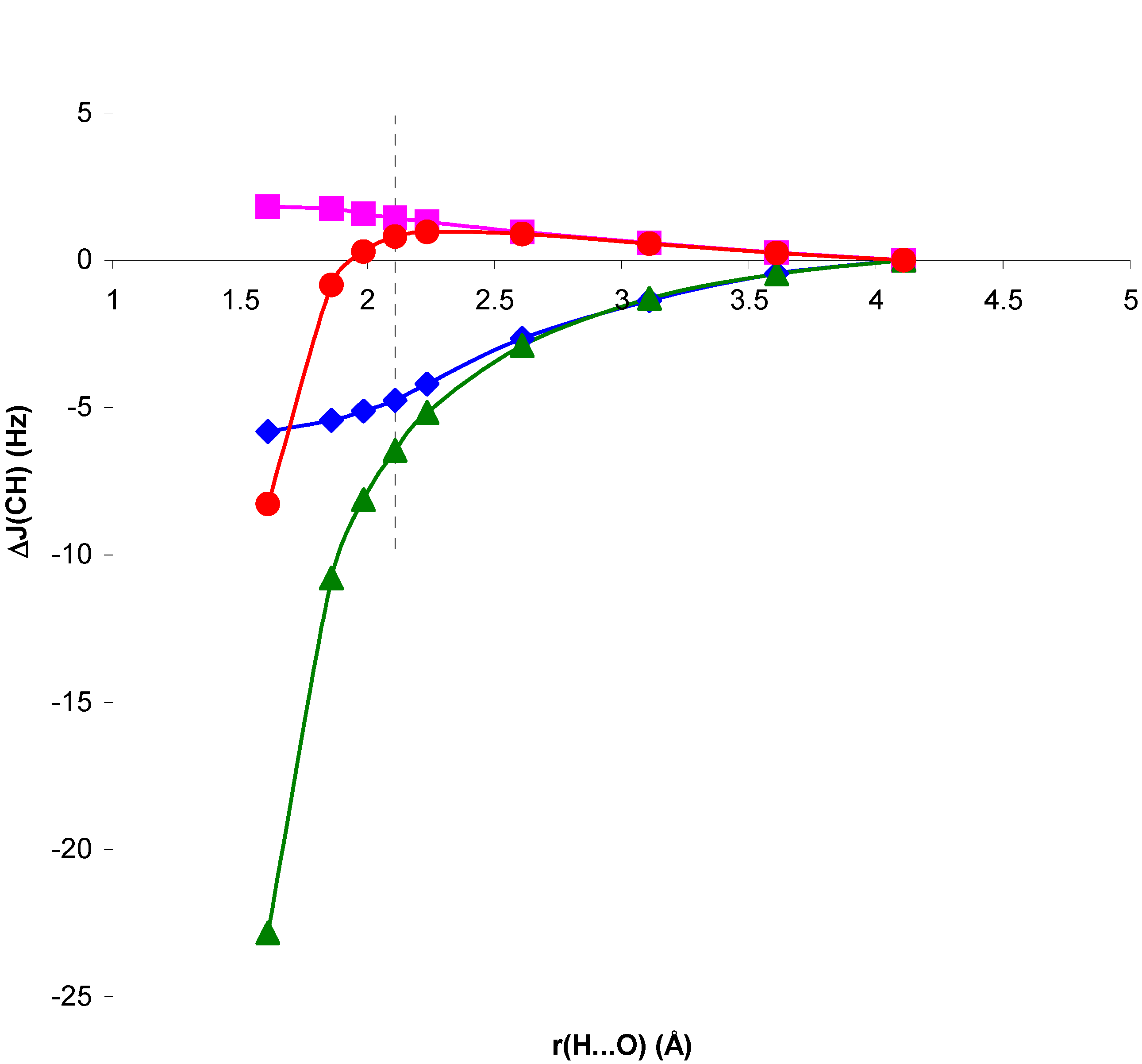
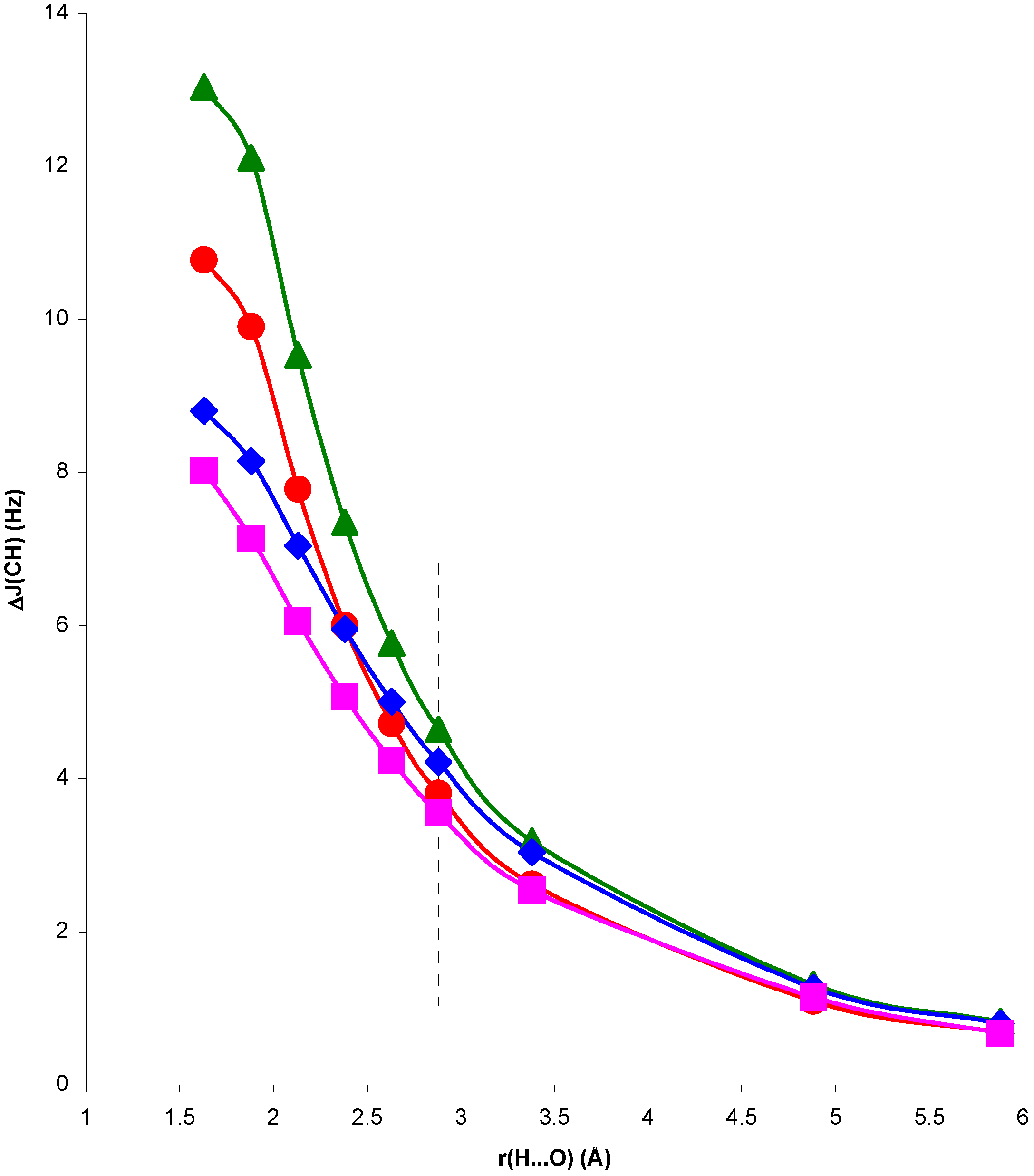
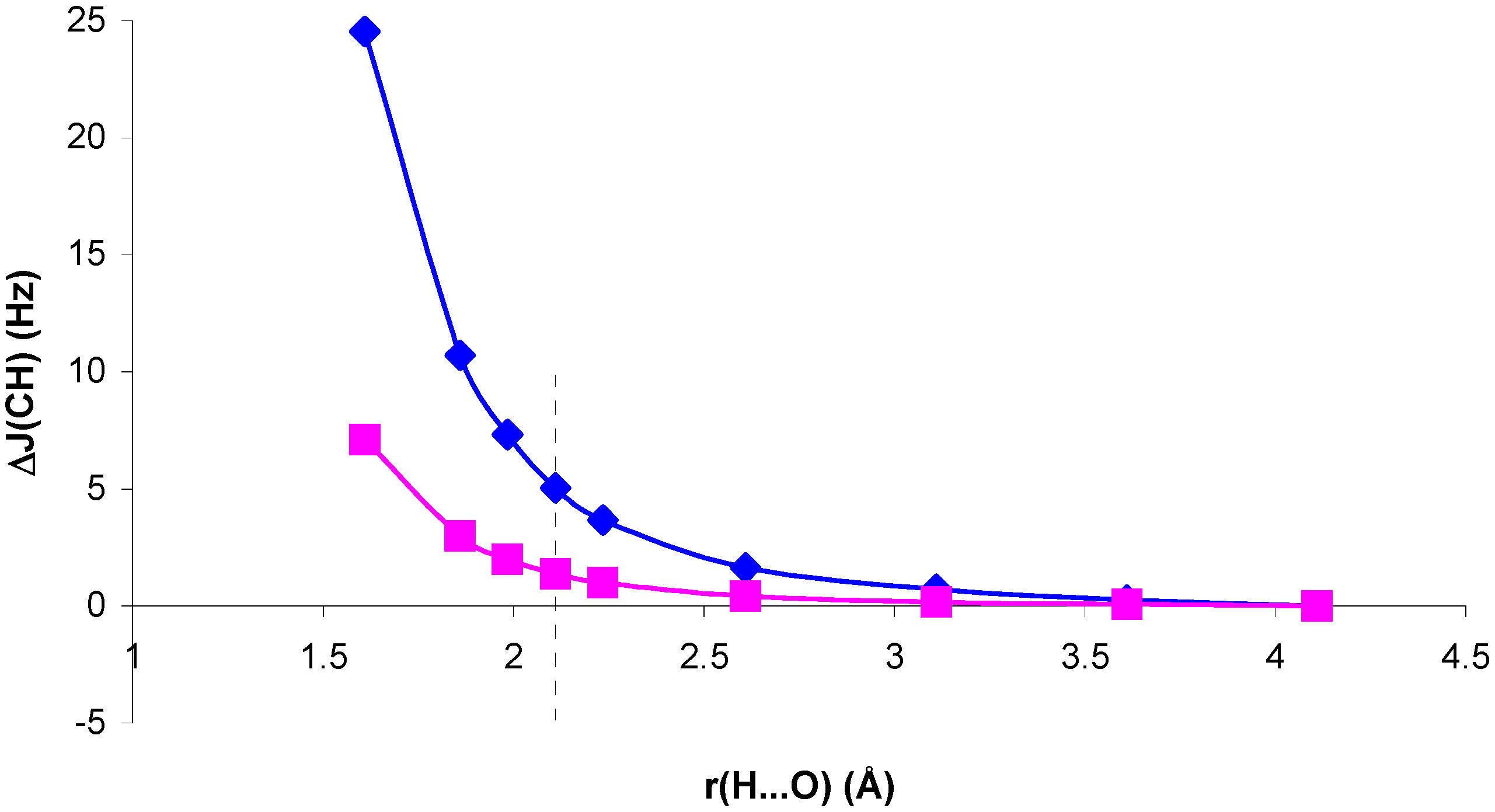
Figure 1.
Change in
1J(CH) for the NCH...OH
2 complex, ΔJ
C, and in the presence of the H
2O electric field, ΔJ
E, with respect to
1J(CH) for the isolated NCH molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
E (RPA),
![Ijms 04 00203 i003]()
ΔJ
E (SOPPA),
![Ijms 04 00203 i004]()
ΔJ
C (RPA),
![Ijms 04 00203 i005]()
ΔJ
C (SOPPA). The dotted line indicates the equilibrium distance.
Figure 1.
Change in
1J(CH) for the NCH...OH
2 complex, ΔJ
C, and in the presence of the H
2O electric field, ΔJ
E, with respect to
1J(CH) for the isolated NCH molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
E (RPA),
![Ijms 04 00203 i003]()
ΔJ
E (SOPPA),
![Ijms 04 00203 i004]()
ΔJ
C (RPA),
![Ijms 04 00203 i005]()
ΔJ
C (SOPPA). The dotted line indicates the equilibrium distance.
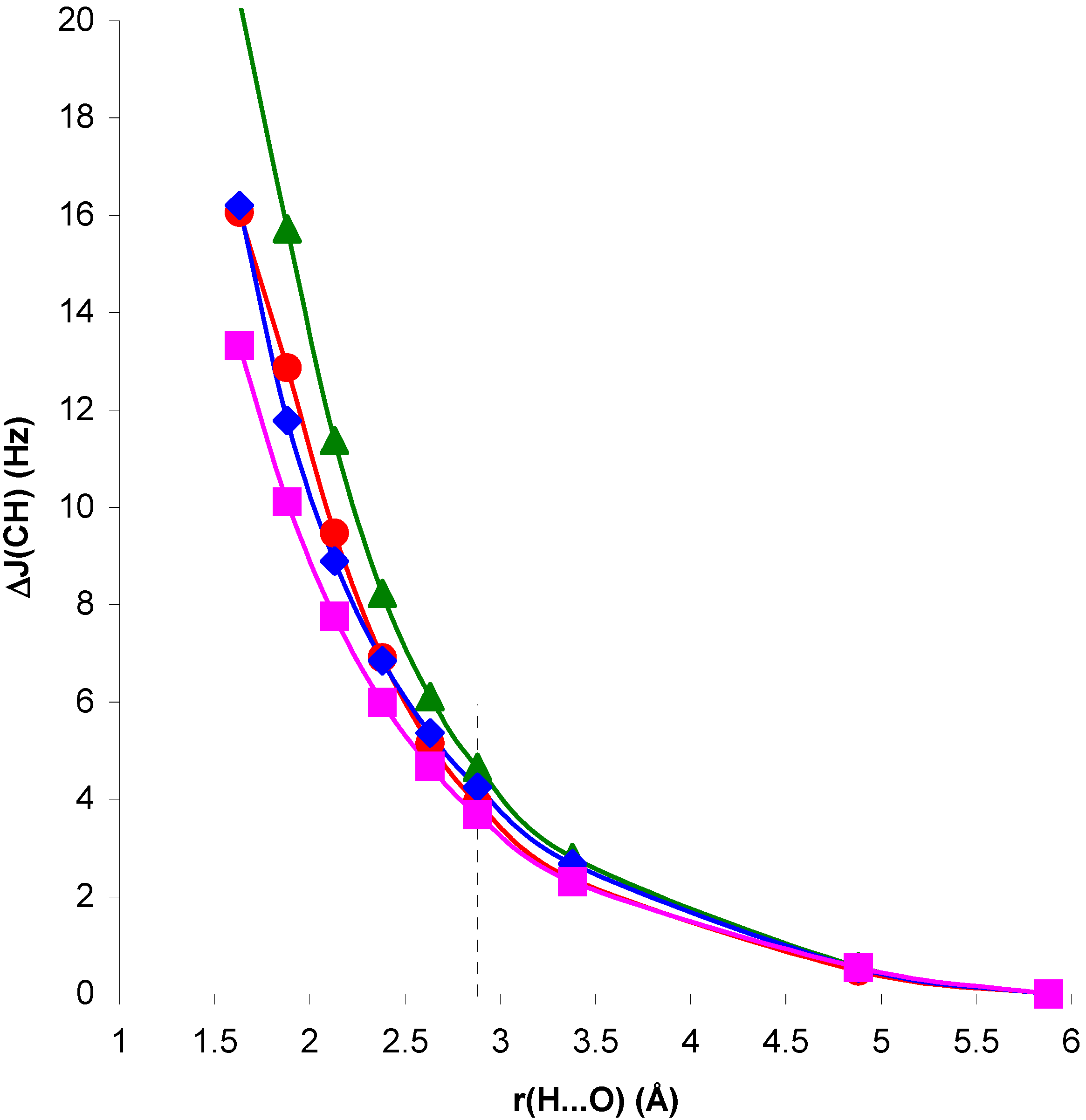
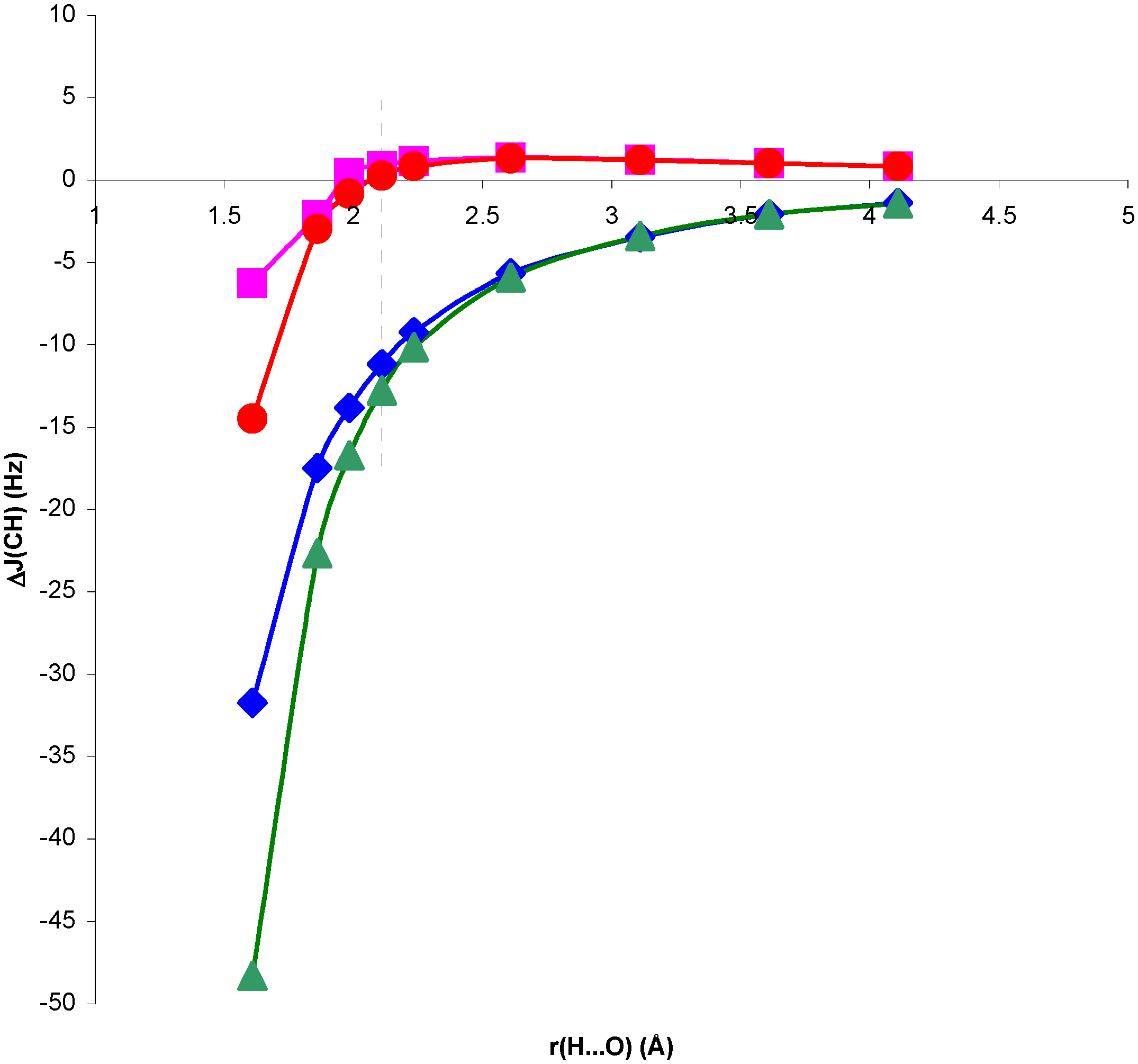
Figure 2.
Change in
1J(CH) for the CH
4...OH
2 complex, ΔJ
C, and in the presence of the H
2O electric field, ΔJ
E, with respect to
1J(CH) for the isolated CH
4 molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
E (RPA),
![Ijms 04 00203 i003]()
ΔJ
E (SOPPA),
![Ijms 04 00203 i004]()
ΔJ
C (RPA),
![Ijms 04 00203 i005]()
ΔJ
C (SOPPA). The dotted line indicates the equilibrium distance.
Figure 2.
Change in
1J(CH) for the CH
4...OH
2 complex, ΔJ
C, and in the presence of the H
2O electric field, ΔJ
E, with respect to
1J(CH) for the isolated CH
4 molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
E (RPA),
![Ijms 04 00203 i003]()
ΔJ
E (SOPPA),
![Ijms 04 00203 i004]()
ΔJ
C (RPA),
![Ijms 04 00203 i005]()
ΔJ
C (SOPPA). The dotted line indicates the equilibrium distance.
Comparison of RPA and SOPPA results in
figure 1 and
figure 2 shows the importance of correlation effects in defining ΔJ
C and ΔJ
E in systems I and II. For system II, ΔJ
C and ΔJ
E behave similarly when calculated at the RPA and SOPPA levels. SOPPA values reflect a smoother change in
1J(CH) than RPA ones as the H...O distance decreases. However, the increase obtained for H...O distances close to equilibrium is significant and measurable. Results for system I exhibit a quite different behaviour. The qualitative trend of
1J(CH) is dramatically changed when correlated values are considered for both ΔJ
C and ΔJ
E. The competition of two different effects seems to define the trend observed for SOPPA values. For H...O distances larger than equilibrium a smooth increase in
1J(CH) for decreasing H...O distances is observed. This trend is opposite to that obtained at both RPA and TDA levels [
13,
15]. For distances shorter than equilibrium an effect yielding a decrease of
1J(CH) seems to dominate, and ΔJ
C and ΔJ
E follow different trends. This is in line with previous findings for the magnetic shielding constants in systems I and II [
14]. As a result, for distances close to equilibrium the two effects nearly cancel each other and ΔJ
C and ΔJ
E exhibit small positive values. This result is in line with that found by Pecul et al. [
16] for the C
2H
2...H
2O complex. Such small change can easily be hidden by other effects in a larger molecular environment. Therefore, SOPPA results suggest that
1J(CH) can be a probe for the presence of C-H...O interactions in systems of type II, but only minor changes can be expected in systems of type I at distances close to or larger than equilibrium.
In order to deepen the previous analysis it is interesting to see the role played by geometric changes in defining ΔJ
C and ΔJ
E. To this end
1J(CH) was calculated for the isolated molecules NCH and CH
4 at the geometry they adopt upon complexation for every H...O distance. The difference between this value and that of the isolated molecule at its equilibrium geometry is identified as ΔJ
G and it is plotted as a function of the H...O distance in
figure 3 and
figure 4 for systems I and II respectively, at both RPA and SOPPA levels. The differences ΔJ
CG= ΔJ
C-ΔJ
G and ΔJ
EG=ΔJ
E-ΔJ
G reflect the complex and electric field changes in
1J(CH) at every fixed optimized geometry, respectively. The corresponding values are plotted in
figure 5 and
figure 6 for systems I and II, respectively.
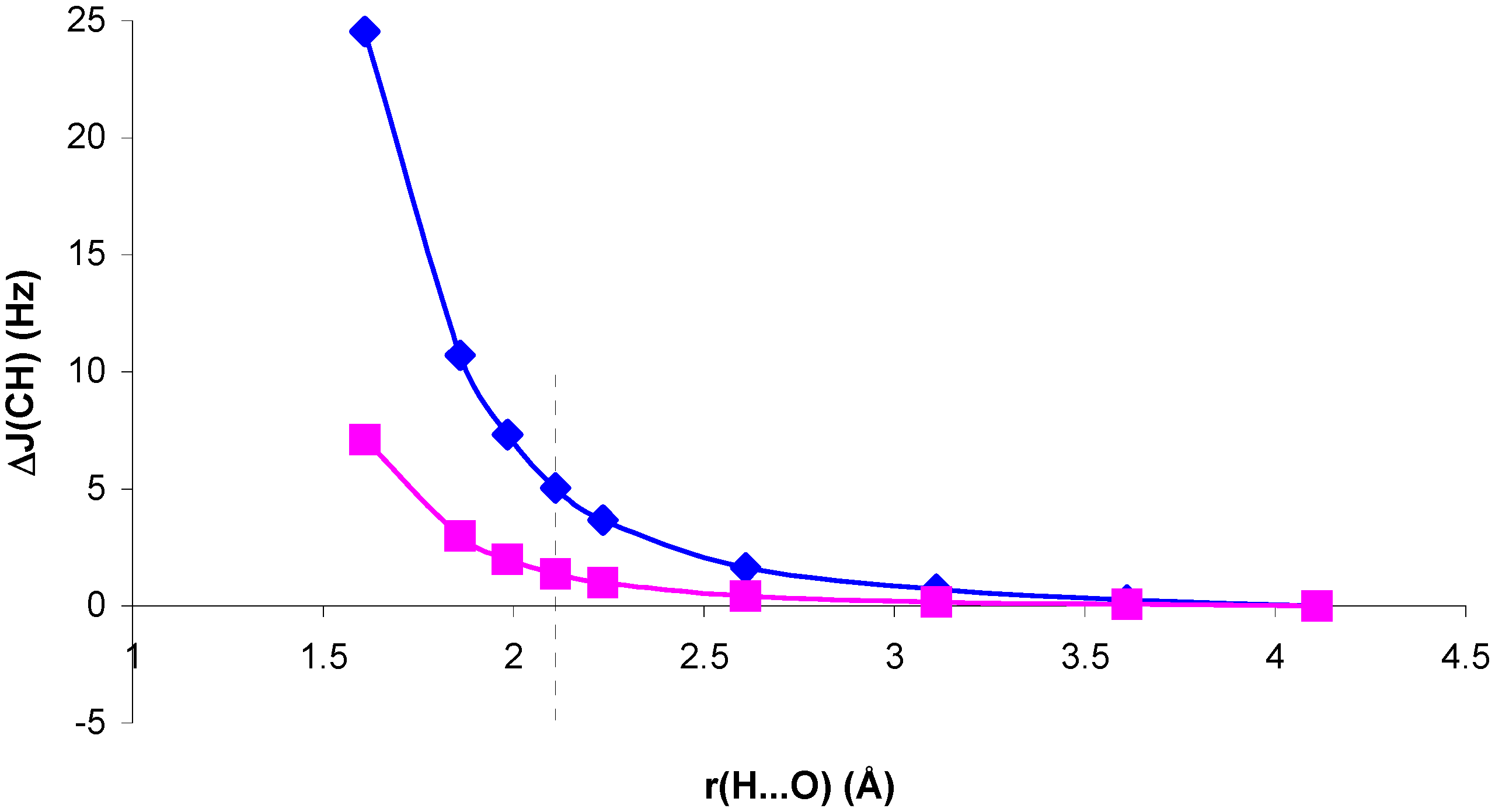
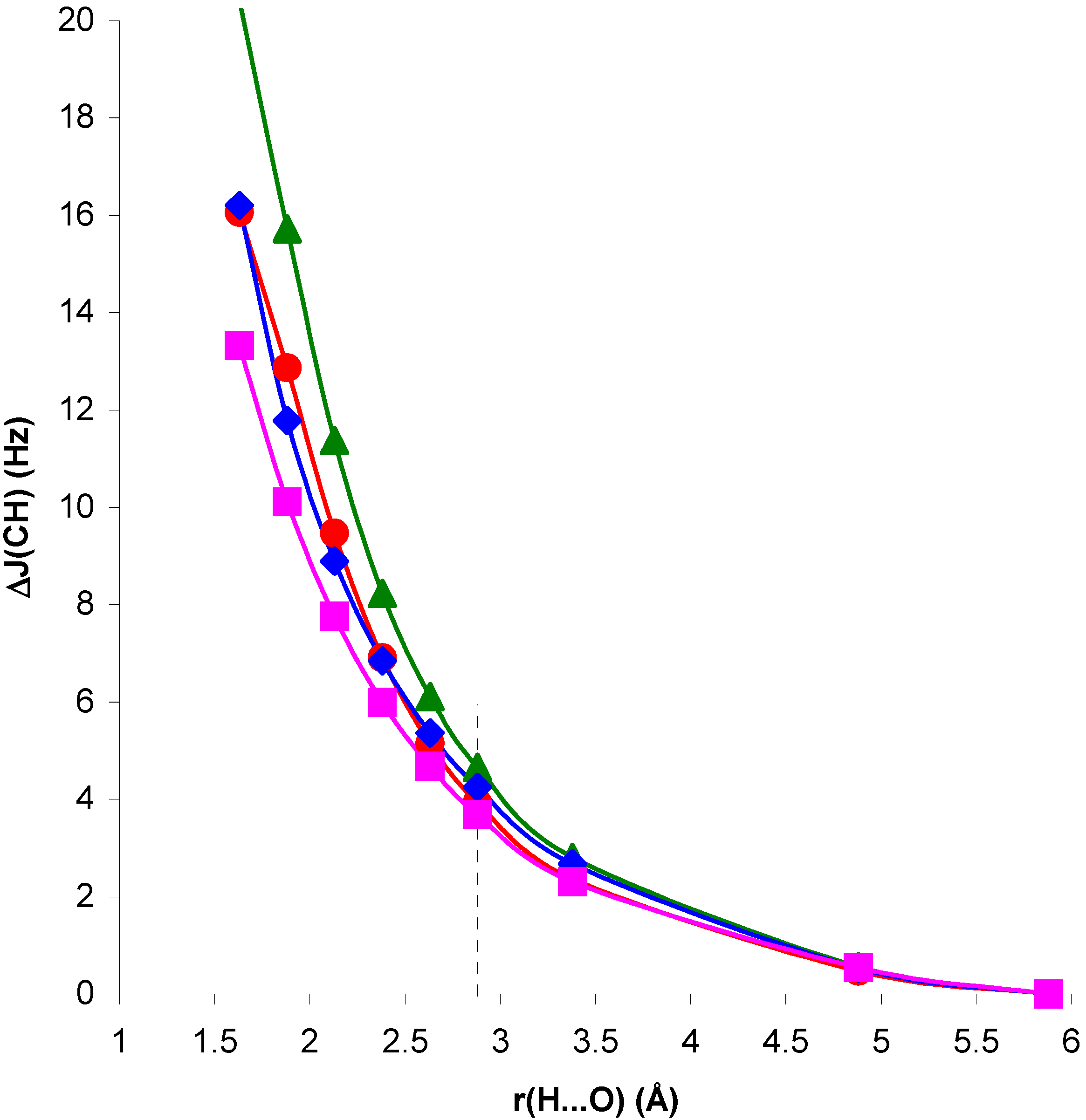
Figure 3.
Geometric change in
1J(CH) for the NCH molecule with the complex geometry, ΔJ
G, with respect to
1J(CH) for the isolated NCH molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
G (RPA),
![Ijms 04 00203 i003]()
ΔJ
G (SOPPA). The dotted line indicates the equilibrium distance.
Figure 3.
Geometric change in
1J(CH) for the NCH molecule with the complex geometry, ΔJ
G, with respect to
1J(CH) for the isolated NCH molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
G (RPA),
![Ijms 04 00203 i003]()
ΔJ
G (SOPPA). The dotted line indicates the equilibrium distance.
Figure 4.
Geometric change in
1J(CH) for the CH
4 molecule with the complex geometry, ΔJ
G, with respect to
1J(CH) for the isolated CH
4 molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
G (RPA),
![Ijms 04 00203 i003]()
ΔJ
G (SOPPA). The dotted line indicates the equilibrium distance.
Figure 4.
Geometric change in
1J(CH) for the CH
4 molecule with the complex geometry, ΔJ
G, with respect to
1J(CH) for the isolated CH
4 molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
G (RPA),
![Ijms 04 00203 i003]()
ΔJ
G (SOPPA). The dotted line indicates the equilibrium distance.
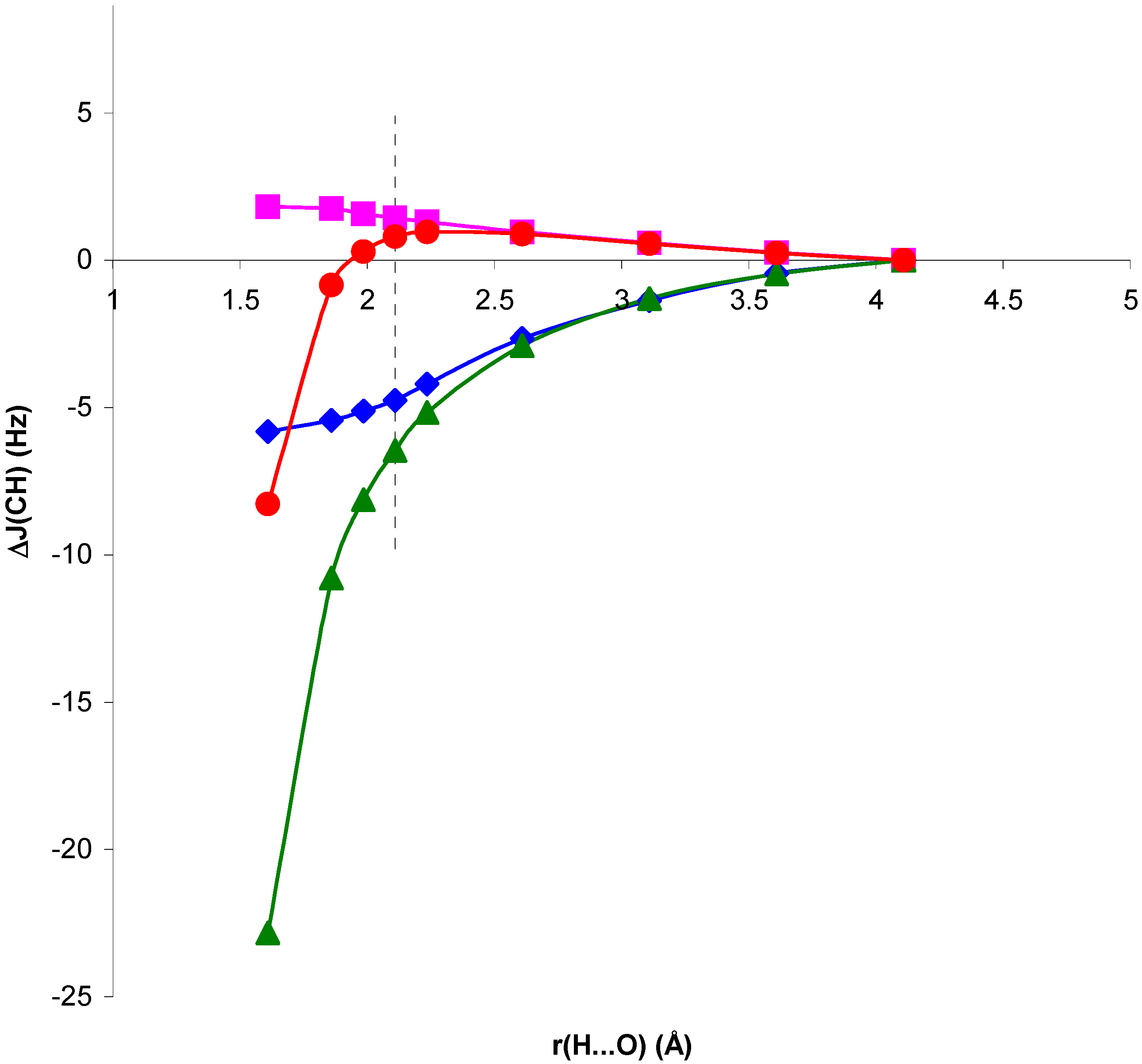
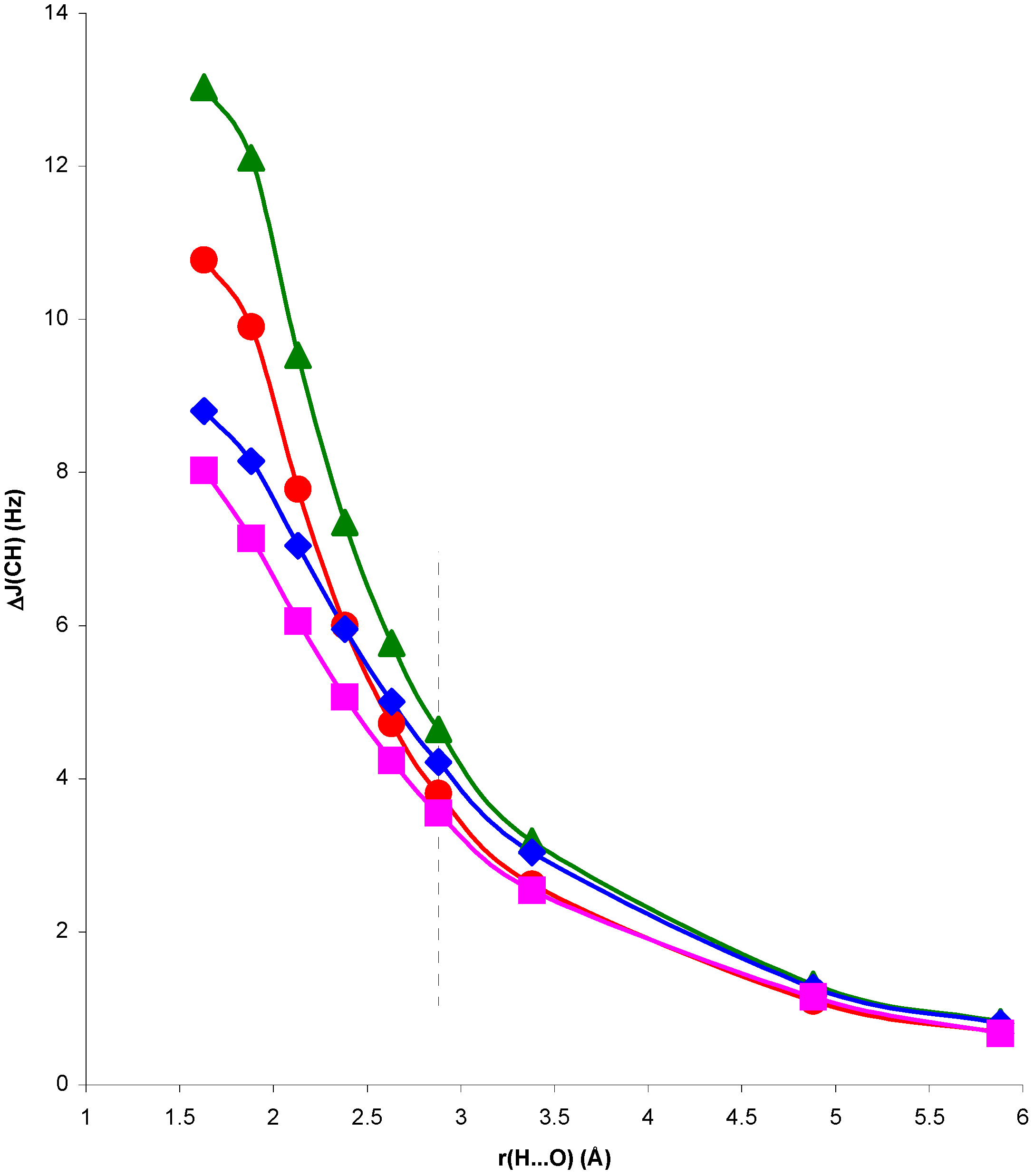
Figure 5.
Complex and electric field changes in
1J(CH) for the NCH...OH
2 complex, ΔJ
CG and ΔJ
EG respectively, at every optimized geometry, with respect to
1J(CH) for the isolated NCH molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
EG (RPA),
![Ijms 04 00203 i003]()
ΔJ
EG (SOPPA),
![Ijms 04 00203 i004]()
ΔJ
CG (RPA),
![Ijms 04 00203 i005]()
ΔJ
CG (SOPPA). The dotted line indicates the equilibrium distance.
Figure 5.
Complex and electric field changes in
1J(CH) for the NCH...OH
2 complex, ΔJ
CG and ΔJ
EG respectively, at every optimized geometry, with respect to
1J(CH) for the isolated NCH molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
EG (RPA),
![Ijms 04 00203 i003]()
ΔJ
EG (SOPPA),
![Ijms 04 00203 i004]()
ΔJ
CG (RPA),
![Ijms 04 00203 i005]()
ΔJ
CG (SOPPA). The dotted line indicates the equilibrium distance.
Figure 6.
Complex and electric field changes in
1J(CH) for the CH
4...OH
2 complex, ΔJ
CG and ΔJ
EG rspectively, at every optimized geometry, with respect to
1J(CH) for the isolated CH
4 molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
EG (RPA),
![Ijms 04 00203 i003]()
ΔJ
EG (SOPPA),
![Ijms 04 00203 i004]()
ΔJ
CG (RPA),
![Ijms 04 00203 i005]()
ΔJ
CG(SOPPA). The dotted line indicates the equilibrium distance.
Figure 6.
Complex and electric field changes in
1J(CH) for the CH
4...OH
2 complex, ΔJ
CG and ΔJ
EG rspectively, at every optimized geometry, with respect to
1J(CH) for the isolated CH
4 molecule, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i002]()
ΔJ
EG (RPA),
![Ijms 04 00203 i003]()
ΔJ
EG (SOPPA),
![Ijms 04 00203 i004]()
ΔJ
CG (RPA),
![Ijms 04 00203 i005]()
ΔJ
CG(SOPPA). The dotted line indicates the equilibrium distance.
In both cases, for distances larger than and close to equilibrium SOPPA values of ΔJ
G are very small and ΔJ
CG, ΔJ
EG follow closely the trend of ΔJ
C, ΔJ
E. For H...O distances shorter than equilibrium, in system II ΔJ
G is positive, i.e. the geometric contribution enhances the trend of ΔJ
CG and ΔJ
EG. In system I the opposite occurs: ΔJ
G is positive but ΔJ
CG and ΔJ
EG are negative. The overall trend, however, is defined by this last contribution. Therefore, the role played by polarization of the electronic system in the presence of the H
2O molecule is essential to define the trend of the
1J(CH) coupling in system I for distances shorter than equilibrium. The corresponding values seem to be largely exaggerated at the RPA level. For this reason this effect is the dominating one for the whole range of H...O distances within this approach [
13].
Comparing RPA and SOPPA results for system I the following rationalization can be carried out. In Ref.[
13], CLOPPA-IPPP decomposition of
1J(CH) into its σ and π-transmitted components carried out at the RPA level showed that there is a small increase in the σ component for decreasing H...O distances. The π component is large and positive and decreases strongly (ca. 10 Hz) for H...O distances between infinity and equilibrium. It has been recognized since long time that the π-transmitted component of triplet response properties in unsaturated compounds can be largely exaggerated at the RPA level. Therefore it can be argued that the reason why SOPPA values of ΔJ exhibit a different pattern to RPA ones originates in the relative importance of the π-transmitted component. π electrons are more weakly bonded than σ ones and exhibit a larger sensitivity to changes in molecular environment. It is thus not surprising that ΔJ
G on one hand and ΔJ
CG and ΔJ
EG on the other behave so differently in system I: the presence of the H
2O molecule (or electric field) is essential to define the polarization of the electronic π system. Such polarization, in turn, affects the π component of
1J(CH) significantly, overcoming the geometric effect. However, this effect is largely exaggerated at the RPA level. In SOPPA values it can be expected that the relative importance of the π-transmitted component is reduced to a more realistic proportion of the total
1J(CH) value. This explains the trend found for ΔJ
C and ΔJ
E within this approach.
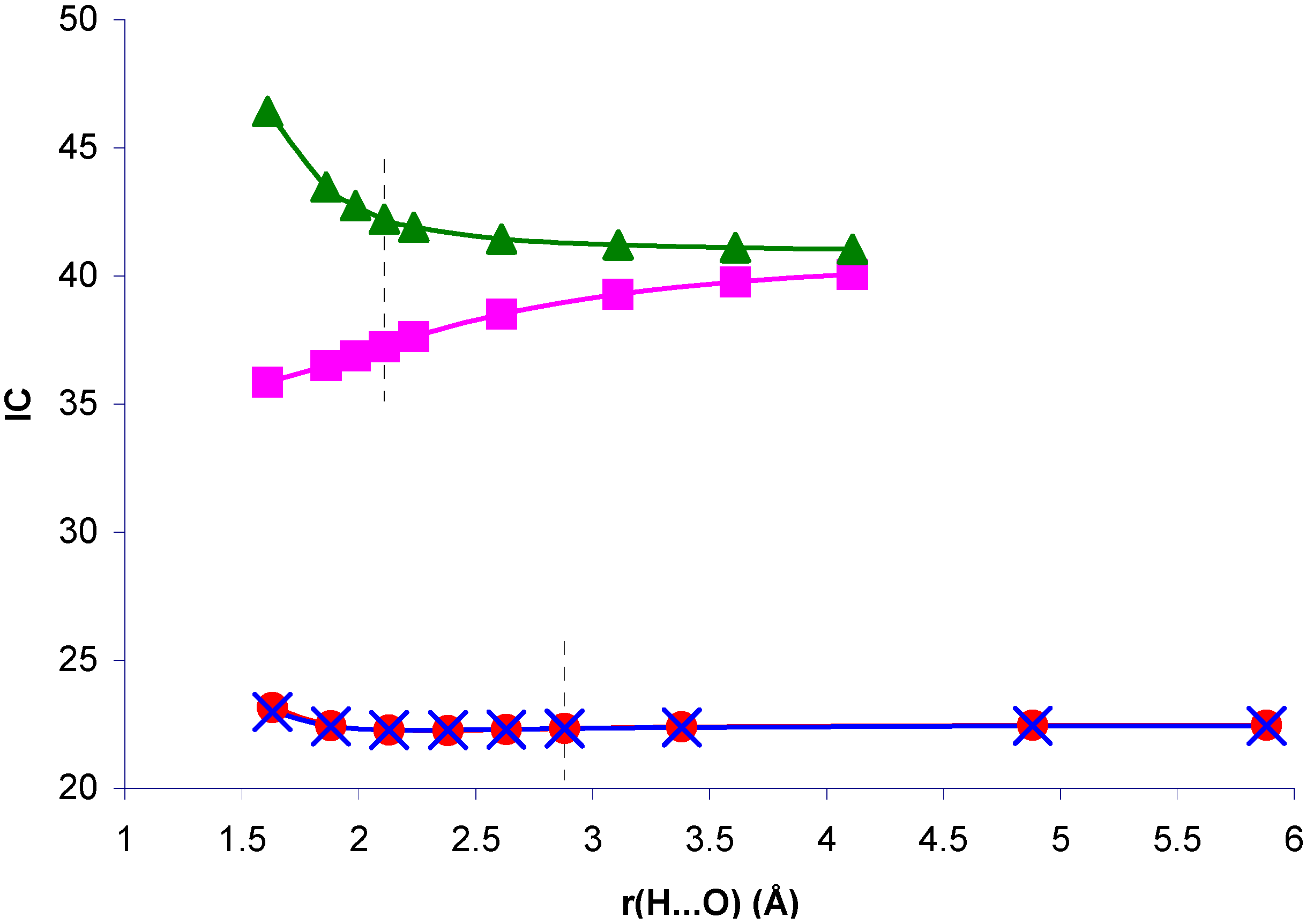
The trend of the relative importance of correlation effects as a function of the H...O distance for systems I and II exhibits interesting features. We define a correlation index (IC):
as a measure of this effect. In
figure 7 IC as a function of the H...O distance is presented for
1J(CH) in systems I and II and the geometric effect on the
1J(CH) values of the isolated molecules.
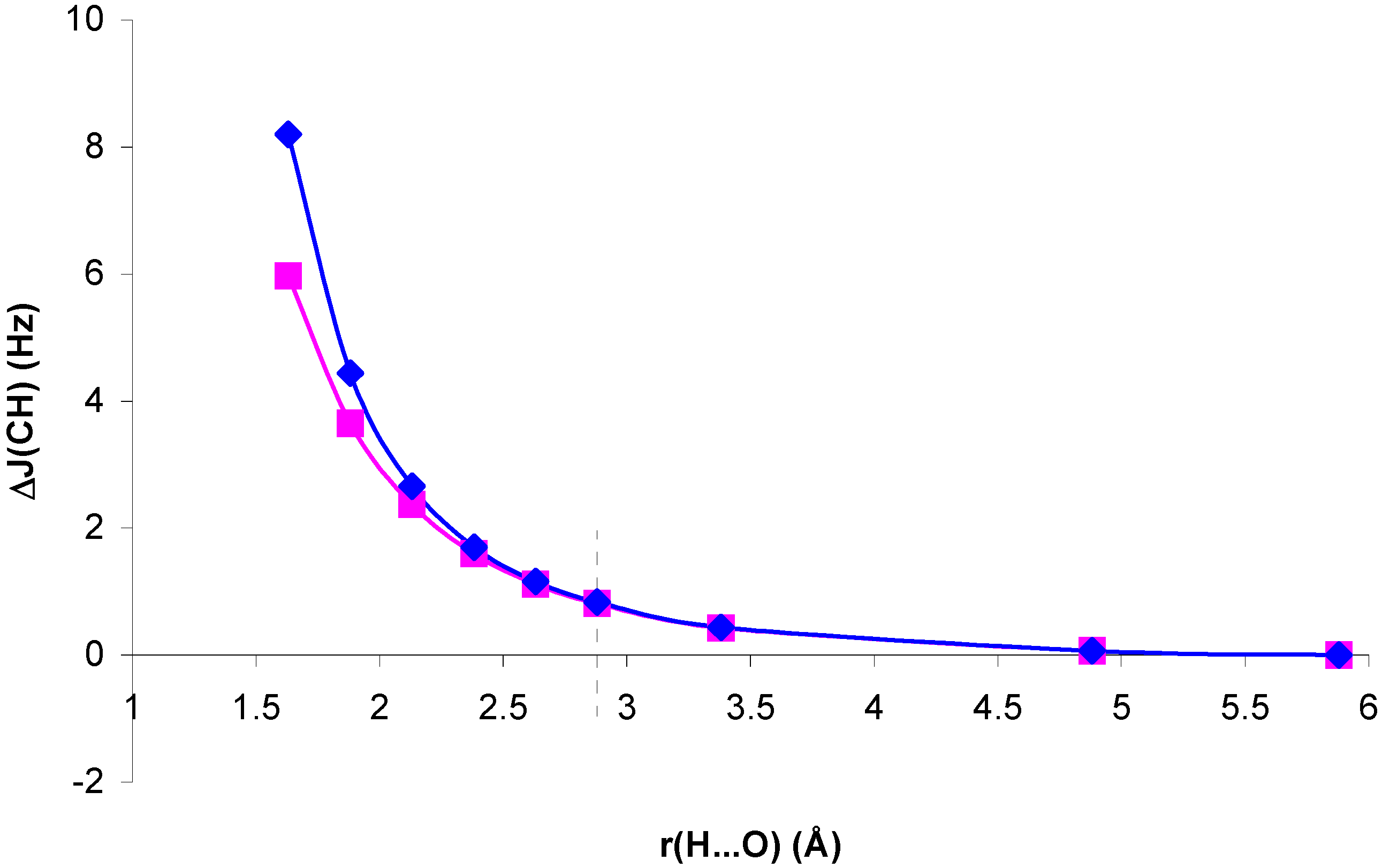
Figure 7.
Correlation index for
1J(CH), IC, in the NCH...OH
2 and CH
4...OH
2 complexes, and in the isolated NCH and CH
4 molecules with the complex geometry, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i003]()
IC (NCH...OH
2),
![Ijms 04 00203 i006]()
IC (CH
4...OH
2),
![Ijms 04 00203 i004]()
IC (NCH),
![Ijms 04 00203 i005]()
IC (CH
4). The dotted lines indicate the equilibrium distance in each case.
Figure 7.
Correlation index for
1J(CH), IC, in the NCH...OH
2 and CH
4...OH
2 complexes, and in the isolated NCH and CH
4 molecules with the complex geometry, as a function of the H...O distance, r(H...O).
![Ijms 04 00203 i003]()
IC (NCH...OH
2),
![Ijms 04 00203 i006]()
IC (CH
4...OH
2),
![Ijms 04 00203 i004]()
IC (NCH),
![Ijms 04 00203 i005]()
IC (CH
4). The dotted lines indicate the equilibrium distance in each case.
Correlation effects are very important to define
1J(CH) in system II, but IC is nearly insensitive to the presence of the H
2O molecule. It follows closely its geometric component. For system I, the decrease of IC for decreasing H...O distances is remarkable. A similar pattern is found if the H
2O molecule is replaced by its electric field. When this field is absent, IC increases, i.e., the geometric effect on IC follows the opposite trend. Therefore, the reduction of IC can be ascribed to the polarization of the electronic distribution in the presence of the H
2O field. This means that there is a coupling mechanism in I which is sensitive to correlation effects and to the mentioned electronic polarization. On one hand, as mentioned earlier, the π-transmitted component of
1J(CH) seems to be very sensitive to correlation effects. On the other hand, CLOPPA decomposition of the polarizability tensor has shown quantitatively that π electrons are highly polarizable [
17]. Therefore it can be speculated that it is the π-transmitted component which defines the trend of IC in system I. The reduction of IC as a consequence of electron polarization can be rationalized on the following grounds. Polarization of π electrons renders the π MOs less polarizable. This can be explicitly verified by calculating the polarizability of the NCH molecule in complex I by means of the IPPP CLOPPA approach. There is a decrease from 21.6 a.u. to 20.5 a.u. It can be expected that electrons occupying less polarizable orbitals will be less sensitive to any type of perturbation, including electron-electron interactions yielding correlation effects on J couplings. This rationalization is consistent with the observed trend of IC. On the other hand, in system II it can be argued that correlation effects originate in coupling mechanisms involving orbitals which are not affected by polarization of the electronic distribution to the same extent. Therefore in that case IC is not affected by such polarization and it has a similar value for the whole range of H...O distances considered.
Intermolecular couplings
The Fermi contact (FC) contribution to intermolecular
1hJ(
1H
17O) and
2hJ(
13C
17O) couplings in systems I and II were calculated at both RPA and SOPPA levels for the respective equilibrium H...O distances. Values thus obtained were negligibly small for both couplings in system II. Values corresponding to system I are displayed in
Table 1. The difference in intermolecular couplings in systems I and II can be ascribed to the much longer equilibrium H...O distance in the second case.
Table 1.
2hJ(CO) and 1hJ(HO) in system I calculated within the RPA and SOPPA approximations.
Table 1.
2hJ(CO) and 1hJ(HO) in system I calculated within the RPA and SOPPA approximations.
| | 2hJ(CO)(Hz) | 1hJ(HO)(Hz) |
| FC (RPA) | -13.64 | 6.35 |
| FC (SOPPA) | -10.50 | 3.54 |
| SD (SOPPA) | -0.04 | -0.10 |
| PSO(SOPPA) | 0.06 | 0.67 |
Intermolecular couplings in system I displayed in
Table 1 exhibit interesting features. On one hand,
2hJ(CO) is large, negative and correlation effects are small. The corresponding spin dipolar (SD) and paramagnetic spin orbital (PSO) contributions are negligibly small. On the other hand,
1hJ(HO) is positive and significantly smaller than
2hJ(CO) in absolute value. Correlation effects yield ca. 3 Hz. This is a large correction on a relative scale, but small in absolute value. The corresponding PSO contribution, although small, is significant in a relative scale. It can be concluded that the main differences between
2hJ(CO) and
1hJ(HO) originate in the FC contribution and its value is reasonably well reproduced at the RPA level. It is interesting to point out that such difference is even larger if reduced couplings are considered, as the gyromagnetic factor of
1H is about four times larger than that of
13C. These results are in line with those found by Pecul et al. for the C
2H
2...H
2O complex [
16]. Absolute values of intermolecular couplings are smaller in that case. This could be explained by the larger equilibrium H...O distance (2.187 Å).
It is interesting to analyze the transmission mechanisms involved in
2hJ(CO) and
1hJ(HO). To this end a CLOPPA decomposition of the FC contribution was carried out at the RPA level in terms of LMOs. The most important two-indices coupling pathways given by every pair of occupied LMOs are presented in
Table 2.
It is seen that coupling pathways involving the C-H bond and an LMO of the H
2O molecule yield contributions to both couplings of similar value and opposite signs. Rather unexpectedly, coupling pathways involving only occupied LMOs of the H
2O molecule yield non-zero contributions only to
2hJ(CO). As these contributions are also negative, the absolute value of
2hJ(CO) is increased. LMOs describing the O lone pairs can be classified as follows: one in-plane sp-type LMO, which will be referred to as lpσ and one of pure p-type, which is referred to as lpπ. As a consequence of the
Table 2.
Main Jij coupling pathways (i,j, occupied LMOs) for 2hJ(CO) and 1hJ(HO) in system I.
Table 2.
Main Jij coupling pathways (i,j, occupied LMOs) for 2hJ(CO) and 1hJ(HO) in system I.
| i | j | 2hJij(CO)(Hz) | 1hJij(HO)(Hz) |
| lpσ(O) | C-H | -6.98 | 5.92 |
| lpσ(O) | lpσ(O) | -1.99 | -0.08 |
| lpσ(O) | O-Ha(b) | -1.24 | 0.05 |
| O-Ha(b) | C-H | -0.59 | 0.53 |
| C-H | C-H | 0.51 | -0.27 |
| Σ Jij | -12.12 | 6.73 |
attractive interaction yielding a weak hydrogen bond of type C-H...O, the sp-type O lone pair extends towards the C and H nuclei. The existence of a contact mechanism involving occupied LMOs of the H
2O electronic system is thus a direct consequence of this effect. However, a non-zero FC contribution to
1hJ(HO) would also be expected on these grounds. Coupling pathways involving only LMOs of the NCH environment are all very small.
The reasons of the difference between
1hJ(HO) and
2hJ(CO) must be traced looking at the distribution of vacant LMOs. The set of vacant MOs was localized as explained in section “Method of calculation”. Within this classification of vacant LMOs the main four-indices coupling pathways J
ia,jb defining
1hJ(HO) and
2hJ(CO) can be obtained. As localization of MOs is not perfect, there are very many terms yielding values of a few Hz. We have looked at those within the local fragment C-H...O. The total sum of coupling pathways involving occupied LMOs C-H, lpσ(O),O-H
a and O-H
b (where H
a and H
b identify the two H atoms of the H
2O molecule) and vacant LMOs C-H*, s(O)*, lpσ(O)*, O-H
a* and O-H
b* and C-H...O* yields
2hJ(CO)=-15.45 Hz and
1hJ(HO)= 5.19 Hz. If the C-H...O* vacant LMO is excluded from the sum, results change to
2hJ(CO)=-4.81 Hz and
1hJ(HO)= 6.04 Hz. It is thus seen that the C-H...O* LMO yields very important contributions in the first case, but it contributes negligibly small values in the second one. In
Table 3 the largest J
ia,jb terms within the fragment are displayed in order to identify the main transmission mechanisms involved.
Individual four-indices coupling pathways displayed in
Table 3 show the above mentioned trend clearly. Looking at results in
Table 3, it must be kept in mind that for given indices i,a,j,b the propagator element P
ia,jb is the same for both couplings and therefore the difference in the values of the corresponding coupling pathway J
ia,jb depends on the “perturbators” V
ia and V
jb at each nucleus. The role of C-H...O* in
1hJ(HO) and
2hJ(CO) is quite different. All coupling pathways involving this vacant LMO are negligibly small for
1hJ(HO), but for
2hJ(CO) such terms yield the largest negative values. It can be concluded that C-H...O* has sufficiently low energy to make the corresponding propagator elements large enough to couple LMOs from the NCH and H
2O environments. The particular shape of this LMO is responsible for the different contributions to both couplings. It is an antibonding like LMO with opposite amplitudes in the C-H and O regions. In particular the corresponding amplitude at the H nucleus is negligibly small. A coupling pathway of strictly local character is thus found which is
Table 3.
Main “coupling pathways” Jia,jb involving the C-H and lpσ(O) occupied LMOs and vacant LMOs within the local fragment C-H...O in system I. Values in Hz.
Table 3.
Main “coupling pathways” Jia,jb involving the C-H and lpσ(O) occupied LMOs and vacant LMOs within the local fragment C-H...O in system I. Values in Hz.
| i | j | a | b | 2hJia,jb(CO) | 1hJia,jb(HO) |
| lpσ(O) | lpσ(O) | CH...O* | CH...O* | -7.74 | -0.03 |
| | | s(O)* | CH...O* | 3.81 | 0.01 |
| | | O-H* | O-H* | 3.77 | 4.68 |
| | | lpσ(O)* | lpσ(O)* | -3.37 | -1.91 |
| | | C-H* | O-H* | -1.66 | -5.13 |
| | | s(O)* | O-H* | -1.53 | -2.53 |
| lpσ(O) | C-H | CH...O* | CH...O* | -4.93 | -0.04 |
| | | O-H* | CH...O* | -3.48 | -0.03 |
| | | s(O)* | C-H | 3.05 | -4.43 |
| | | lpσ(O)* | CH...O* | 2.71 | 0.02 |
| | | lpσ(O)* | C-H* | -2.59 | 7.84 |
| | | O-H* | O-H | 2.21 | 5.65 |
operative in C-H...O interactions and is very efficient for
2hJ(CO) but does not contribute to
1hJ(HO). The argument is valid for the reduced coupling constants, and it explains the larger absolute value of
2hK(CO) than
1hK(HO). Coupling pathways involving the C-H→C-H* excitation yield contributions of opposite signs to both couplings. This can be directly ascribed to the general fact that the bonding and antibonding LMOs have opposite relative phases at the C and H nuclei. As the second excitation to both couplings entering a given coupling pathway J
ia,jb is centered at the O nucleus, the corresponding values necessarily carry opposite signs. This kind of coupling pathway is the one usually found when a through-space mechanism operates. In fact the leading contribution to
1hJ(HO) is of this type. The corresponding value is positive for this coupling and negative for
2hJ(CO).


 ΔJE (RPA),
ΔJE (RPA),  ΔJE (SOPPA),
ΔJE (SOPPA),  ΔJC (RPA),
ΔJC (RPA),  ΔJC (SOPPA). The dotted line indicates the equilibrium distance.
ΔJC (SOPPA). The dotted line indicates the equilibrium distance.

 IC (CH4...OH2),
IC (CH4...OH2), 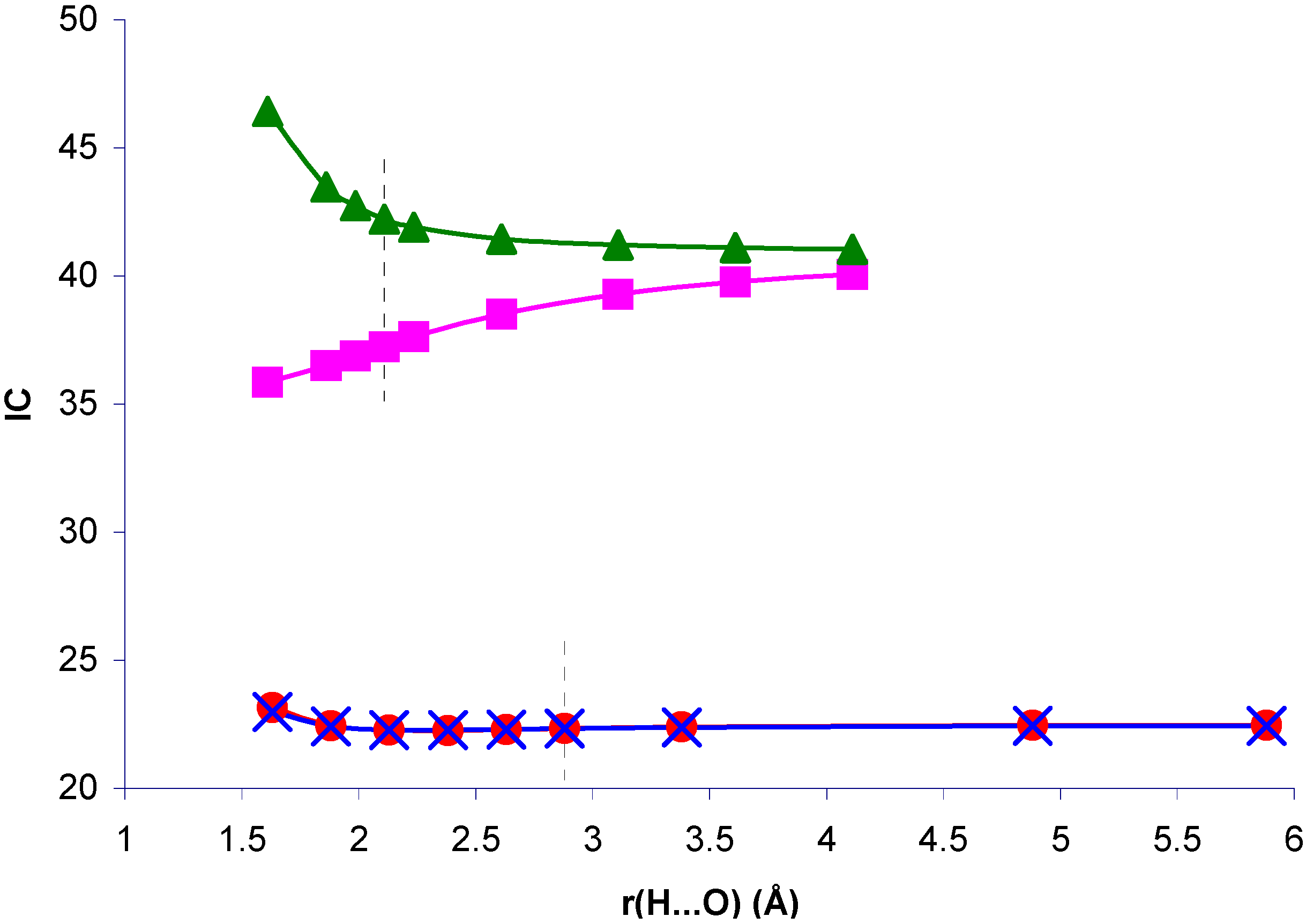

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}