
Transcriptome Analysis Reveals Association of E-Class AmMADS-Box Genes with Petal Malformation in Antirrhinum majus L.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
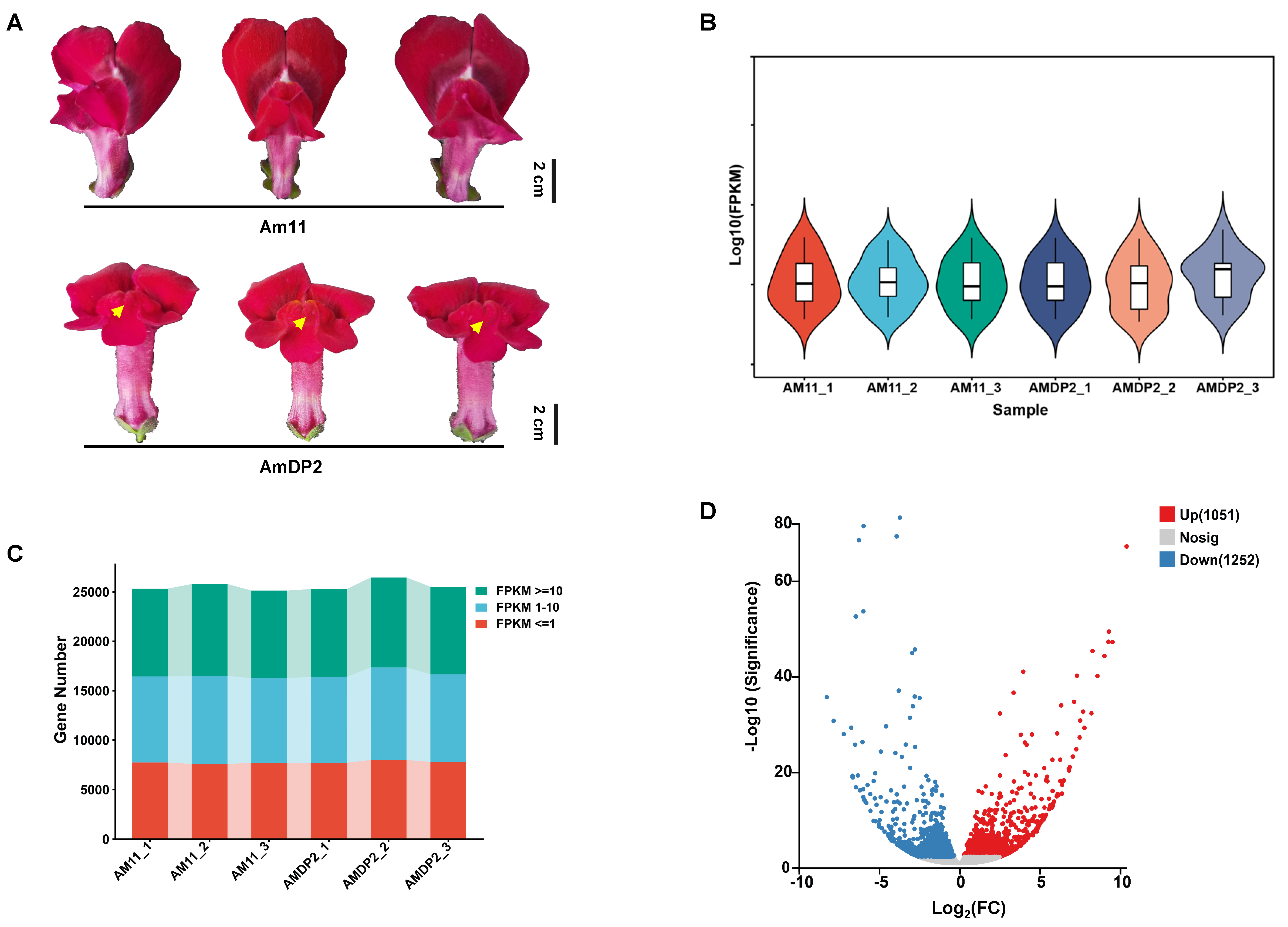
2.1. Sequencing Read Filtering and Identification of Differentially Expressed Genes (DEGs)
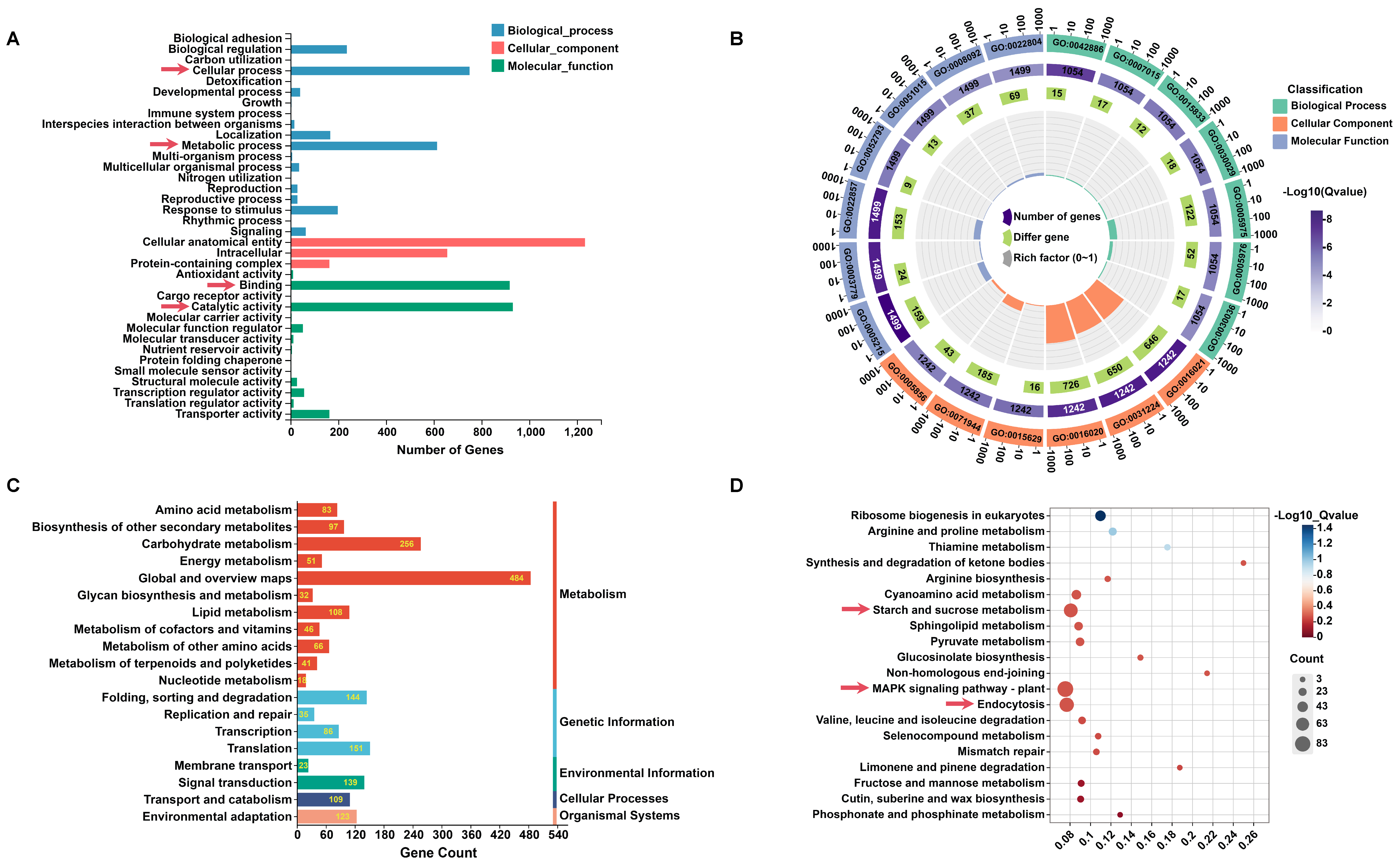
2.2. GO and KEGG Pathway Enrichment of DEGs
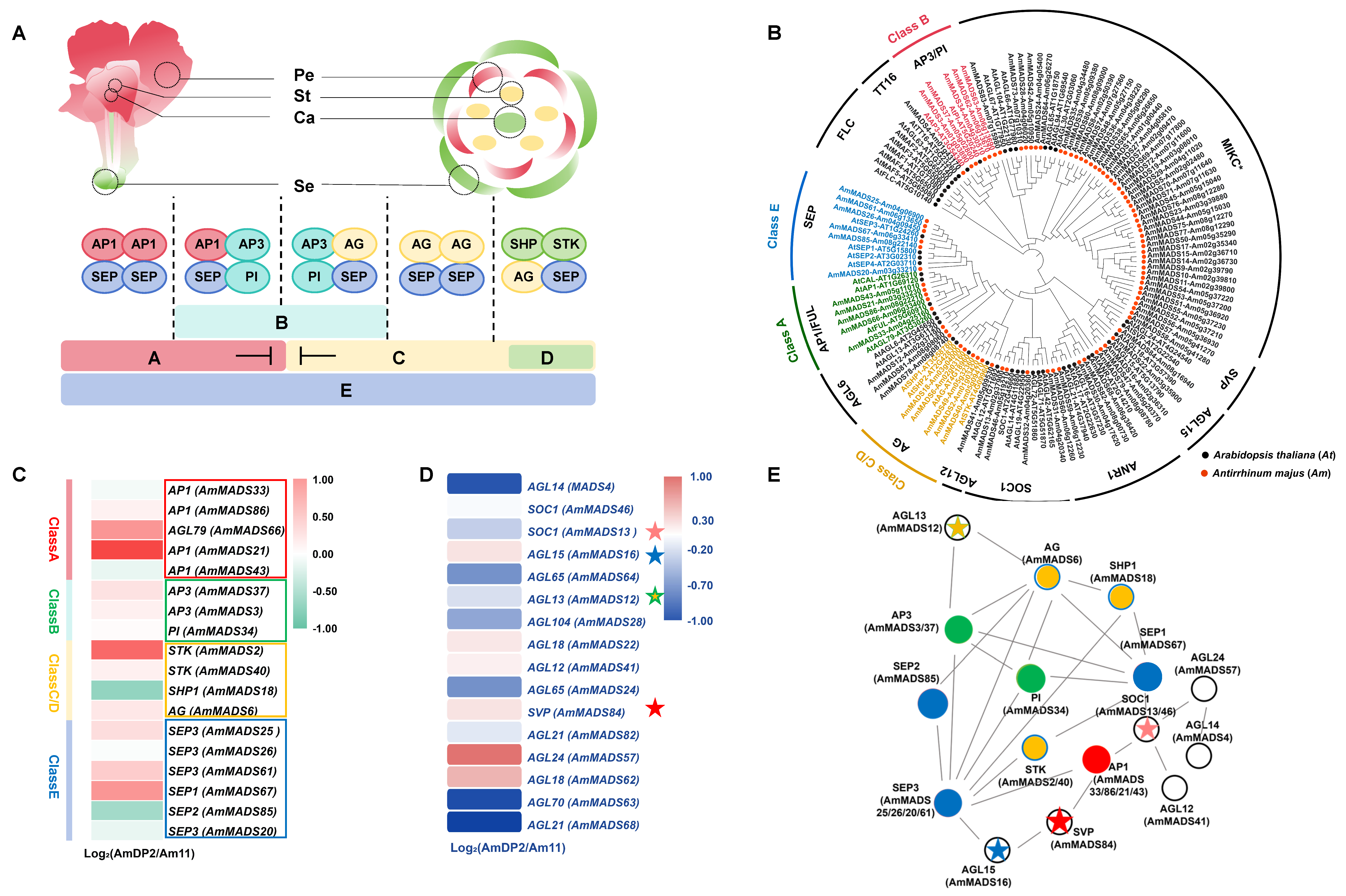
2.3. Identification and Evolutionary Conservation of MIKC-Type AmMADS Genes
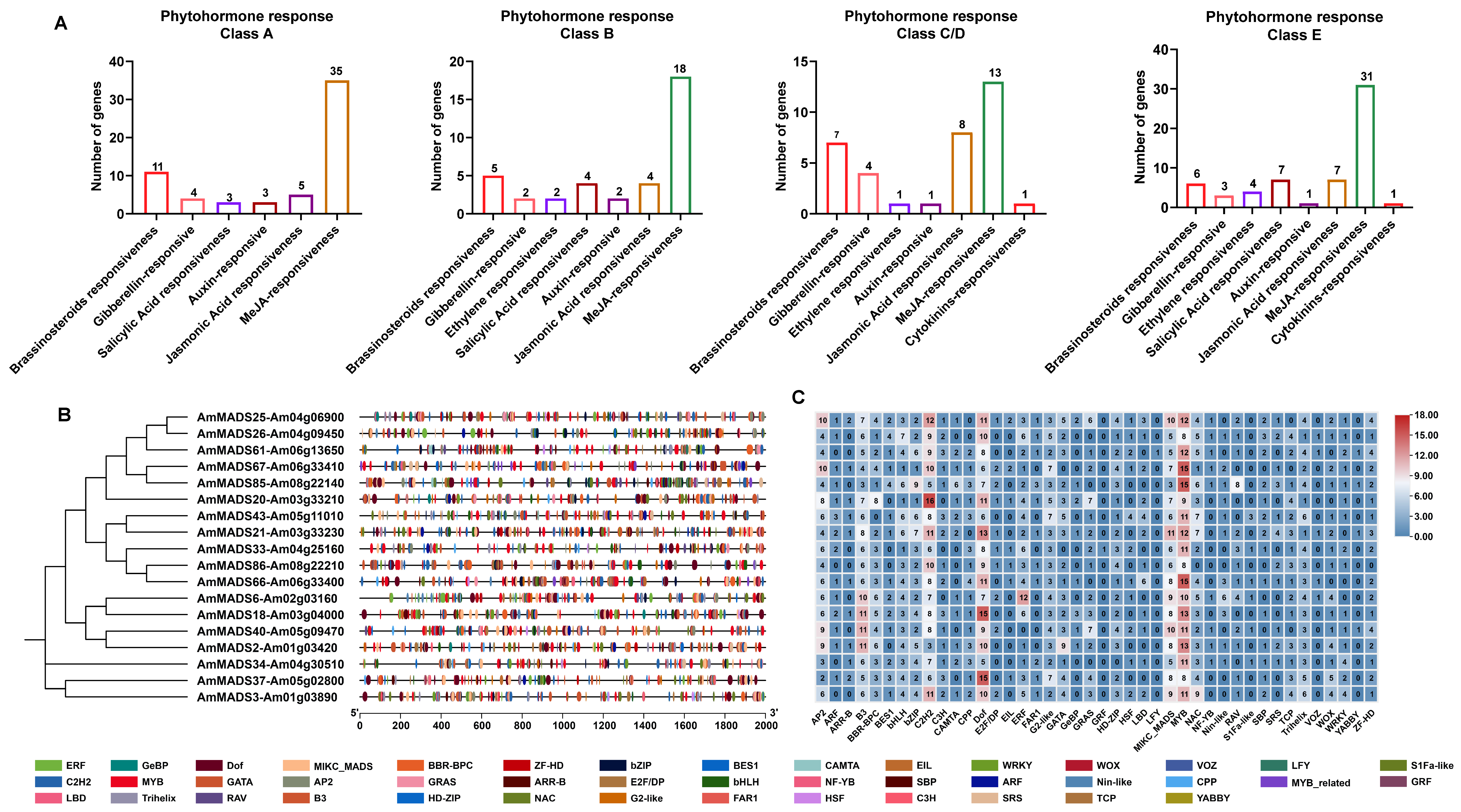
2.4. Cis-Acting Element and TFBS Analysis in ABCDE Model DEGs Promoters
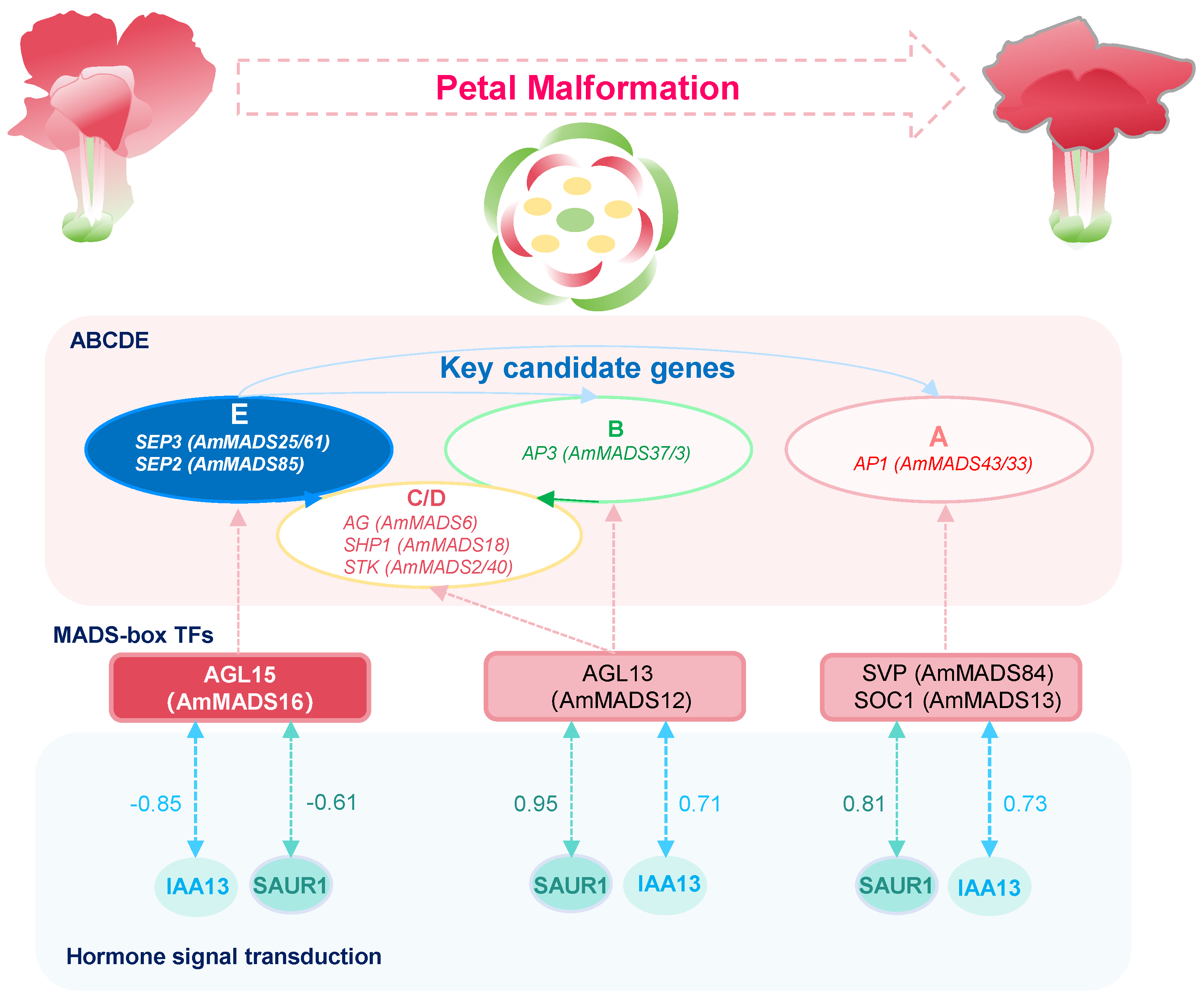
2.5. Co-Expression Networks Among Phytohormones, TFs, and ABCDE Model DEGs
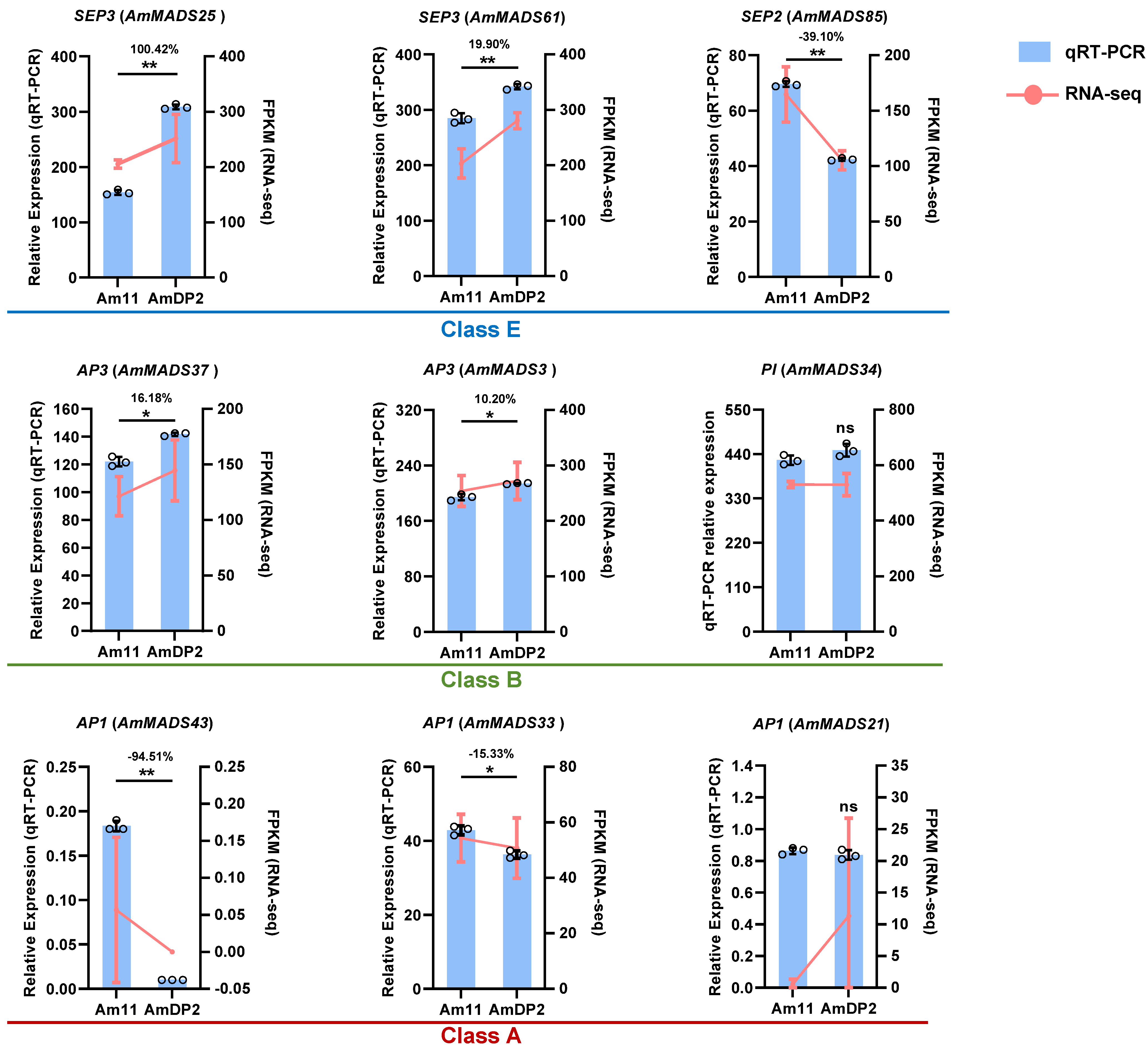
2.6. Validation of Key ABCDE Model Genes by qRT-PCR
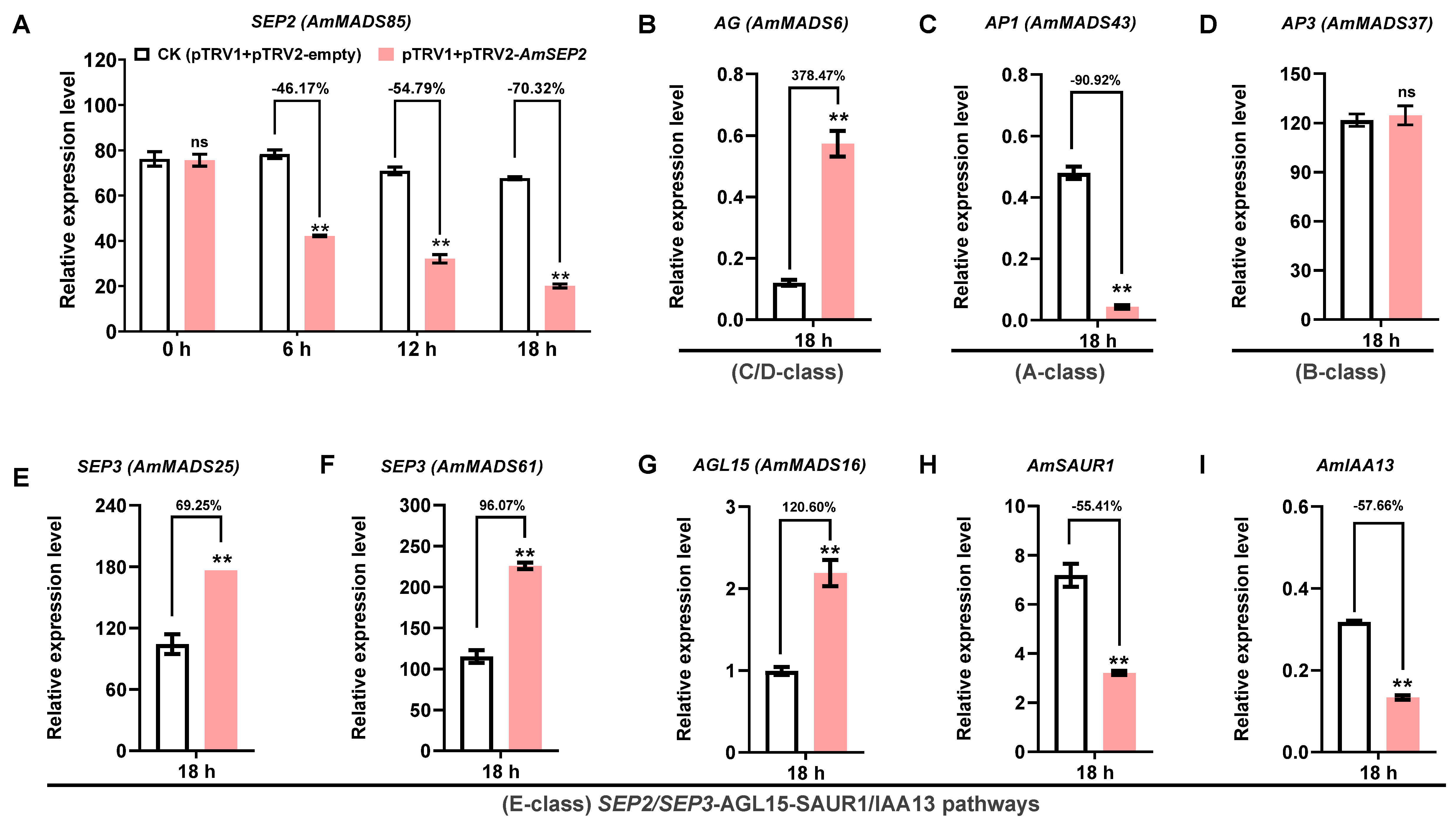
2.7. Silencing of AmSEP2 Promotes Petal Malformation in A. majus: Gene Network Interactions
3. Discussion
3.1. Integrated Complex Approaches as a Powerful Strategy for Functional Gene Discovery
3.2. E-Class AmMADS-Box Genes: Central Factor in Petal Malformation of A. majus
3.3. Complex Regulatory Networks Underlying Petal Malformation in A. majus
4. Material and Methods
4.1. Plant Materials and Sample Preparation
4.2. cDNA Library Construction and RNA-Seq
4.3. Sequencing Data Filtering and DEG Identification
4.4. GO and KEGG Functional Enrichment Analysis
4.5. Identification of MIKC-Type AmMADS-box Genes
4.6. Chromosomal Localization and Phylogenetic Analysis
4.7. Protein Interaction and Subcellular Localization
4.8. Cis-Acting Element and TFBS Analysis
4.9. WGCNA for Co-Expression Network Analysis
4.10. Correlation Heatmap of ABCDE Model Genes, Phytohormones, and TFs Genes
4.11. Nucleic Acid Isolation and qRT-PCR Analysis
4.12. VIGS Vector Construction and Transient Transformation
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saedler, H.; Huijser, P. Molecular biology of flower development in Antirrhinum majus (snapdragon). Gene 1993, 135, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Schwarz-Sommer, Z.; Davies, B.; Hudson, A. An everlasting pioneer: The story of Antirrhinum research. Nat. Rev. Genet. 2003, 4, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.L.; Hu, H.Z.; Shi, S.B.; Yuan, X.M.; Yan, B.; Chen, L.Q. An update on the function, biosynthesis and regulation of floral volatile terpenoids. Horticulturae 2021, 7, 451. [Google Scholar] [CrossRef]
- Shan, H.Y.; Cheng, J.; Zhang, R.; Yao, X.; Kong, H.Z. Developmental mechanisms involved in the diversification of flowers. Nat. Plants 2019, 5, 917–923. [Google Scholar] [CrossRef]
- Irish, V.F. The Arabidopsis petal: A model for plant organogenesis. Trends Plant Sci. 2008, 13, 430–436. [Google Scholar] [CrossRef]
- Fu, X.H.; Shan, H.Y.; Yao, X.; Cheng, J.; Jiang, Y.C.; Yin, X.F.; Kong, H.Z. Petal development and elaboration. J. Exp. Bot. 2022, 73, 3308–3318. [Google Scholar] [CrossRef]
- Bowman, J.L.; Moyroud, E. Reflections on the ABC model of flower development. Plant Cell 2024, 36, 1334–1357. [Google Scholar] [CrossRef]
- Theißen, G. Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant Biol. 2001, 4, 75–85. [Google Scholar] [CrossRef]
- Díaz, J.; Álvarez-Buylla, E.R. Spatio-temporal dynamics of the patterning of Arabidopsis flower meristem. Front. Plant Sci. 2021, 15, 585139. [Google Scholar] [CrossRef]
- Krizek, B.A.; Fletcher, J.C. Molecular mechanisms of flower development: An armchair guide. Nat. Rev. Genet. 2005, 6, 688–698. [Google Scholar] [CrossRef]
- Thomson, B.; Wellmer, F. Molecular regulation of flower development. Curr. Top. Dev. Biol. 2019, 131, 185–210. [Google Scholar] [PubMed]
- Theißen, G.; Melzer, R.; Rümpler, F. MADS-domain transcription factors and the floral quartet model of flower development: Linking plant development and evolution. Development 2016, 143, 3259–3271. [Google Scholar] [CrossRef] [PubMed]
- Salamah, A.; Prihatiningsih, R.; Rostina, I.; Dwiranti, A. Comparative morphology of single and double flowers in Hibiscus rosa-sinensis L. (Malvaceae): A homeosis study. AIP Conf. Proc. 2018, 2023, 020136. [Google Scholar]
- Ó’Maoiléidigh, D.S.; Graciet, E.; Wellmer, F. Gene networks controlling Arabidopsis thaliana flower development. New Phytol. 2014, 201, 16–30. [Google Scholar] [CrossRef]
- Causier, B.; Schwarz-Sommer, Z.; Davies, B. Floral organ identity: 20 years of ABCs. Semin. Cell Dev. Biol. 2010, 21, 73–79. [Google Scholar] [CrossRef]
- Honma, T.; Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 2001, 409, 525–529. [Google Scholar] [CrossRef]
- Hugouvieux, V.; Blanc-Mathieu, R.; Janeau, A.; Paul, M.; Lucas, J.; Xu, X.C.; Ye, H.L.; Lai, X.L.; Hir, S.L.; Guillotin, A.; et al. SEPALLATA-driven MADS transcription factor tetramerization is required for inner whorl floral organ development. Plant Cell 2024, 36, 3435–3450. [Google Scholar] [CrossRef]
- Zhou, Y.Z.; Xu, Z.D.; Yong, X.; Ahmad, S.; Yang, W.R.; Cheng, T.R.; Wang, J.; Zhang, Q.X. SEP-class genes in Prunus mume and their likely role in floral organ development. BMC Plant Biol. 2017, 17, 10. [Google Scholar] [CrossRef]
- Qi, F.T.; Liu, Y.T.; Luo, Y.L.; Cui, Y.M.; Lu, C.F.; Li, H.; Huang, H.; Dai, S.L. Functional analysis of the ScAG and ScAGL11 MADS-Box transcription factors for anthocyanin biosynthesis and bicolour pattern formation in Senecio cruentus ray florets. Hortic. Res. 2022, 9, uhac071. [Google Scholar] [CrossRef]
- Weigel, D.; Meyerowltzt, E.M. The ABCs of floral homeotic genes review. Cell 1994, 78, 203–209. [Google Scholar] [CrossRef]
- Jack, T. Relearning our ABCs: New twists on an old model. Trends Plant Sci. 2001, 6, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.R.; Liu, H.L.; Du, H.Y.; Wang, S.J.; Yang, W.M.; Chi, Y.J.; Wang, J.; Huang, F.; Yu, D.Y. Soybean MADS-box gene GmAGL1 promotes flowering via the photoperiod pathway. BMC Genom. 2018, 19, 51. [Google Scholar]
- Jia, H.; Chen, R.; Cong, B.; Cao, K.; Sun, C.; Luo, D. Characterization and transcriptional profiles of two rice MADS-Box genes. Plant Sci. 2000, 155, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.M.; Wang, X.; Wang, Y.W.; Hu, R.B.; Zhang, X.M.; Chen, J.X.; Fu, Y.F. Genome-wide expression analysis of soybean MADS genes showing potential function in the seed development. PLoS ONE 2013, 8, e62288. [Google Scholar] [CrossRef]
- Kaufmann, K.; Melzer, R.; Theissen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Sun, J.Z.; Liu, Y.C.; Zheng, Y.H.; Xue, Y.G.; Fan, Y.H.; Ma, X.F.; Ji, Y.J.; Liu, G.Y.; Zhang, X.M.; Li, Y.; et al. The MADS-box transcription factor GmFULc promotes GmZTL4 gene transcription to modulate maturity in soybean. J. Integr. Plant Biol. 2024, 66, 1603–1619. [Google Scholar]
- Su, Y.L.; Liu, J.X.; Liang, W.Q.; Dou, Y.H.; Fu, R.F.; Li, W.Q.; Feng, C.Z.; Gao, C.X.; Zhang, D.B.; Kang, Z.S.; et al. Wheat AGAMOUS LIKE 6 transcription factors function in stamen development by regulating the expression of Ta APETALA3. Development 2019, 146, dev177527. [Google Scholar] [CrossRef]
- Yang, H.W.; Akagi, T.; Kawakatsu, T.; Tao, R. Gene networks orchestrated by MeGl: A single-factor mechanism underlying sex determination in persimmon. Plant J. 2019, 98, 97–111. [Google Scholar] [CrossRef]
- Ji, X.T.; Wang, M.L.; Xu, Z.Z.; Wang, K.; Sun, D.Y.; Niu, L.X. PlMYB308 regulates flower senescence by modulating ethylene biosynthesis in Herbaceous Peony. Front. Plant Sci. 2022, 13, 872442. [Google Scholar] [CrossRef]
- Xiang, X.J.; Sun, L.P.; Yu, P.; Yang, Z.F.; Zhang, P.P.; Zhang, Y.X.; Wu, W.X.; Chen, D.B.; Zhan, X.D.; Khan, R.M.; et al. The MYB transcription factor Baymax1 plays a critical role in rice male fertility. Theor. Appl. Genet. 2021, 134, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.; Mitsuda, N.; Goto, K.J.; Oshima, Y.; Ohme-Takagi, M.; Otagaki, S.; Matsumoto, S.; Shiratake, K. The petal-specific InMYB1 promoter functions by recognizing petaloid cells. Plant Cell Physiol. 2016, 57, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, Y.Q.; Wang, Y.; Li, Y.H.; He, J.N.; Li, Y.J.; Du, L.S.; Gao, Y.R.; Ma, N.; Gao, J.P.; et al. Disruption of transcription factor RhMYB123 causes the transformation of stamen to malformed petal in rose (Rosa hybrida). Plant Cell Rep. 2022, 41, 2293–2303. [Google Scholar] [CrossRef]
- Jia, P.; Xing, L.B.; Zhang, C.G.; Zhang, D.; Ma, J.J.; Zhao, C.P.; Han, M.Y.; Ren, X.L.; An, N. MdKNOX19, a class II knotted-like transcription factor of apple, plays roles in ABA signalling/sensitivity by targeting ABI5 during organ development. Plant Sci. 2021, 302, 110701. [Google Scholar] [CrossRef]
- Varaud, E.; Brioudes, F.; Szécsi, J.; Leroux, J.; Brown, S.; Perrot-Rechenmann, C.; Bendahmane, M. AUXIN RESPONSE FACTOR8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell 2011, 23, 973–983. [Google Scholar] [CrossRef]
- Liu, C.J.; Zhang, J.; Zhang, N.; Shan, H.Y.; Su, K.M.; Zhang, J.S.; Meng, Z.; Kong, H.Z.; Chen, Z.D. Interactions among proteins of floral MADS-box genes in basal eudicots: Implications for evolution of the regulatory network for flower development. Mol Biol Evol. 2010, 27, 1598–1611. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, 638–646. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.F.; Liu, Y.; Yu, J.; Yao, G.L.; Yang, H.G.; Yang, D.M.; Wu, Y.G. An integrated transcriptome and metabolome analysis reveals the gene network regulating flower development in Pogostemon Cablin. Front. Plant Sci. 2023, 14, 1201486. [Google Scholar] [CrossRef]
- Yu, Q.X.; Jin, X.L.; Liu, C.X.; Wen, Y.F. An integrated analysis of transcriptome and miRNA sequencing provides insights into the dynamic regulations during flower morphogenesis in petunia. Horticulturae 2022, 8, 284. [Google Scholar] [CrossRef]
- Zhang, X.N.; Lin, S.G.; Peng, D.; Wu, Q.S.; Liao, X.Z.; Xiang, K.L.; Wang, Z.H.; Tembork, L.R.; Bendahmane, M.; Bao, M.Z.; et al. Integrated multi-omic data and analyses reveal the pathways underlying key ornamental traits in carnation flowers. Plant Biotechnol. J. 2022, 20, 1182–1196. [Google Scholar] [CrossRef]
- Yang, J.; Wang, J.; Yang, D.M.; Xia, W.N.; Wang, L.; Wang, S.; Zhao, H.Q.; Chen, L.Q.; Hu, H.Z. Genome-wide analysis of CSL family genes involved in petiole elongation, floral petalization, and response to salinity stress in Nelumbo nucifera. Int. J. Mol. Sci. 2024, 25, 12531. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Wu, M.F.; Winter, C.M.; Berns, M.C.; Nole-Wilson, S.; Yamaguchi, A.; Coupland, G.; Krizek, B.A.; Wagner, D. A molecular framework for auxin-mediated initiation of flower primordia. Dev. Cell 2013, 24, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Pan, B.Z.; Li, L.; Yang, C.X.; Xu, Z.F. Developmental basis for flower sex determination and effects of cytokinin on sex determination in Plukenetia volubilis (Euphorbiaceae). Plant Reprod. 2020, 33, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Cheng, L.; Chen, C.L.; Wu, Y.X.; Wang, H.; Zhang, J.Q.; Zhu, Y.F.; Wang, Y.X. Microstructural observation on pistil abortion of ‘Li Guang’ apricot and transcriptome reveal the mechanism of endogenous hormones involved in pistil abortion. Sci. Hortic. 2022, 293, 110749. [Google Scholar] [CrossRef]
- Zhao, Y.F.; Broholm, S.K.; Wang, F.; Rijpkema, A.S.; Lan, T.Y.; Albert, V.A.; Teeri, T.H.; Elomaa, P. TCP and MADS-box transcription factor networks regulate heteromorphic flower type identity in Gerbera Hybrida. Plant Physiol. 2020, 184, 1455–1468. [Google Scholar] [CrossRef]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E.; Yanofsky, M.F. B and C fforal organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef]
- Ditta, G.; Pinyopich, A.; Robles, P.; Pelaz, S.; Yanofsky, M.F. The SEP4 gene of Arabidopsis thaliana functions in fforal organ and meristem identity. Curr Biol. 2004, 14, 1935–1940. [Google Scholar] [CrossRef]
- Morel, P.; Chambrier, P.; Boltz, V.; Chamot, S.; Rozier, F.; Rodrigues Bento, S.; Trehin, C.; Monniaux, M.; Zethof, J.; Vandenbussche, M. Divergent functional diversification patterns in the SEP/AGL6/AP1 MADS-Box transcription factor superclade. Plant Cell 2019, 31, 3033–3056. [Google Scholar] [CrossRef]
- Tao, Z.; Shen, L.; Liu, C.; Liu, L.; Yan, Y.Y.; Yu, H. Genome-wide identification of SOC1 and SVP targets during the floral transition in Arabidopsis. Plant J. 2012, 70, 549–561. [Google Scholar] [CrossRef]
- Mencia, R. From the archives: The shape of flowers, timely flowering, and floral organ longevity. Plant Cell 2025, 37, koaf028. [Google Scholar] [CrossRef]
- Hsu, W.H.; Yeh, T.J.; Huang, K.Y.; Li, J.Y.; Chen, H.Y.; Yang, C.H. AGAMOUS-LIKE13, a putative ancestor for the E functional genes, specifies male and female gametophyte morphogenesis. Plant J. 2014, 77, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.E.; Heck, G.R.; Perry, S.E.; Patterson, S.E.; Bleecker, A.B.; Fang, S.C. The embryo MADS domain factor AGL15 acts postembryonically: Inhibition of perianth senescence and abscission via constitutive expression. Plant Cell 2000, 12, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.J.; Lehti-Shiu, M.D.; Fernandez, D.E. The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition in Arabidopsis. Plant J. 2007, 50, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Serivichyaswat, P.; Ryu, H.S.; Kim, W.; Kim, S.; Chung, K.S.; Kim, J.J.; Ahn, J.H. Expression of the floral repressor miRNA156 is positively regulated by the AGAMOUS-like proteins AGL15 and AGL18. Mol. Cells 2015, 38, 259–266. [Google Scholar] [CrossRef]
- Li, H.F.; Liang, W.Q.; Yin, C.S.; Zhu, L.; Zhang, D.B. Genetic interaction of OsMADS3, DROOPING LEAF, and OsMADS13 in specifying rice floral organ identities and meristem determinacy. Plant Physiol. 2011, 156, 263–274. [Google Scholar] [CrossRef]
- Lu, R.; Hu, S.Q.; Feng, J.; Liu, Z.F.; Kang, C. The AP2 transcription factor BARE RECEPTACLE regulates floral organogenesis via auxin pathways in woodland strawberry. Plant Cell 2024, 36, 4970–4987. [Google Scholar] [CrossRef]
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef]
- Weiss, D.; Van Der Luit, A.; Knegt, E.; Vermeer, E.; Mol, J.N.M.; Kooter, J.M. Identification of endogenous gibberellins in Petunia flowers (induction of anthocyanin biosynthetic gene expression and the antagonistic effect of abscisic acid). Plant Physiol. 1995, 107, 695–702. [Google Scholar] [CrossRef]
- Attia, K.A.; Abdelkhalik, A.F.; Ammar, M.H.; Wei, C.; Yang, J.S.; Lightfoot, D.A.; El-Sayed, W.M.; El-Shemy, H.A. Antisense phenotypes reveal a functional expression of OsARF1, an auxin response factor, in transgenic rice. Curr. Issues Mol. Biol. 2009, 11, 29–34. [Google Scholar] [CrossRef]
- Cheng, C.X.; Yu, Q.; Wang, Y.R.; Wang, H.; Dong, Y.H.; Ji, Y.Q.; Zhou, X.F.; Li, Y.H.; Jiang, C.Z.; Gan, S.S.; et al. Ethylene-regulated asymmetric growth of the petal base promotes flower opening in rose (Rosa hybrida). Plant Cell 2021, 33, 1229–1251. [Google Scholar] [CrossRef]
- Cheng, H.; Qin, L.J.; Lee, S.; Fu, X.D.; Richards, D.E.; Cao, D.N.; Luo, D.; Harberd, N.P.; Peng, J.R. Gibberellin regulates Arabidopsis floral development via suppression of DELLA protein function. Development 2004, 131, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Domagalska, M.A.; Schomburg, F.M.; Amasino, R.M.; Vierstra, R.D.; Nagy, F.; Davis, S.J. Attenuation of brassinosteroid signaling enhances FLC expression and delays flowering. Development 2007, 134, 2841–2850. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Lee, H.; Lee, J.; Kim, H.; Ryu, H. ABSCISIC ACID-INSENSITIVE 3 is involved in brassinosteroid-mediated regulation of flowering in plants. Plant Physiol. Biochem. 2019, 139, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ou, Y.; Zhang, Z.C.; Li, J.M.; He, Y.H. Brassinosteroid signaling recruits histone 3 lysine-27 demethylation activity to FLOWERING LOCUS C chromatin to inhibit the floral transition in Arabidopsis. Mol. Plant 2018, 10, 1135–1146. [Google Scholar] [CrossRef]
- Zuriaga, E.; Romero, C.; Blanca, J.M.; Badenes, M.L. Resistance to Plum Pox Virus (PPV) in apricot (Prunus armeniaca L.) is associated with down-regulation of two MATHd genes. BMC Plant Biol. 2018, 18, 25. [Google Scholar] [CrossRef]
- Zong, W.B.; Song, Y.A.; Xiao, D.D.; Guo, X.T.; Li, F.Q.; Sun, K.L.; Tang, W.J.; Xie, W.H.; Luo, Y.; Liang, S.; et al. Dominance complementation of parental heading date alleles of Hd1, Ghd7, DTH8, and PRR37 confers transgressive late maturation in hybrid rice. Plant J. 2024, 118, 2108–2123. [Google Scholar] [CrossRef]
- Zhu, K.; Chen, H.; Mei, X.; Lu, S.; Xie, H.; Liu, J.; Chai, L.; Xu, Q.; Wurtzel, E.T.; Ye, J.; et al. Transcription factor CsMADS3 coordinately regulates chlorophyll and carotenoid pools in Citrus hesperidium. Plant Physiol. 2023, 193, 519–536. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, X.; Fan, J.; Li, J.; Ren, S.; Gu, X.; Li, P.; Xu, M.; Xu, J.; Lei, W.; et al. Natural variation in BnaA07.MKK9 confers resistance to Sclerotinia stem rot in oilseed rape. Nat Commun. 2024, 15, 5059. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gappedread alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef]
- Chen, C.J.; Wu, Y.; Li, J.W.; Wang, X.; Zeng, Z.H.; Xu, J.; Liu, Y.L.; Feng, J.T.; Chen, H.; He, Y.H.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, D.; Chen, Y.; He, Y.; Song, J.; Jiang, Y.; Yang, M.; Zheng, X.; Wang, L.; Hu, H. Transcriptome Analysis Reveals Association of E-Class AmMADS-Box Genes with Petal Malformation in Antirrhinum majus L. Int. J. Mol. Sci. 2025, 26, 4450. https://doi.org/10.3390/ijms26094450
Yang D, Chen Y, He Y, Song J, Jiang Y, Yang M, Zheng X, Wang L, Hu H. Transcriptome Analysis Reveals Association of E-Class AmMADS-Box Genes with Petal Malformation in Antirrhinum majus L. International Journal of Molecular Sciences. 2025; 26(9):4450. https://doi.org/10.3390/ijms26094450
Chicago/Turabian StyleYang, Dongmei, Yiwen Chen, Yutong He, Jiayi Song, Ye Jiang, Meiyun Yang, Xingyan Zheng, Li Wang, and Huizhen Hu. 2025. "Transcriptome Analysis Reveals Association of E-Class AmMADS-Box Genes with Petal Malformation in Antirrhinum majus L." International Journal of Molecular Sciences 26, no. 9: 4450. https://doi.org/10.3390/ijms26094450
APA StyleYang, D., Chen, Y., He, Y., Song, J., Jiang, Y., Yang, M., Zheng, X., Wang, L., & Hu, H. (2025). Transcriptome Analysis Reveals Association of E-Class AmMADS-Box Genes with Petal Malformation in Antirrhinum majus L. International Journal of Molecular Sciences, 26(9), 4450. https://doi.org/10.3390/ijms26094450