Introduction
The concept of atomic and ionic radii has been found to be very useful in understanding, explaining, correlating and even predicting many physico-chemical properties of atoms, ions and molecules. The periodicity of chemical and physical properties of the elements has been recognized from the date of early history of chemistry. The atomic volume curve of Lother Meyer was a striking example of periodicity of physical properties of elements. The atomic size has been also an important periodic property of the elements. The atomic and ionic radii values are important input in many calculations of size dependent physico–chemical properties of isolated atoms. The crystal chemists have tried for many years to treat atoms and ions as hard spheres and published series of atomic and ionic radii with the only significance that such atomic and ionic radii, when added, reproduce the minimum distance of separation between atoms and ions respectively in crystal lattices. Approximate additivity of atomic and ionic radii were noted by early investigators like Goldschmidt et al [
1], Pauling [
2], Zachariasen [
3], and Bragg [
4]. However, the inter atomic or inter ionic distance depend upon the crystal type, its allotropic modification, co-ordination number, temperature etc. Unfortunately this very important property –– the size concept of atoms and ions is heavily underplayed. The text books dealing with this size aspects of atoms or ions never make the position distinctly clear as to the meaning and purport of the terms ‘atomic’ or ‘ionic’ radii and the term ‘radii’ is kept vague. But invariably the text books display one or the other chart of atomic and ionic radii and recently there has been a proliferation of the tables and data of atomic and ionic radii with bewildering array of terms including bonded, non-bonded, ionic, covalent and van der Waals radii [
5]. But such radii data though useful in reproducing the inter atomic/ionic distances for the particular purposes but not useful in computing the radial dependent absolute properties of atoms. Also, there is no means of verifying the merit of a set of radii determined experimentally or calculated theoretically because, there is no experimental value of the sizes of atoms and ions [
6].
According to quantum mechanical view, the atoms and the ions do not have any rigid shape or size and hence the question of atomic and ionic radii simply does not arise in the true sense of the term. However, chemical experience suggests that the atoms and ions do have an effective size because, the atoms and ions cannot approach each other beyond certain limiting distance under the influence of forces encountered in chemical interactions. The determination of the empirical atomic and ionic radii, on the basis of hard sphere approximation model, has a history stretching back to the work of Bragg
4 in 1920’s to the work of Slater [
7] in the year 1964. Attempts of computing theoretical atomic and ionic radii within the scope of Self Consistent Field (SCF) theory are also reported [
7,
8].
The inherent approximation of the SCF method suggests that each electron have its own one-electron function or orbital in many–electron situation. The fact that each electron in a many electron system should have one-electron function is a pre-quantum mechanical thinking [
9] when it was pointed out that many spectroscopic facts could be rationalized if energy levels were identified with those of a single electron moving in a central field. Slater [
7] argued that there must be a connection between empirically determined atomic and ionic size with the wave functions of atoms and ions and pointed out a good correlation between the atomic radii and the distance of maximum charge density in the outermost electron shell of the atom. To be more precise, according to the suggestion of Slater, the principal maxima of the radial charge density distribution function of the outermost orbital of the atom may be considered as its theoretical atomic radius. Slater [
7] published a set of atomic radii computed through comparison of a large number of crystal data and it is claimed that such crystal radii are very close to the theoretical atomic radii calculated by Liberman, Waber and Cromer through self–consistent field wave functions for all the atoms with relativistic correlations according to the Dirac equations. Waber and Cromer [
8] calculated the theoretical atomic radii of a number of atoms through relativistic Hartree–Fock method. In absence of any experimental data [
6] regarding the absolute size of atoms and ions the question of accuracy of any theoretical or experimental method of computing atomic or ionic size cannot be just tested. Sporadic informations of calculations of atomic size by sophisticated theoretical methods are found to appear in the literature but such data are mostly unpublished. Also it appears that such data has never been applied to compute any property of isolated atom or ion. In the premises, we propose a less sophisticated simple technique of computing theoretical or absolute radii of atoms and ions. The level of theory is semi-empirical. Slater [
10] suggested an analytical form of the radial part of one-electron function that provides with a scope of computing a number of one–electron and radial properties including the scope of computation of theoretical atomic and ionic radii. The computation of theoretical atomic and ionic radii becomes tremendously simplified if Slater Orbitals are used in such calculation.
Slater [
10] proposed a much simpler analytical form for the radial part of the one-electron function or orbital now widely known as Slater–type orbitals, STO’s. The analytical form of normalized radial part of atomic orbital R
nl(r) is given by,
Where, Z= atomic number and S is the screening constant and n* is the effective principal quantum number. The quantity, (Z–S) is further identified as effective nuclear charge, Z*.
In terms of Slater orbitals the theoretical atomic or ionic radii,
The eqn. (3) is the formula of computation of theoretical atomic and ionic radii in terms of Slater orbital. This shows that theoretical radii of atoms and ions can be easily computed only if ξ is known and one can know ξ, if the effective principal quantum number of the outermost shell of the atom (n
*) and the screening constant (S) are known. Although the SCF ξ is also available [
11], Slater [
10] laid down a set of empirical rules for calculating S and n
* to give good approximations to the best atomic orbitals of this type. The goal of this work has been to explore the possibility of developing a simple but effective method of computing the absolute radii atoms and ions relying upon Slater’s definition of absolute radii and using Slater’s orbitals. It is well known that orbital exponent, ξ obtained by Slater’s empirical rules are found useful in SCF calculation giving satisfactory results although use of SCF optimized ξ is simultaneously in use [
11].
In the present venture we have computed the theoretical atomic radii of as many as 103 elements of the periodic table by semi-empirical method using Slater’s orbital and involving Slater’s empirical rule for the computation of ξ. Since there is no scope of comparing the atomic and ionic radii with the experimental values, we have tested the range of validity of calculated sizes in a number of ways. The periodicity exhibited by the computed radii is compared with the periodicity inherent in the periodic table. In order to explore the internal consistency between the calculated sizes and the size dependent properties of the atoms, a comparative study of size vis-a-vis the electronegativity and the experimental quantity like first ionization potential of the atoms is made. A comparative study of the relative magnitudes of the sizes of the atoms of present calculation and the radii published by other workers – both theoretical and experimental is also made. The computed theoretical radii are used to calculate a number of size dependent intrinsic physical properties of isolated atoms viz., (1) the diamagnetic part of magnetic susceptibility (χdia) of 54 elements, and (2) the atomic polarizability (α), and (3) the global hardness (η) of as many as 103 elements of the periodic table. We have compared such computed size dependent properties of the atoms with available results of such quantities.
Method of Computation
The radial charge density distribution function is defined [
5,
12,
13] as 4πr
2 R
2 or simply r
2R
2 where R is the radial function. According to Slater [
7], theoretical atomic radius is the maximum of the radial charge density distribution function of the topmost electron of the atom or ion.
Radial charge density distribution function ρ(r) is given by
Now, putting the normalized form of the R from the eqn.(1) into eqn.(4) we get
The parameters n and ξ are explained above.
Differentiating the left-hand side with respect to r, and equating the result with zero, we get the maximum of the radial charge density distribution function, the theoretical atomic radii,
Equating the right hand side of the eqn. (6) equal to zero and replacing r by r
max, we obtain,
From which it follows that
From this relation we obtain the formula for computing the theoretical atomic or ionic radii already noted in eqn (3) above, i.e. the theoretical atomic or ionic radii r = r
max
the relevant quantities and parameters are defined above.
The screening constant (S) may be calculated from Slater’s rule and are profusely available in any standard textbook of physical and inorganic chemistry. The values of n
* for principal quantum number n up to 6 and Z
* for about 26 elements are published by Pople [
14]. For the rest of the atoms, n
* are calculated by simple extrapolation. and we have got the extrapolated value of n
* = 4.3 for principal quantum number 7. The Z
* and ξ for required atoms are calculated and for that matter we have relied upon the ground state electron–configuration published by Shriver and Atkins [
15]. In this work we report the theoretical radii of atoms only. The calculated orbital exponents and atomic radii are shown in
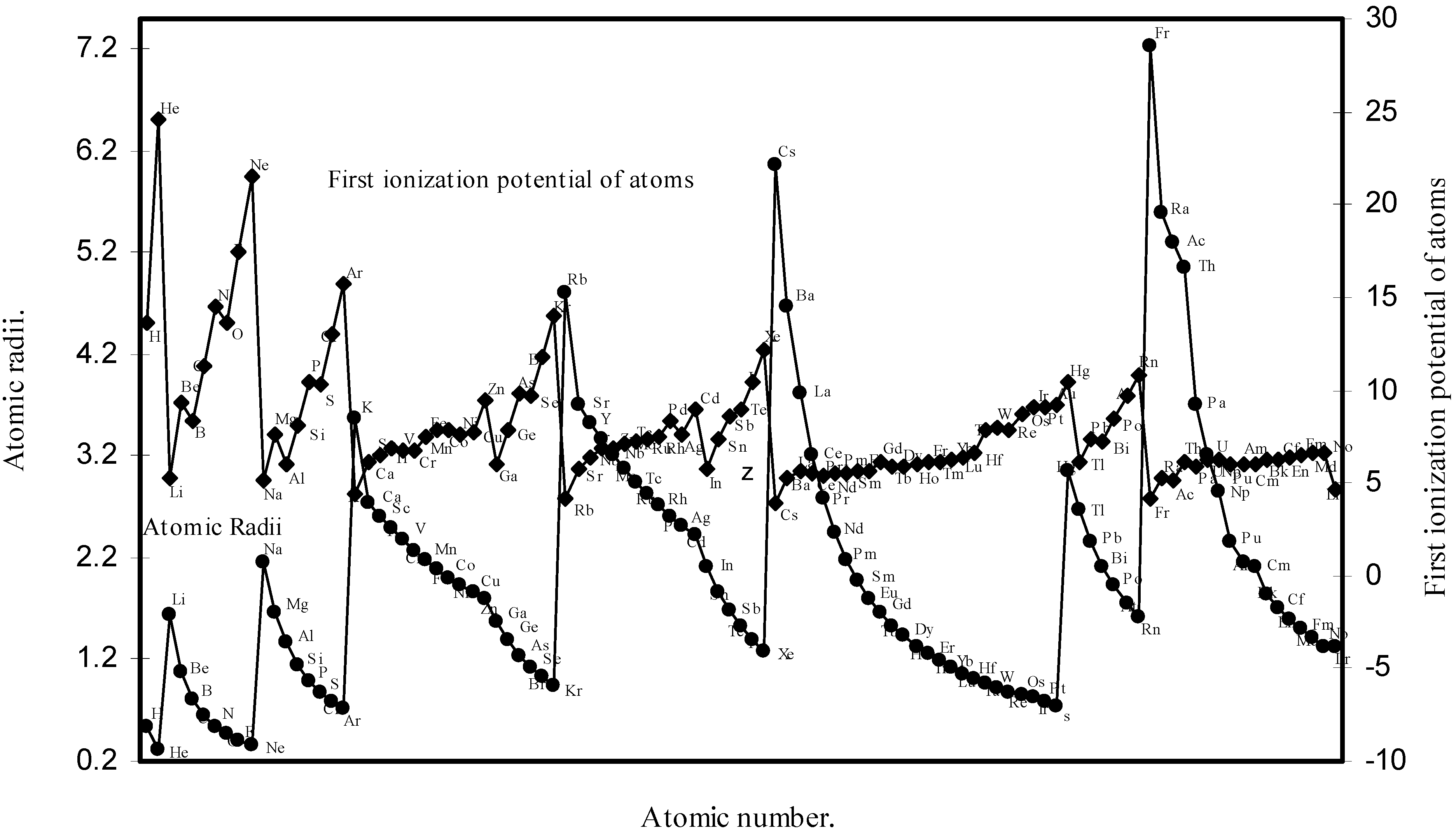
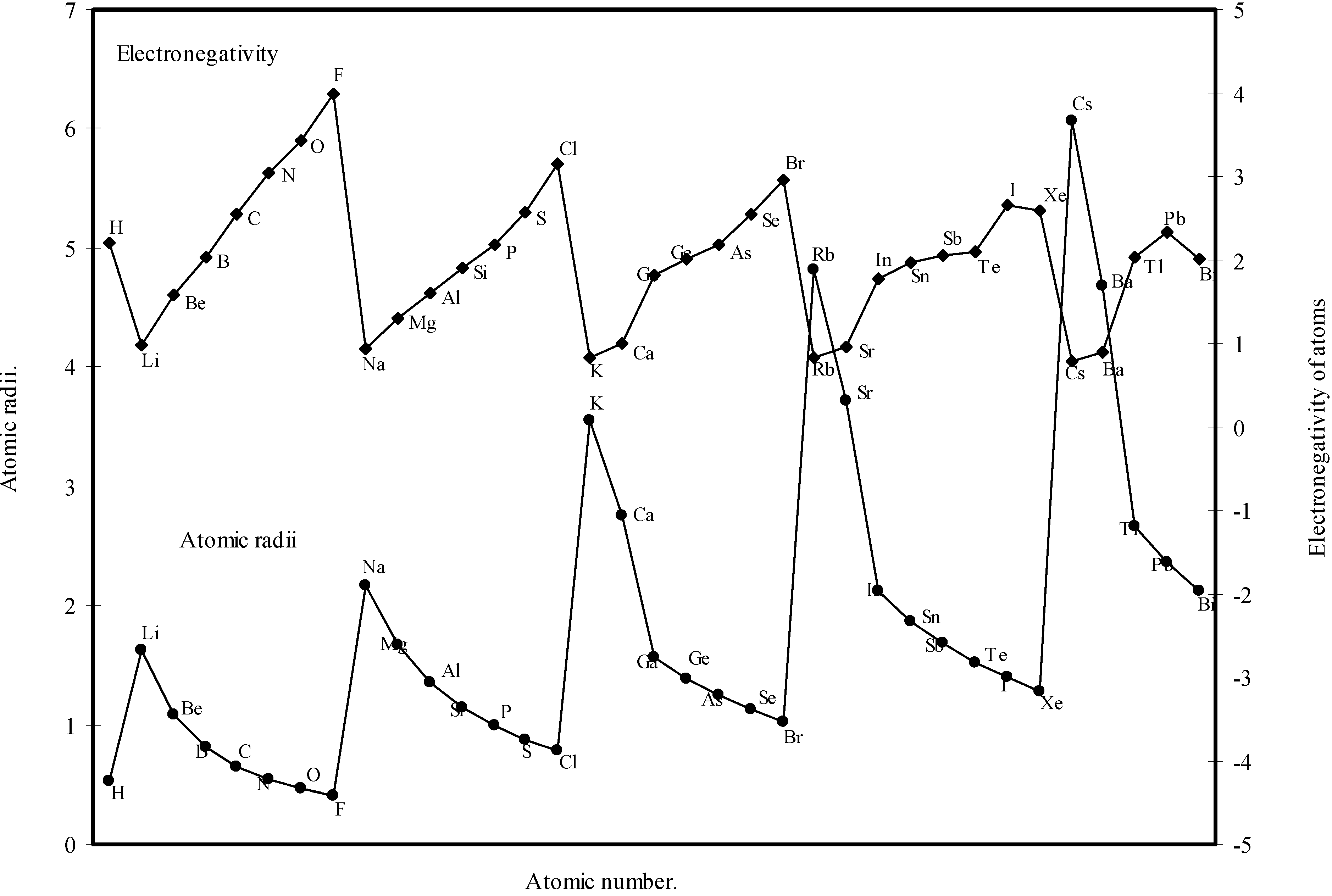
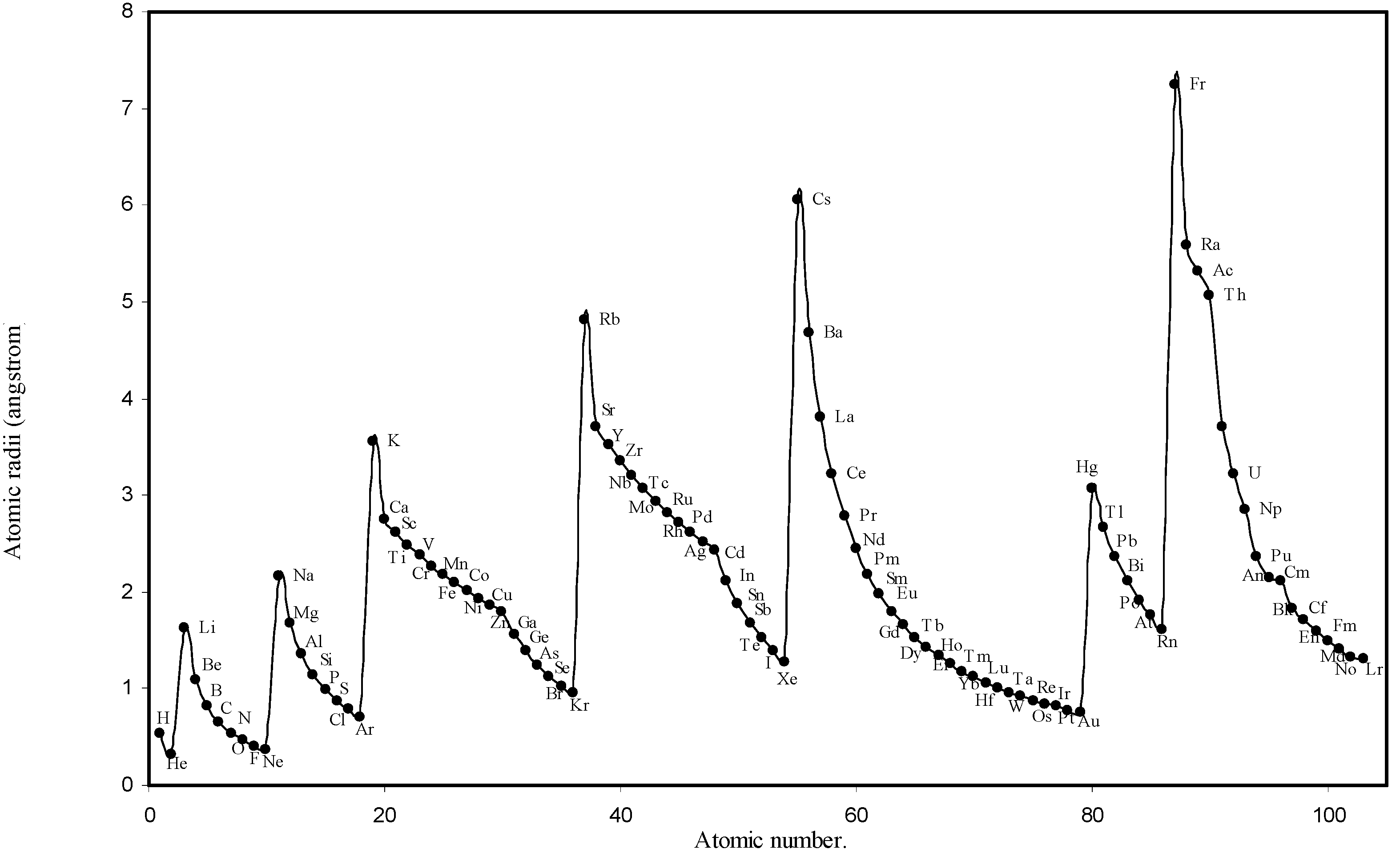
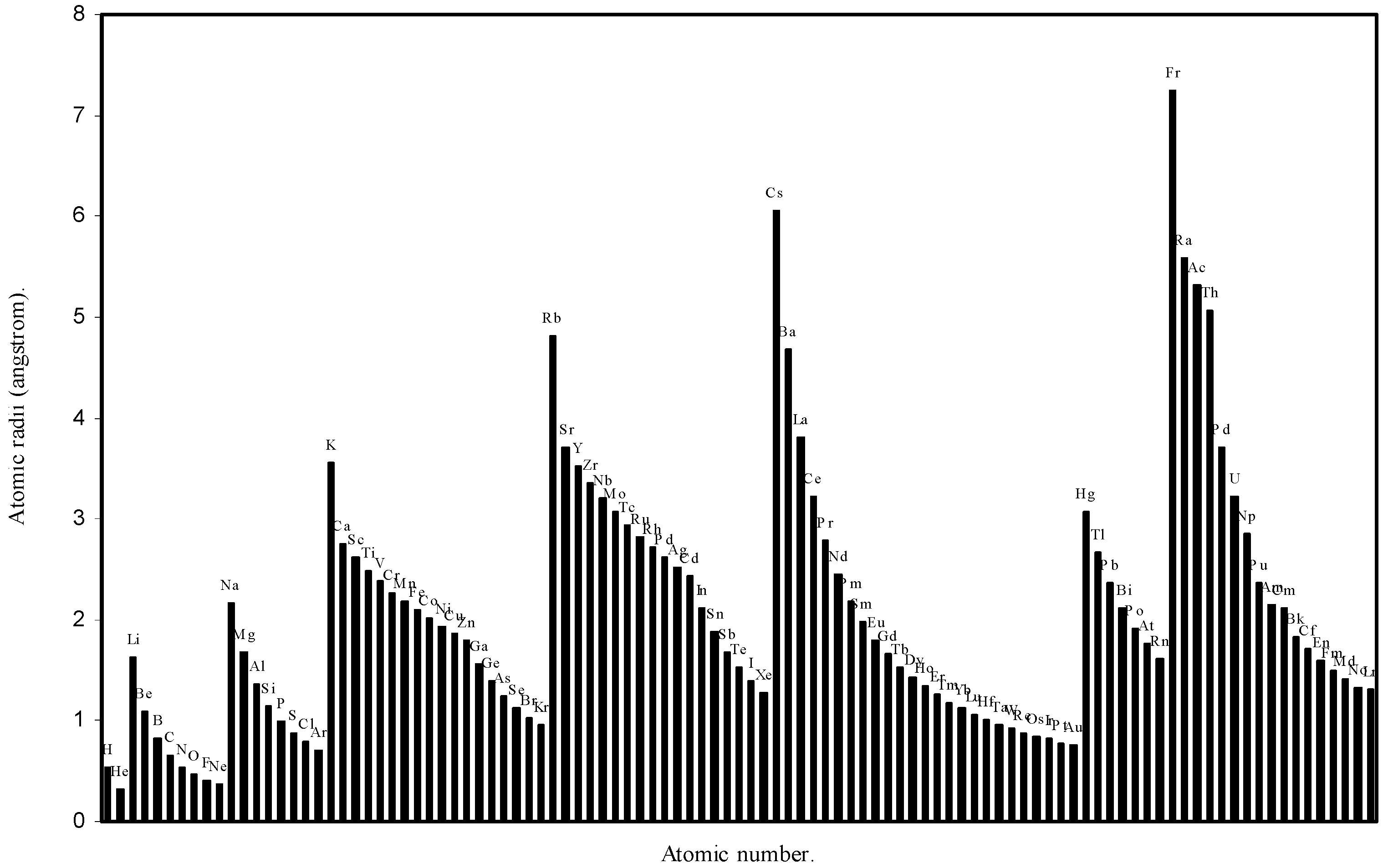
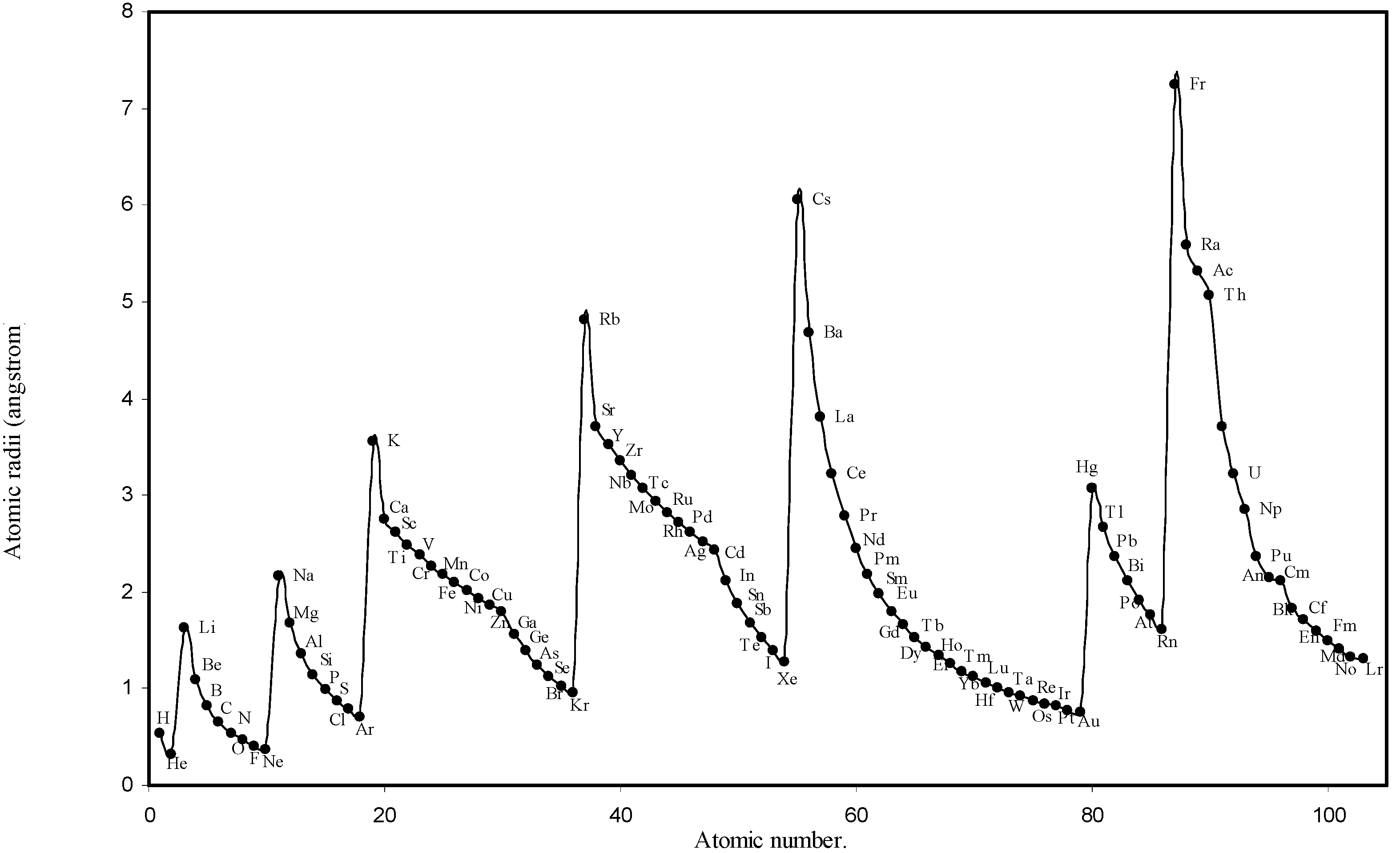
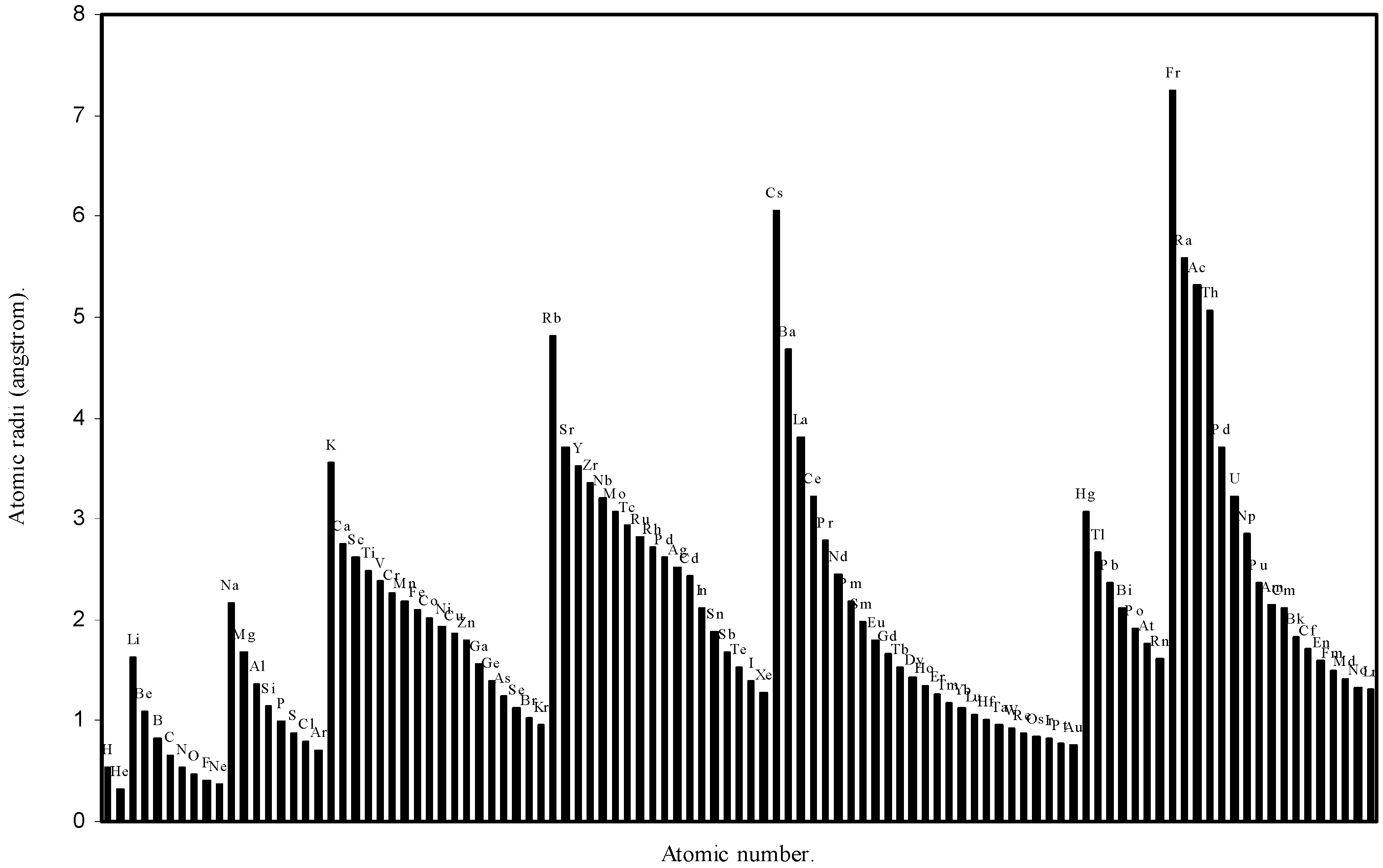
Table 1. The computed theoretical atomic radii are plotted as a function of atomic number in
Figure 1 and
Figure 2. To explore the effectiveness of the present method in exhibiting the well known
Figure 1.
Plot of atomic radii (angstrom) as a function of atomic number.
Figure 1.
Plot of atomic radii (angstrom) as a function of atomic number.
Figure 2.
Plot of atomic radii (angstrom) as a function of atomic number.
Figure 2.
Plot of atomic radii (angstrom) as a function of atomic number.
Table 1.
Calculated orbital exponents (ξ) and radii of atoms.
Table 1.
Calculated orbital exponents (ξ) and radii of atoms.
| Atoms | Orbital Exponent | Atomic Radii (Å) | Atoms | Orbital Exponent | Atomic Radii (Å) | Atoms | Orbital Exponent | Atomic Radii (Å) |
|---|
| H | 1 | 0.5292 | Kr | 2.2297 | 0.9493 | Lu | 3 | 1.0583 |
| He | 1.7 | 0.3113 | Rb | 0.55 | 4.8106 | Hf | 3.15 | 1.0079 |
| Li | 0.65 | 1.6282 | Sr | 0.7125 | 3.7135 | Ta | 3.3095 | 0.9594 |
| Be | 0.975 | 1.0855 | Y | 0.75 | 3.5278 | W | 3.4643 | 0.9165 |
| B | 1.3 | 0.8141 | Zr | 0.7875 | 3.3598 | Re | 3.619 | 0.8773 |
| C | 1.625 | 0.6513 | Nb | 0.825 | 3.2071 | Os | 3.7738 | 0.8413 |
| N | 1.95 | 0.5427 | Mo | 0.8625 | 3.0677 | Ir | 3.9286 | 0.8182 |
| O | 2.275 | 0.4652 | Tc | 0.9 | 2.9398 | Pt | 4.0833 | 0.7776 |
| F | 2.6 | 0.4071 | Ru | 0.9375 | 2.8222 | Au | 4.2381 | 0.7492 |
| Ne | 2.925 | 0.3618 | Rh | 0.975 | 2.7137 | Hg | 4.3929 | 3.0636 |
| Na | 0.7333 | 2.1649 | Pd | 1.0125 | 2.6132 | Tl | 1.1905 | 2.667 |
| Mg | 0.95 | 1.6711 | Ag | 1.05 | 2.5199 | Pb | 1.3452 | 2.3603 |
| Al | 1.1667 | 1.3607 | Cd | 1.0875 | 2.433 | Bi | 1.5 | 2.1167 |
| Si | 1.3833 | 1.1476 | In | 1.25 | 2.1167 | Po | 1.6548 | 1.9187 |
| P | 1.6 | 0.9922 | Sn | 1.4125 | 1.8732 | At | 1.8095 | 1.7546 |
| S | 1.8167 | 0.8738 | Sb | 1.575 | 1.6799 | Rn | 1.9643 | 1.6164 |
| Cl | 2.0333 | 0.7807 | Te | 1.7375 | 1.5228 | Fr | 0.5116 | 7.2404 |
| Ar | 2.25 | 0.7056 | I | 1.9 | 1.3926 | Ra | 0.6628 | 5.5887 |
| K | 0.5946 | 3.5598 | Xe | 2.0625 | 1.2828 | Ac | 0.6977 | 5.3091 |
| Ca | 0.7703 | 2.7479 | Cs | 0.5238 | 6.0615 | Th | 0.7326 | 5.0569 |
| Sc | 0.8108 | 2.6106 | Ba | 0.6786 | 4.6788 | Pa | 1 | 3.7042 |
| Ti | 0.8514 | 2.4861 | La | 0.8333 | 3.8102 | U | 1.1512 | 3.2177 |
| V | 0.8919 | 2.3732 | Ce | 0.9881 | 3.2133 | Np | 1.3023 | 2.8443 |
| Cr | 0.9324 | 2.2701 | Pr | 1.1429 | 2.778 | Pu | 1.5698 | 2.3596 |
| Mn | 0.973 | 2.1754 | Nd | 1.2976 | 2.4468 | Am | 1.7209 | 2.1525 |
| Fe | 1.0135 | 2.0885 | Pm | 1.4524 | 2.1861 | Cm | 1.7558 | 2.1097 |
| Co | 1.0541 | 2.008 | Sm | 1.6071 | 1.9756 | Bk | 2.0233 | 1.8308 |
| Ni | 1.0946 | 1.9337 | Eu | 1.7619 | 1.802 | Cf | 2.1744 | 1.7035 |
| Cu | 1.1351 | 1.8648 | Gd | 1.9167 | 1.6565 | En | 2.3256 | 1.5928 |
| Zn | 1.1757 | 1.8004 | Tb | 2.0714 | 1.5328 | Fm | 2.4767 | 1.4956 |
| Ga | 1.3514 | 1.5663 | Dy | 2.2262 | 1.4262 | Md | 2.6279 | 1.4096 |
| Ge | 1.527 | 1.3862 | Ho | 2.381 | 1.3335 | No | 2.7791 | 1.3329 |
| As | 1.7027 | 1.2431 | Er | 2.5357 | 1.2521 | Lr | 2.814 | 1.3164 |
| Se | 1.8784 | 1.1269 | Tm | 2.6905 | 1.1801 | | | |
| Br | 2.0541 | 1.0305 | Yb | 2.8452 | 1.1159 | | | |
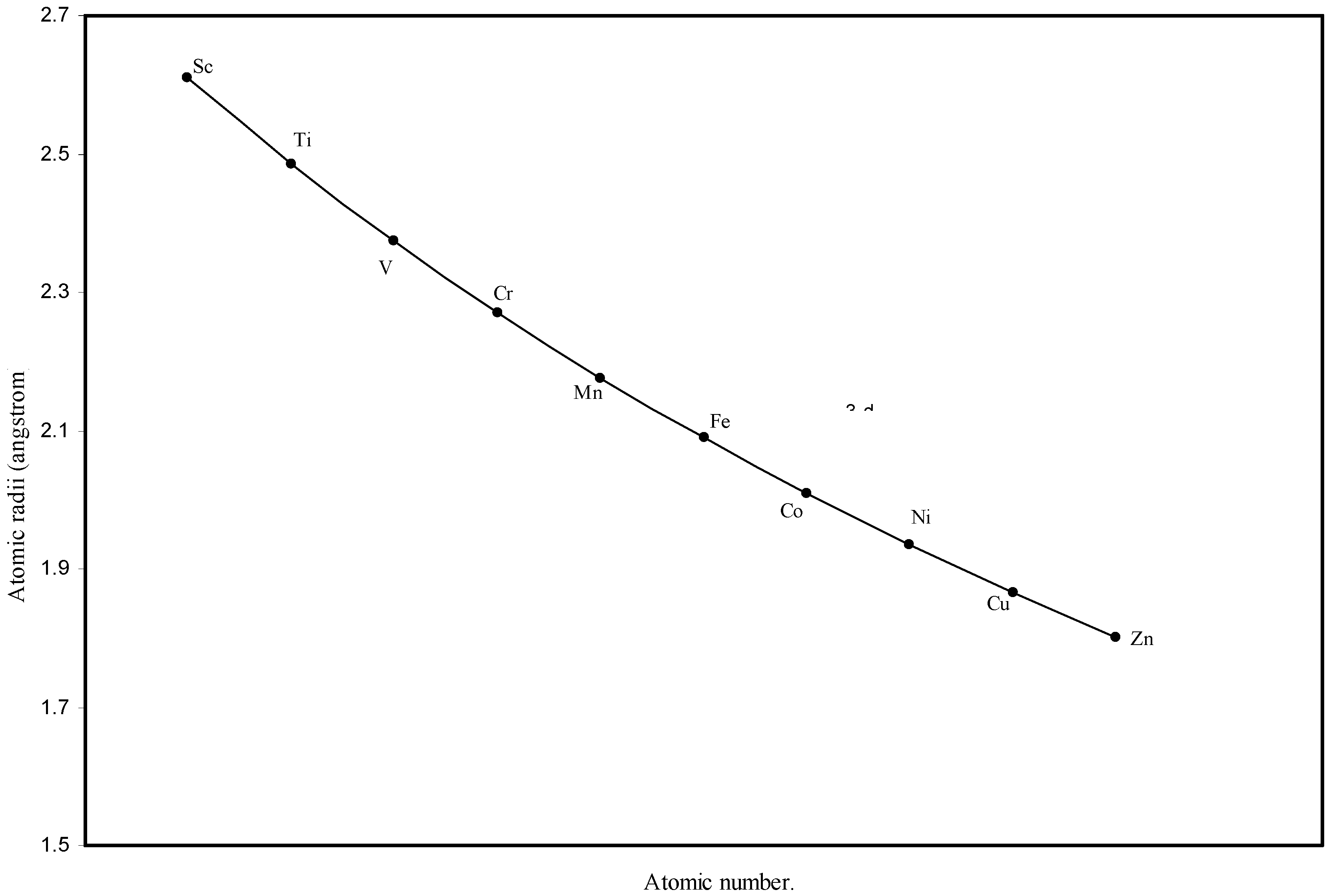
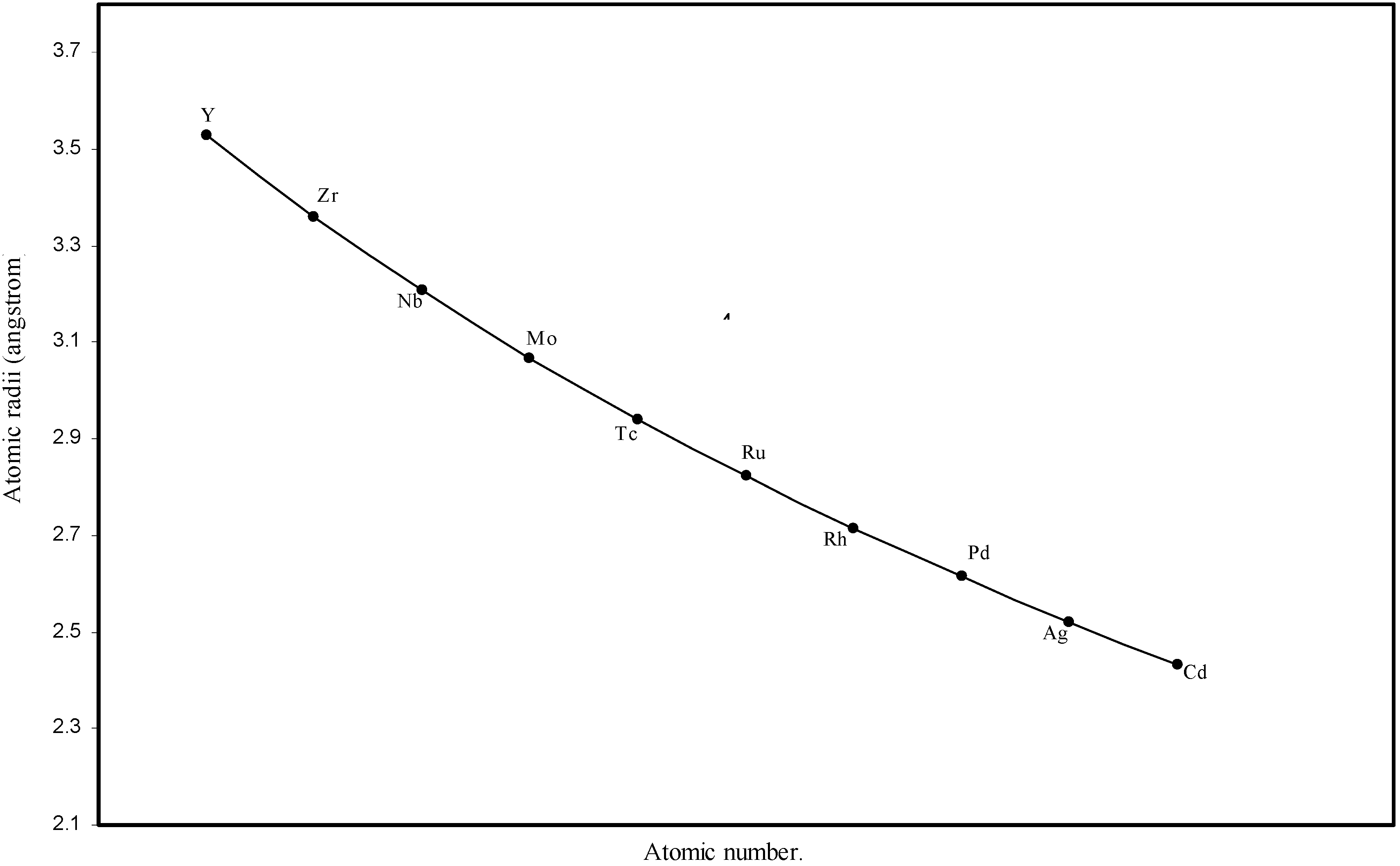
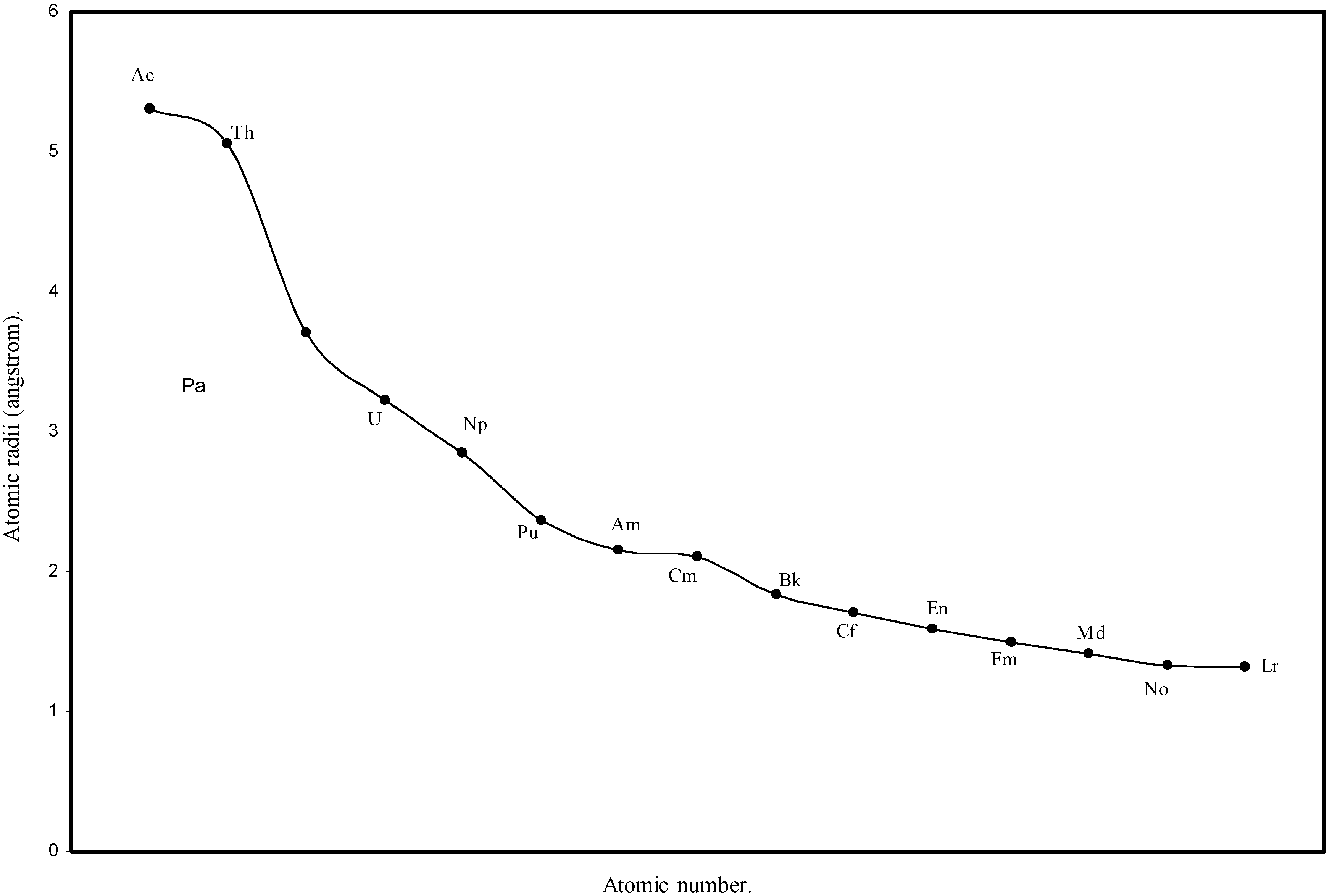
d-block and f-block contractions in the periodic table, the computed radii of 3d, 4d, 4f and 5f elements are separately plotted as a function of the atomic numbers in
Figs. 3,
Figs. 4,
Figs. 5, and
Figs. 6, respectively. The methods of computation of absolute size dependent physical properties like (A) the diamagnetic part of magnetic susceptibilities (χ
dia), (B) atomic polarizabilities (α), and (C) the global hardness (η) are stated below.
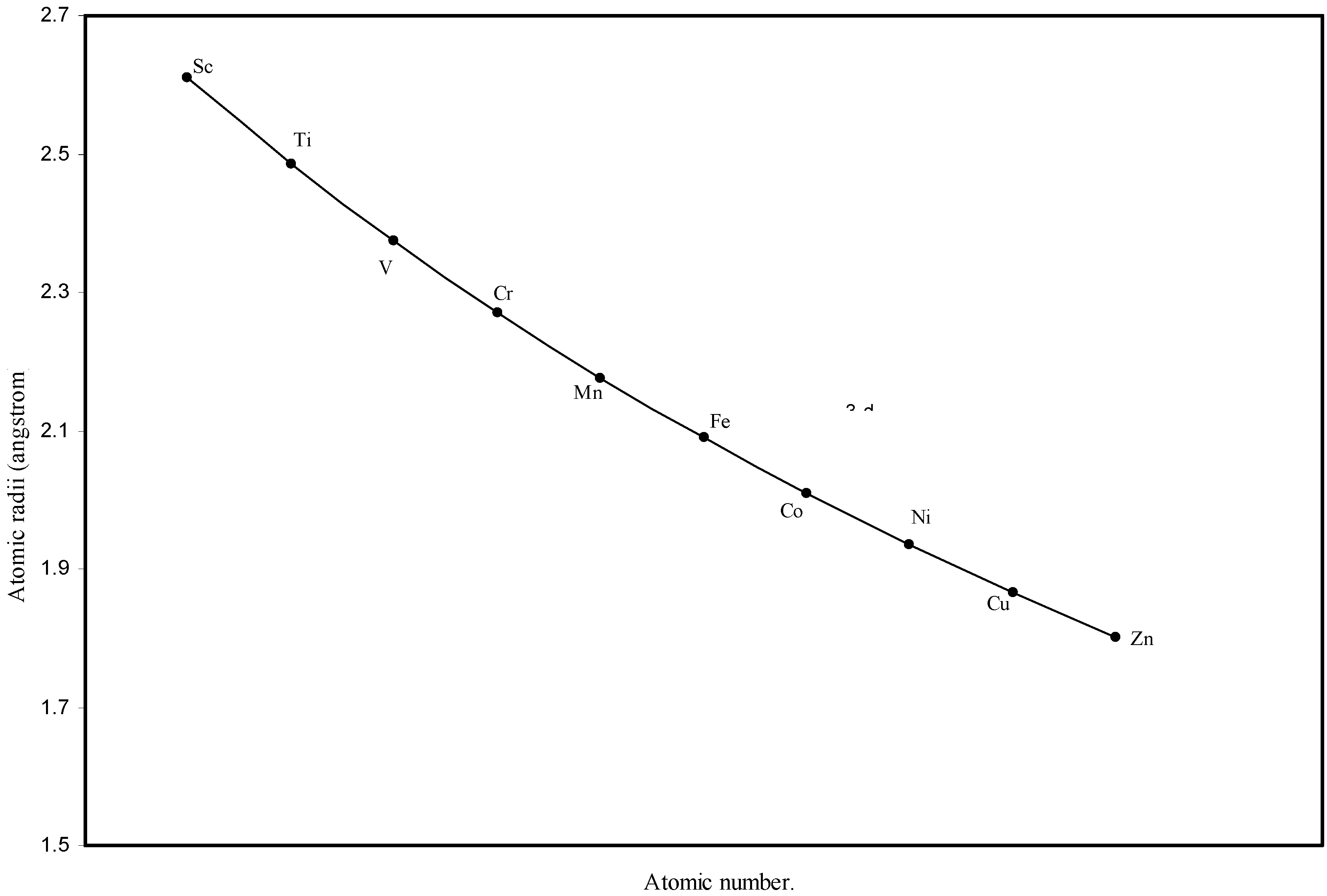
Figure 3.
Plot of atomic radii of first transition sereis (3-d block) elements in angstrom unit as a function of atomic number.
Figure 3.
Plot of atomic radii of first transition sereis (3-d block) elements in angstrom unit as a function of atomic number.
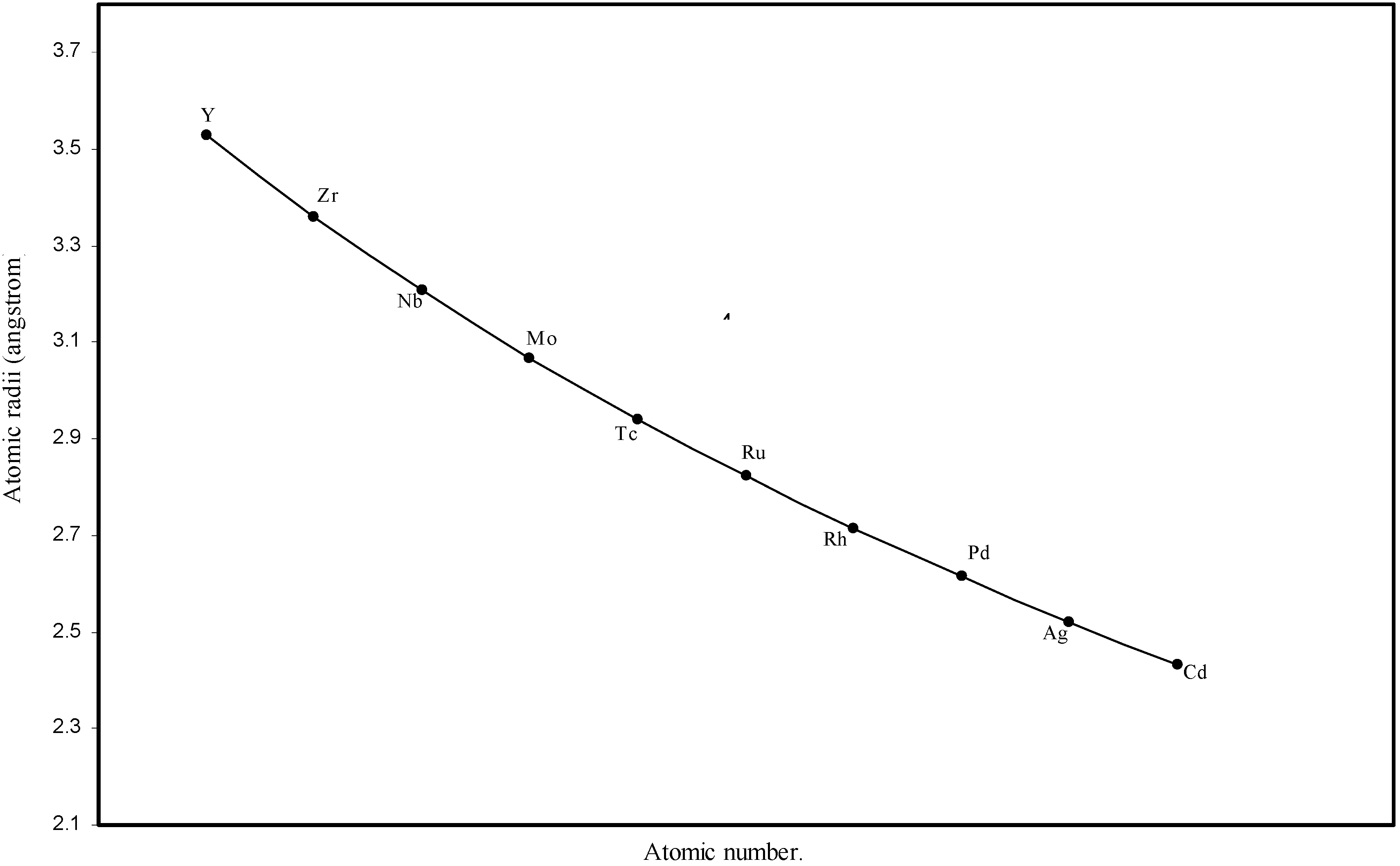
Figure 4.
Plot of atomic radii of second transition series (4-d block) elements in angstrom unit as a function of atomic number.
Figure 4.
Plot of atomic radii of second transition series (4-d block) elements in angstrom unit as a function of atomic number.
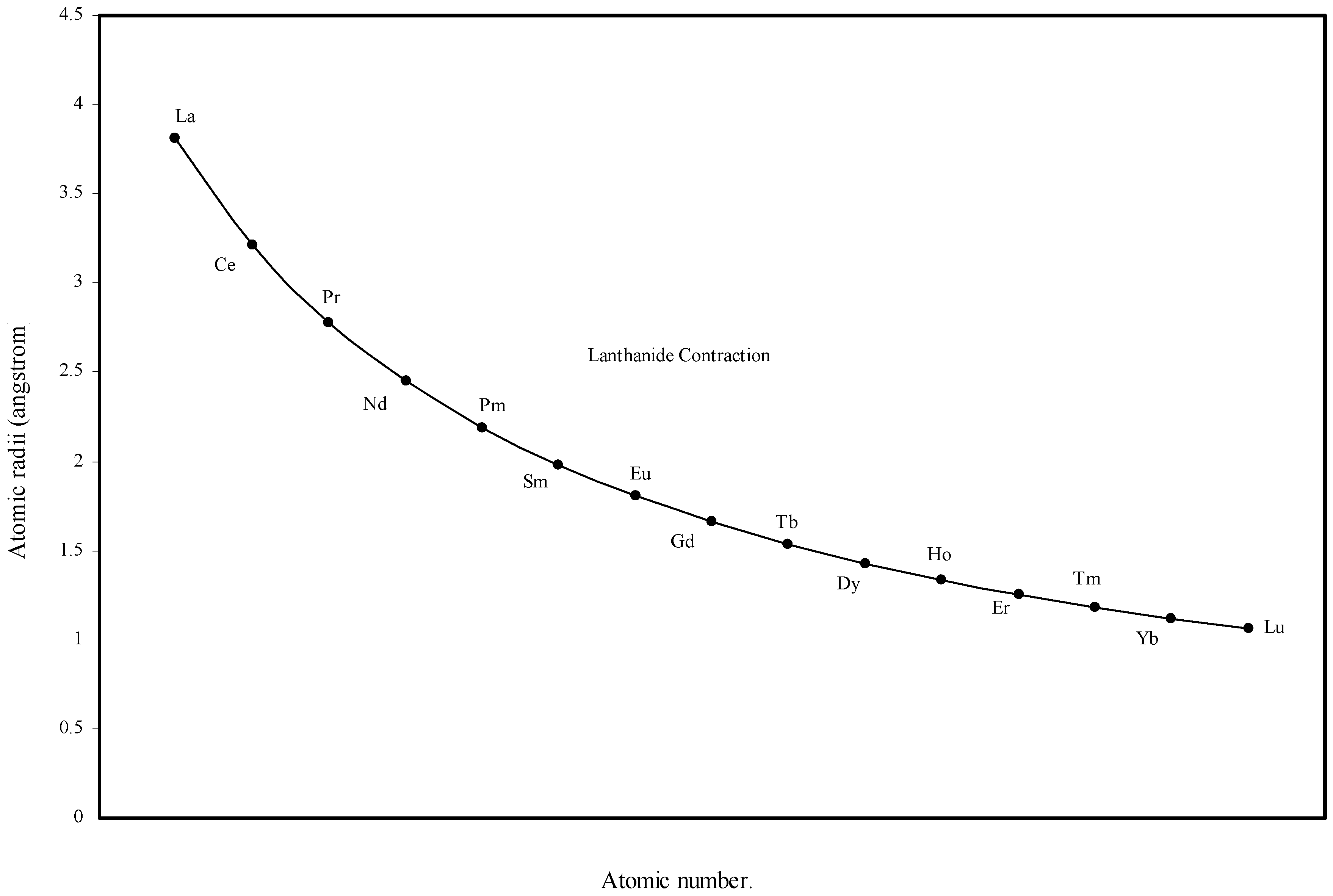
Figure 5.
Plot of atomic radii of lanthanides in angstrom unit as function of atomic number.
Figure 5.
Plot of atomic radii of lanthanides in angstrom unit as function of atomic number.
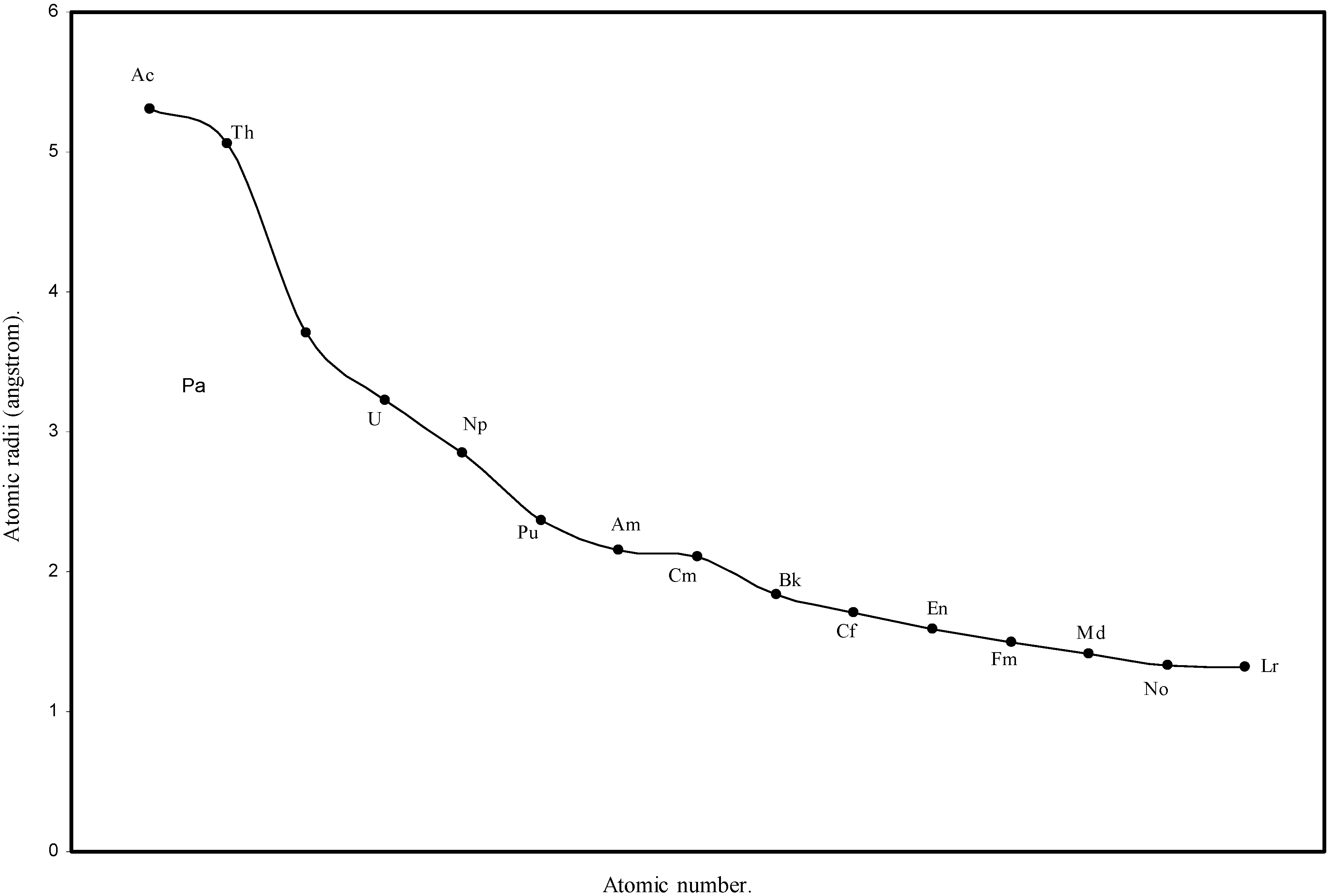
Figure 6.
Plot of atomic radii of actinides in angstrom unit as a function of Atomic number.
Figure 6.
Plot of atomic radii of actinides in angstrom unit as a function of Atomic number.
(A) Diamagnetic part of magnetic susceptibility, χdia.
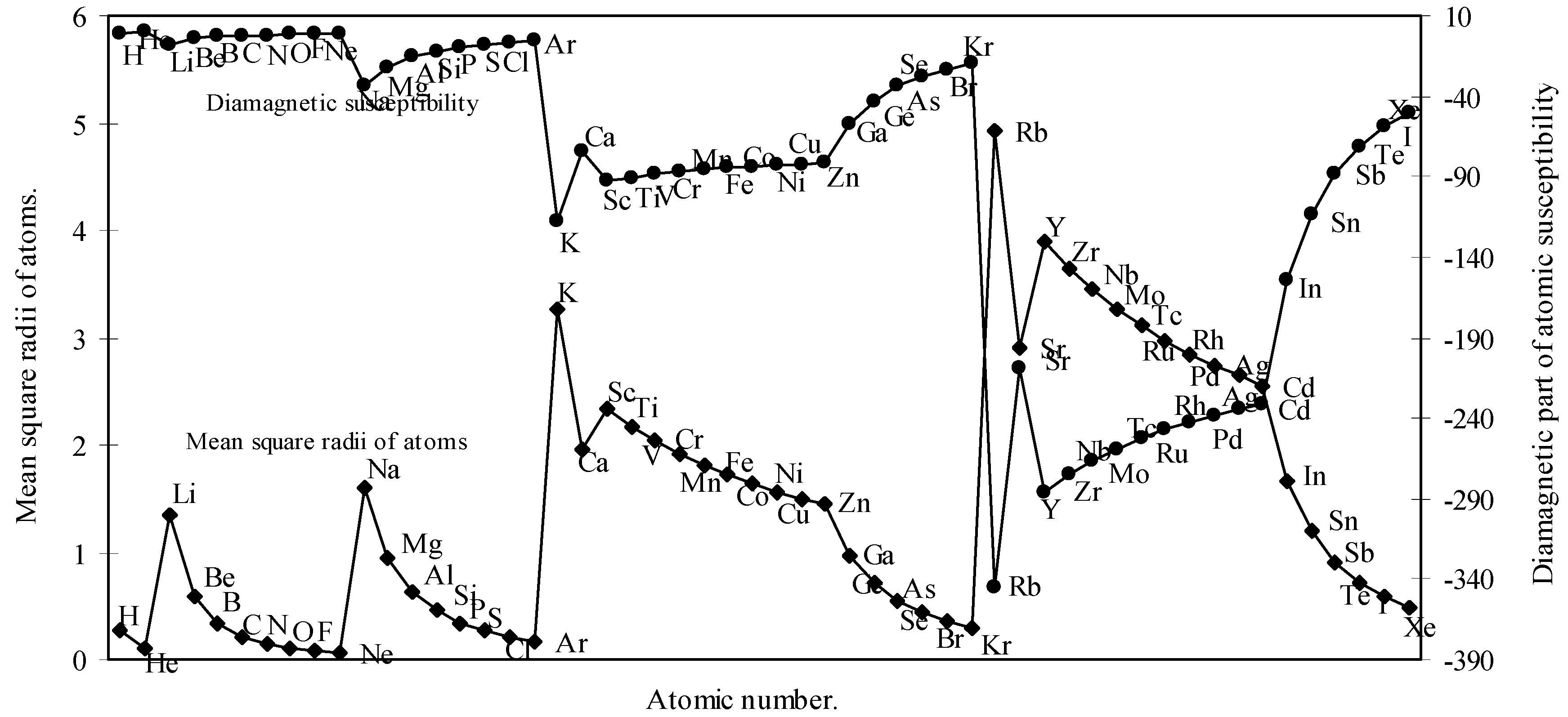
The theoretical determination of atomic and ionic radii provides with a scope of a theoretical calculation of the diamagnetic part of the atomic susceptibility, which occurs even when an atom already has a permanent moment. We have computed the diamagnetic susceptibility of as many as 54 elements starting from hydrogen. Since atoms after element with atomic number 54 are strongly paramagnetic due to L-S coupling, the computation of diamagnetic susceptibility of atoms of such elements is not taken up.
The induced diamagnetic moment of an atom is opposite to the applied field and is proportional to the square of the orbit radius. The classical equation of diamagnetic susceptibility of atom [
16,
17] per mole is given by
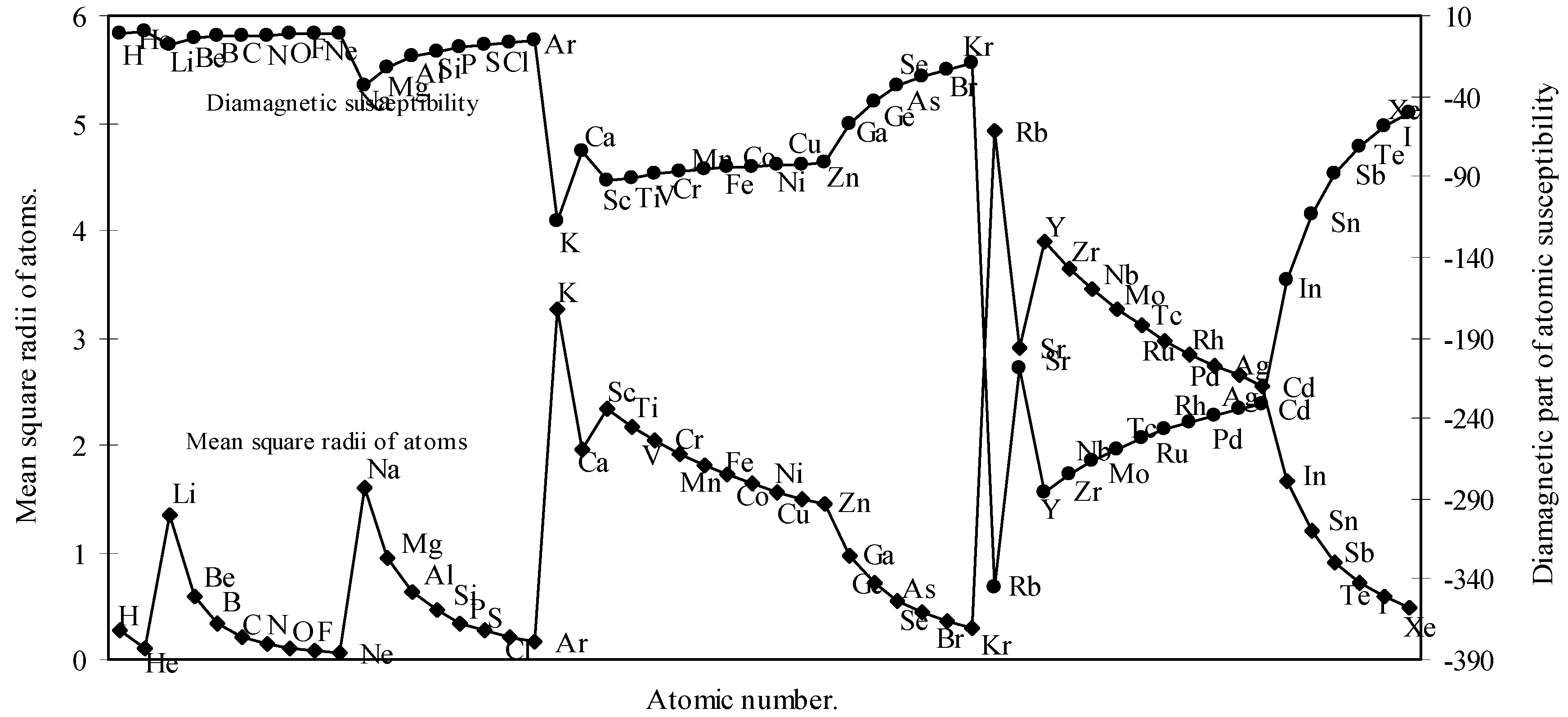
Where, <r2>av is the mean square of the actual orbital radius or the average of the square of all the orbit radii and Σ implies the summation of <r2>av for all n electrons in atom as the total moment is given by the sum over all the electrons in the atom.
There can not be any experimental value for Σ
n <r
2>
av of an atom as such and quantum mechanics can compute it and it is interpreted as the average of the square of the distance from the center for the probability distribution [
18]. However, it is mentioned that a slight modification of the formula (8) by replacing the pre-factor of the RHS by 1.888 in place of 2.84 gives better result [
19]. The modified formula is
We have calculated the <r
2>
av of each atom by calculating the radii of each orbital. We have computed the <r
2>
av for all the 54 elements. With these values of <r
2>
av, the molar diamagnetic susceptibility is computed through eqn.(9). The calculated <r
2>
av and χ
dia are shown in
Table 2. The computed values of <r
2>
av and χ
dia are plotted as a function of atomic number in
Fig.7.
Figure 7.
Plot of mean square radii and diamagnetic part of magnetic susceptibility of atoms as a function of atomic number.
Figure 7.
Plot of mean square radii and diamagnetic part of magnetic susceptibility of atoms as a function of atomic number.
(B) Atomic polarizability, α.
One more important size dependent physical property of the atoms is the atomic polarizability, α. By polarizability, Pearson [
20] meant the ease of deforming the valance electron cloud of a chemical species. A closely related experimental quantity is electric dipole polarizability, α, which actually
Table 2.
Computed mean square radii, < r2 > and diamagnetic part of atomic susceptibility, (χdia) of atoms.
Table 2.
Computed mean square radii, < r2 > and diamagnetic part of atomic susceptibility, (χdia) of atoms.
| Atom | < r2 > X 10–16 sq.cm | χdia x 10–6 c.c | Atoms | < r2 > X 10–16 sq.cm | χdia x 10–6 c.c |
|---|
| H | 0.2801 | -0.5287 | Ni | 1.5669 | -82.8339 |
| He | 0.0969 | -0.3659 | Cu | 1.5013 | -82.2003 |
| Li | 1.3447 | -7.6165 | Zn | 1.4422 | -81.6834 |
| Be | 0.5994 | -4.5267 | Ga | 0.9694 | -56.7355 |
| B | 0.3377 | -3.1882 | Ge | 0.7087 | -42.8176 |
| C | 0.2164 | -2.4513 | As | 0.5452 | -33.912 |
| N | 0.1504 | -1.9872 | Se | 0.4345 | -27.8915 |
| O | 0.1106 | -1.6698 | Br | 0.3553 | -23.4787 |
| F | 0.0847 | -1.4395 | Kr | 0.2965 | -20.1491 |
| Ne | 0.0669 | -1.264 | Rb | 4.9346 | -344.7138 |
| Na | 1.5972 | -33.1713 | Sr | 2.9131 | -208.9958 |
| Mg | 0.9558 | -21.6546 | Y | 3.8844 | -286.0142 |
| Al | 0.6368 | -15.6297 | Zr | 3.6483 | -275.519 |
| Si | 0.4549 | -12.0238 | Nb | 3.444 | -266.593 |
| P | 0.3413 | -9.6652 | Mo | 3.266 | -258.9836 |
| S | 0.2655 | -8.0205 | Tc | 3.1099 | -252.4745 |
| Cl | 0.2125 | -6.8214 | Ru | 2.9723 | -246.9151 |
| Ar | 0.1741 | -5.915 | Rh | 2.8505 | -242.1745 |
| K | 3.2677 | -117.2196 | Pd | 2.7419 | -238.1305 |
| Ca | 1.9664 | -74.2518 | Ag | 2.6448 | -234.6907 |
| Sc | 2.338 | -92.6958 | Cd | 2.5577 | -231.7906 |
| Ti | 2.1789 | -90.5027 | In | 1.6726 | -154.7359 |
| V | 2.0413 | -88.6433 | Sn | 1.2015 | -113.4178 |
| Cr | 1.9214 | -87.0608 | Sb | 0.9035 | -87.9572 |
| Mn | 1.8158 | -85.7077 | Te | 0.7218 | -70.8638 |
| Fe | 1.723 | -84.5766 | I | 0.5866 | -58.7022 |
| Co | 1.6403 | -83.6167 | Xe | 0.4872 | -49.6685 |
describes the linear response of the electron cloud of a chemical species to an external field much lower than what would be needed to ionize the system. The static electric dipole polarizability α can be determined theoretically and experimentally in various ways.
The induced dipole moment in an atom or molecule is proportional to the applied electric field [
13] and the necessary formula can be written as
where,
F = the applied electric field,
d = induced dipole moment, and α is the constant of proportionality and is called polarizability.
It has been shown [
21] that the polarizability α of a conducting sphere of radius r is equal to r
3.
And in terms of its volume V, the same formula looks like
where
The generality of linear volume–polarizability has been assessed [
21] quantum mechanically in case of atoms. Although the formula for the polarizability has been laid down in eqn (11), it is suggested that, due to inhomogeneity of the electron cloud, the actual formula [
22] should be
where, K is the proportionality constant.
For atoms, the value of ‘K’ was determined by Dimitrieva and Plindov [
23] by ‘Atomic Oscillation Theory’ as 0.585. Dutta and Hati [
22] used the value of K= 0.792 which is the arithmetic average of 1 and the value of Dimitrieva and Plindov [
23]. But the exact quantum mechanical calculation [
19] of polarizability of hydrogen atom derives the formula
where a
0 is the Bohr radius.
Comparing eqns. (14) and (15) we see that K = 4.5
Relying more upon the quantum mechanical derivation [
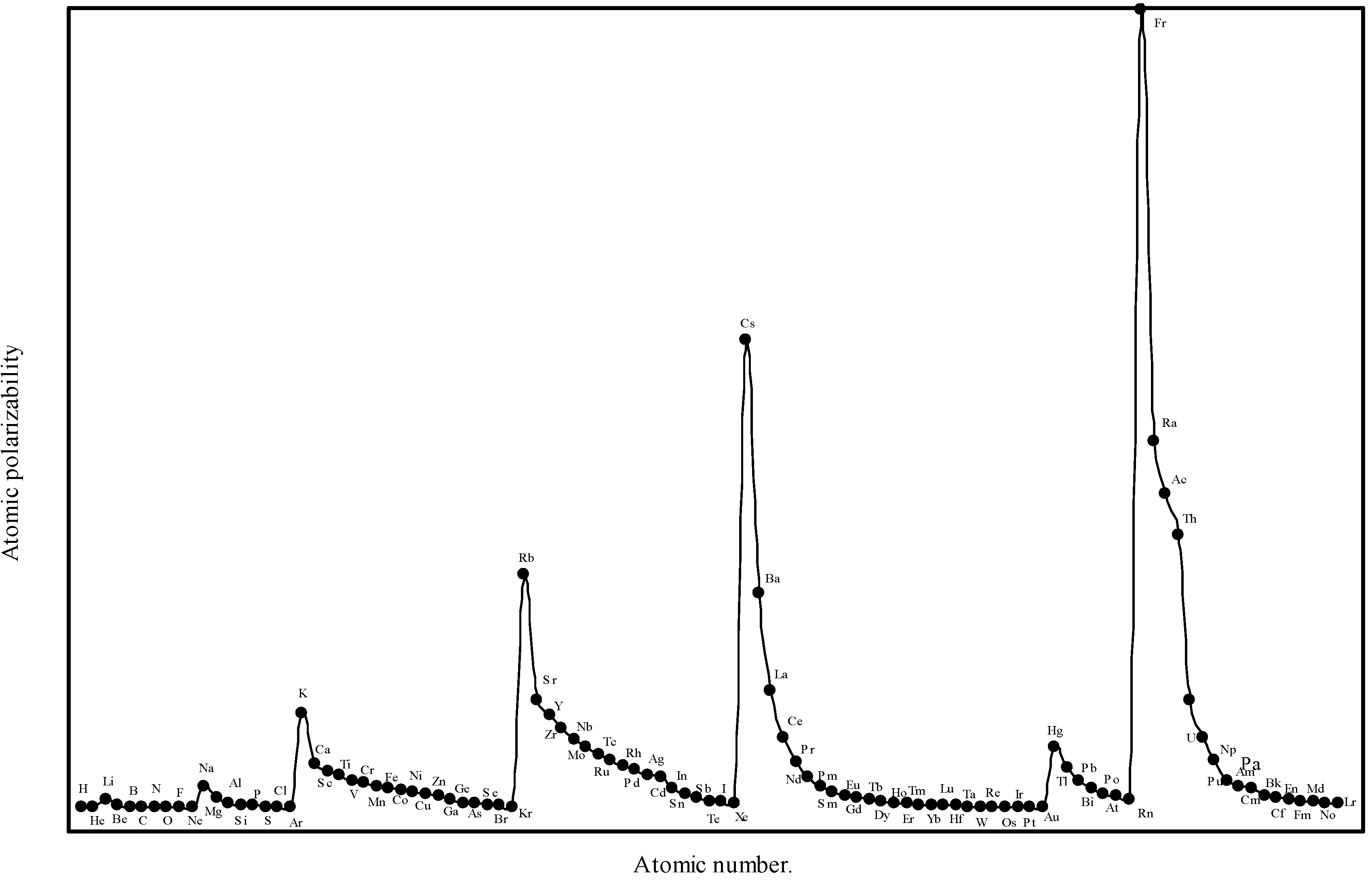
19] of the atomic polarizability, we see that, the proportional constant K in eqn.(14) is neither 1 nor less than 1, but it is equal to 4.5 for hydrogen atom. In absence of any other reliable value of ‘K’, the proportionality constant, we have first tried to obtain an optimized the proportionality constant K. We first computed the atomic polarizability of as much as 8 elements whose experimental polarizabilities are quoted by Purcell in reference [
19] using the above quantum mechanical value of K = 4.5. We find that the calculated values of atomic polarizability of eight elements with K = 4.5 through the theoretical atomic radii are close to the experimental values quoted by Purcell [
19]. We have, therefore, computed the atomic polarizabilities of the rest of the elements with the same value of the proportionality constant K=4.5. The computed atomic polarizability of all the 103 elements are shown in
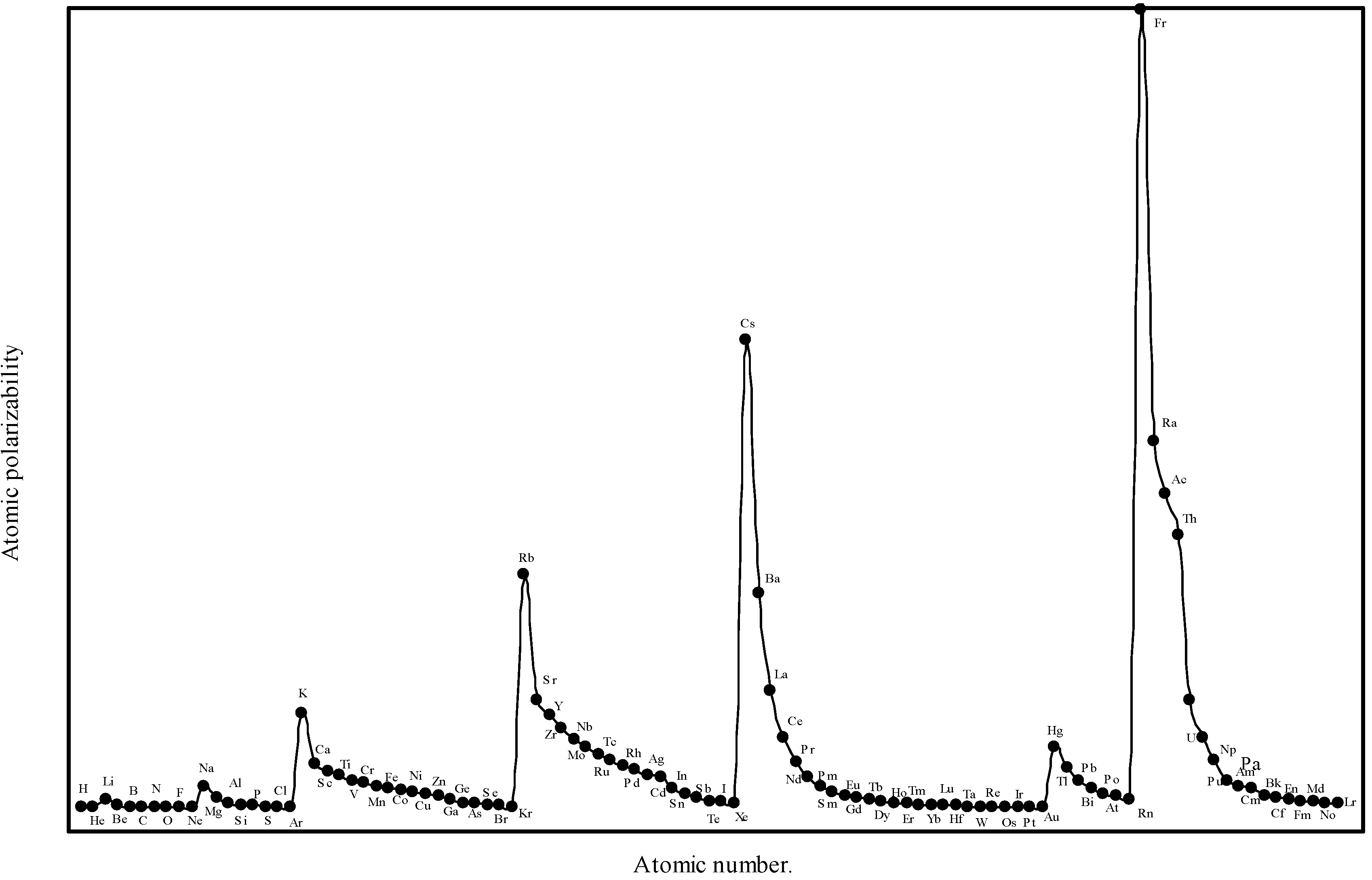
Table 3. We have plotted the theoretically computed atomic polarizability as a function of atomic number in
Figure 8.
Figure 8.
Plot of atomic polarizability (c.c) as a function of atomic number.
Figure 8.
Plot of atomic polarizability (c.c) as a function of atomic number.
Table 3.
Computed atomic polarizability ( α) and global hardness (η) of atoms.
Table 3.
Computed atomic polarizability ( α) and global hardness (η) of atoms.
| Atoms | α x 10–24 c.c | η (ev) | Atoms | α x 10–24 c.c | η (ev) | Atoms | α x 10–24 c.c | η (ev) |
|---|
| H | 0.6669 | 13.588 | Kr | 3.8497 | 7.5748 | Lu | 5.3338 | 6.7947 |
| He | 0.1358 | 22.383 | Rb | 500.9683 | 1.4948 | Hf | 4.6075 | 7.1344 |
| Li | 19.4239 | 4.4164 | Sr | 230.4462 | 1.9364 | Ta | 3.9739 | 7.4951 |
| Be | 5.7558 | 6.6244 | Y | 197.5715 | 2.0383 | W | 3.4643 | 7.8459 |
| B | 2.428 | 8.8328 | Zr | 170.6683 | 2.1402 | Re | 3.0385 | 8.1965 |
| C | 1.2432 | 11.0407 | Nb | 148.4397 | 2.2422 | Os | 2.6796 | 8.5472 |
| N | 0.7193 | 13.25 | Mo | 129.9126 | 2.344 | Ir | 2.3756 | 8.8973 |
| O | 0.453 | 15.4574 | Tc | 114.3315 | 2.446 | Pt | 2.1175 | 9.2474 |
| F | 0.3034 | 17.6634 | Ru | 101.1523 | 2.5479 | Au | 1.8924 | 9.598 |
| Ne | 0.2131 | 19.875 | Rh | 89.9286 | 2.6498 | Hg | 129.646 | 2.3456 |
| Na | 45.659 | 3.3215 | Pd | 80.3028 | 2.7517 | Tl | 85.3653 | 2.6962 |
| Mg | 21 | 4.303 | Ag | 72.005 | 2.8536 | Pb | 59.1717 | 3.0466 |
| Al | 11.337 | 5.2846 | Cd | 64.8095 | 2.9555 | Bi | 42.6767 | 3.3972 |
| Si | 6.8011 | 6.2659 | In | 42.6767 | 3.3972 | Po | 31.7858 | 3.7477 |
| P | 4.3955 | 7.2473 | Sn | 29.5777 | 3.8388 | At | 24.3079 | 4.0983 |
| S | 3.0023 | 8.2293 | Sb | 21.3335 | 4.2805 | Rn | 19.0046 | 4.4487 |
| Cl | 2.1412 | 9.2107 | Te | 15.8906 | 4.7221 | Fr | 1705 | 0.9913 |
| Ar | 1.5808 | 10.191 | I | 12.1532 | 5.0636 | Ra | 785.4977 | 1.2867 |
| K | 202.9969 | 2.02 | Xe | 9.4993 | 5.6056 | Ac | 673.4033 | 1.3544 |
| Ca | 93.3717 | 2.6162 | Cs | 1002.1964 | 1.1863 | Th | 581.6815 | 1.4222 |
| Sc | 80.0633 | 2.7545 | Ba | 460.9098 | 1.5369 | Pa | 228.7156 | 1.9413 |
| Ti | 69.1712 | 2.8924 | La | 248.9177 | 1.8873 | U | 149.9164 | 2.2348 |
| V | 60.1472 | 3.03 | Ce | 149.2883 | 2.2378 | Np | 103.5473 | 2.5281 |
| Cr | 52.6438 | 3.1676 | Pr | 96.4738 | 2.5885 | Pu | 59.1266 | 3.0473 |
| Mn | 46.3265 | 3.3055 | Nd | 65.9186 | 2.9389 | Am | 44.8789 | 3.3407 |
| Fe | 40.9939 | 3.443 | Pm | 47.0135 | 3.2893 | Cm | 42.2547 | 3.4084 |
| Co | 36.4555 | 3.5811 | Sm | 34.6984 | 3.6398 | Bk | 27.5918 | 3.9277 |
| Ni | 32.5372 | 3.7187 | Eu | 26.3316 | 3.9905 | Cf | 22.2453 | 4.2212 |
| Cu | 29.1769 | 3.8561 | Gd | 20.4544 | 4.341 | En | 18.1843 | 4.5146 |
| Zn | 26.2615 | 3.994 | Tb | 16.2057 | 4.6913 | Fm | 15.0542 | 4.808 |
| Ga | 17.2917 | 4.5909 | Dy | 13.0543 | 5.0419 | Md | 12.6038 | 5.1013 |
| Ge | 11.9864 | 5.1874 | Ho | 10.6707 | 5.3924 | No | 10.6563 | 5.3949 |
| As | 8.6443 | 5.7846 | Er | 8.8334 | 5.743 | Lr | 10.2631 | 5.4629 |
| Se | 6.4397 | 6.381 | Tm | 7.3955 | 6.0934 | | | |
| Br | 4.9244 | 6.978 | Yb | 6.253 | 6.4439 | | | |
(C) Global Hardness, η.
The term hardness,η, as applied to atoms and molecules has no reference to its mechanical physical hardness [
24]. We are concerned here about the chemical hardness of atoms. The definition of hardness has evolved with time from qualitative concept to the rigorous quantum mechanical definition [
20]. The concept of hardness was first introduced by Mulliken [
25] and formalized as a qualitative thumb rule of chemical reactivity known as HSAB principle by Pearson [
20]. Using the Density Functional Theory (DFT) as basis, the qualitative HSAB principle of Pearson [
20] was placed on a quantum mechanical basis by Parr, Pearson and others [
26,
27,
28,
29,
30]. In terms of Density Functional Theory, the qualitative idea of hardness and softness was quantified and the terms ‘hardness’ and ‘softness’ were identified as fundamental theoretical parameters. Although the hardness was rigorously defined in terms of the Density Functional Theory, the operational and approximate definition for the purpose of computation was also suggested as follows [
27,
28,
30].
The operational definition of hardness, η
where,
But in case of atoms, Pearson [
31] pointed out that a simple formula of computing hardness of atoms could be derived from simple electrostatic considerations using atomic size. Dutta and Hati [
22] have used the formula derived by Pearson to calculate the global hardness of a series of atoms. The only needed quantity for computing global hardness of atoms is the atomic radius. We therefore venture to use the atomic radii for the instant calculation to compute the global hardness η of atoms. We have derived the formula for computing the global hardness of atoms as follows:
The energy, E (q) of charging a conducting sphere of radius R with charge q is classically given by [
18]
where, q = charge and R = radius.
The implication of the eqn (18) is that the E(q) is the energy in ergs, q is the charge in electrostatic unit and R should be measured in cm. Now increasing q by one unit and decreasing q by one unit, we get the changes in energy which are I and A, the ionization potential and electron affinity of atom respectively.
Now in e.s.u, the one unit of charge, which is associated with the process of ionization I, and the electron affinity A, should be e, the electronic charge. Hence,
Since, the global hardness of atoms η = 1/2 (I – A)
Or,
where, e = the electronic charge in e.s.u.
But the formula derived by Pearson [
31] looks a bit different from the formula in eqn. (19).
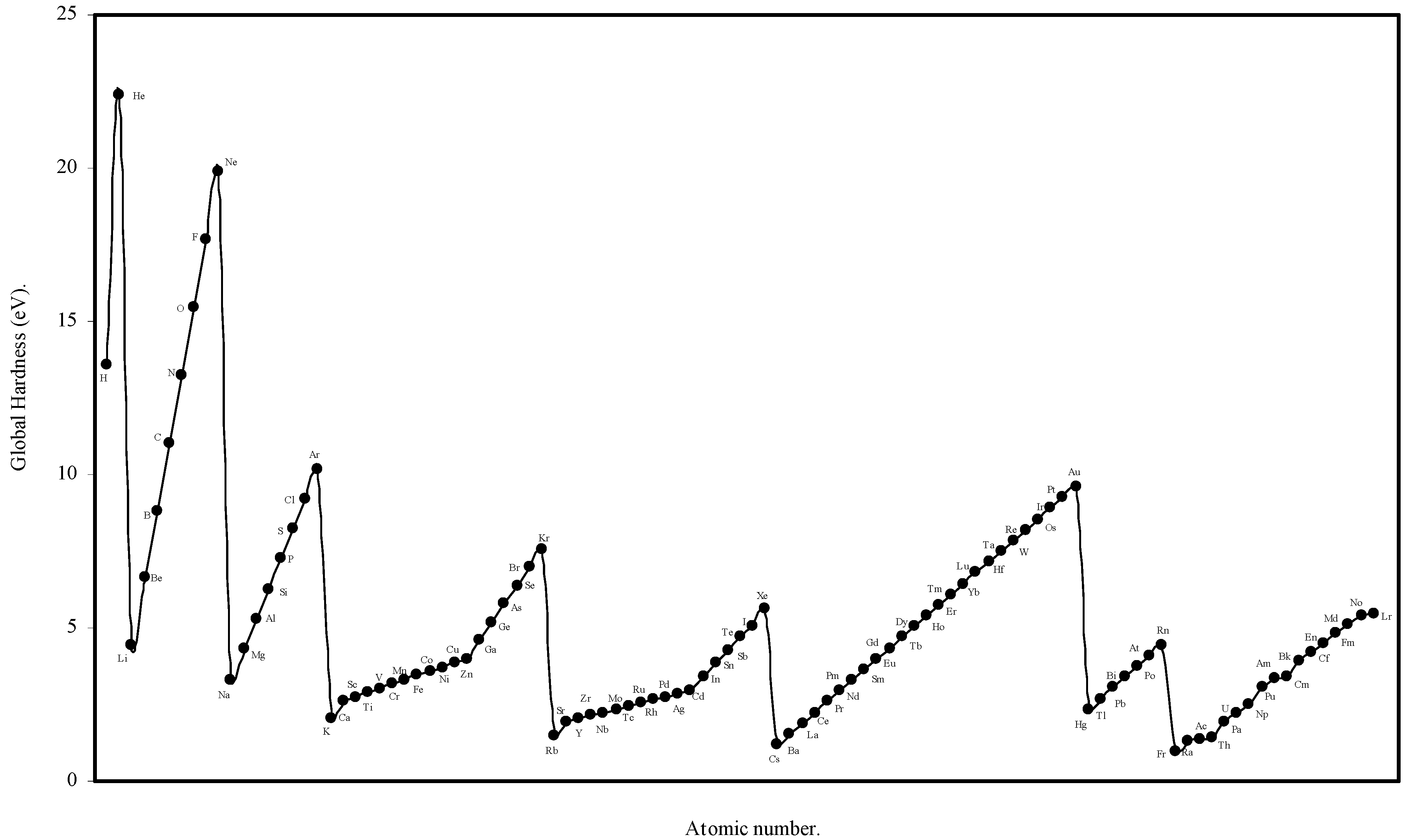
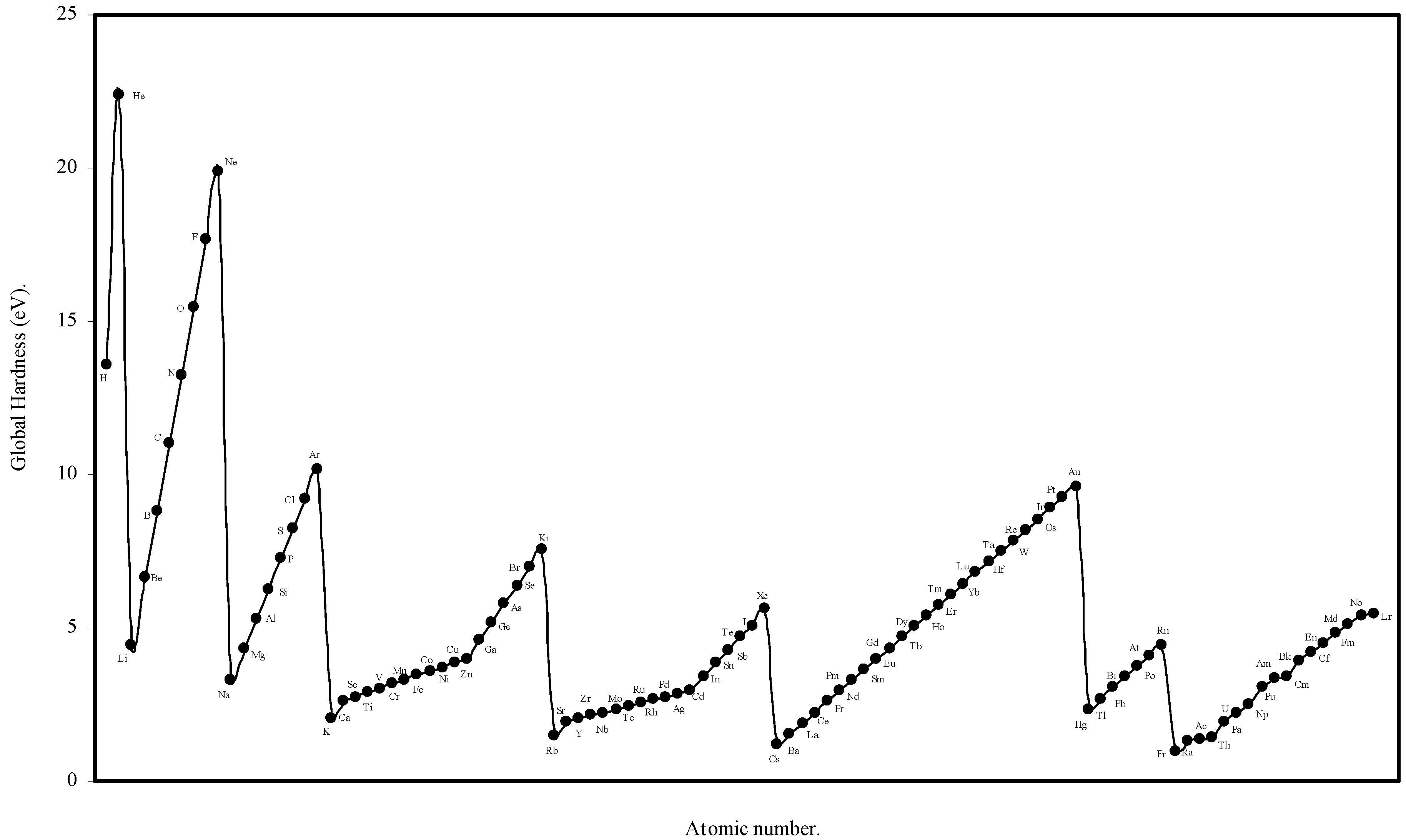
We have computed the global hardness of atoms, η, through eqn (19) and ultimately it is converted to electron volt (eV). The theoretically computed values of global hardness of as many as 103 atoms are tabulated in
Table 3. We have plotted the global hardness of atoms as a function of atomic number in
Figure 9.
Figure 9.
Plot of Global hardness (eV) of atoms as a function of atomic number.
Figure 9.
Plot of Global hardness (eV) of atoms as a function of atomic number.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}