
A Positive Feedback DNA-PK/MYT1L-CXCR1-ERK1/2 Proliferative Signaling Loop in Glioblastoma

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
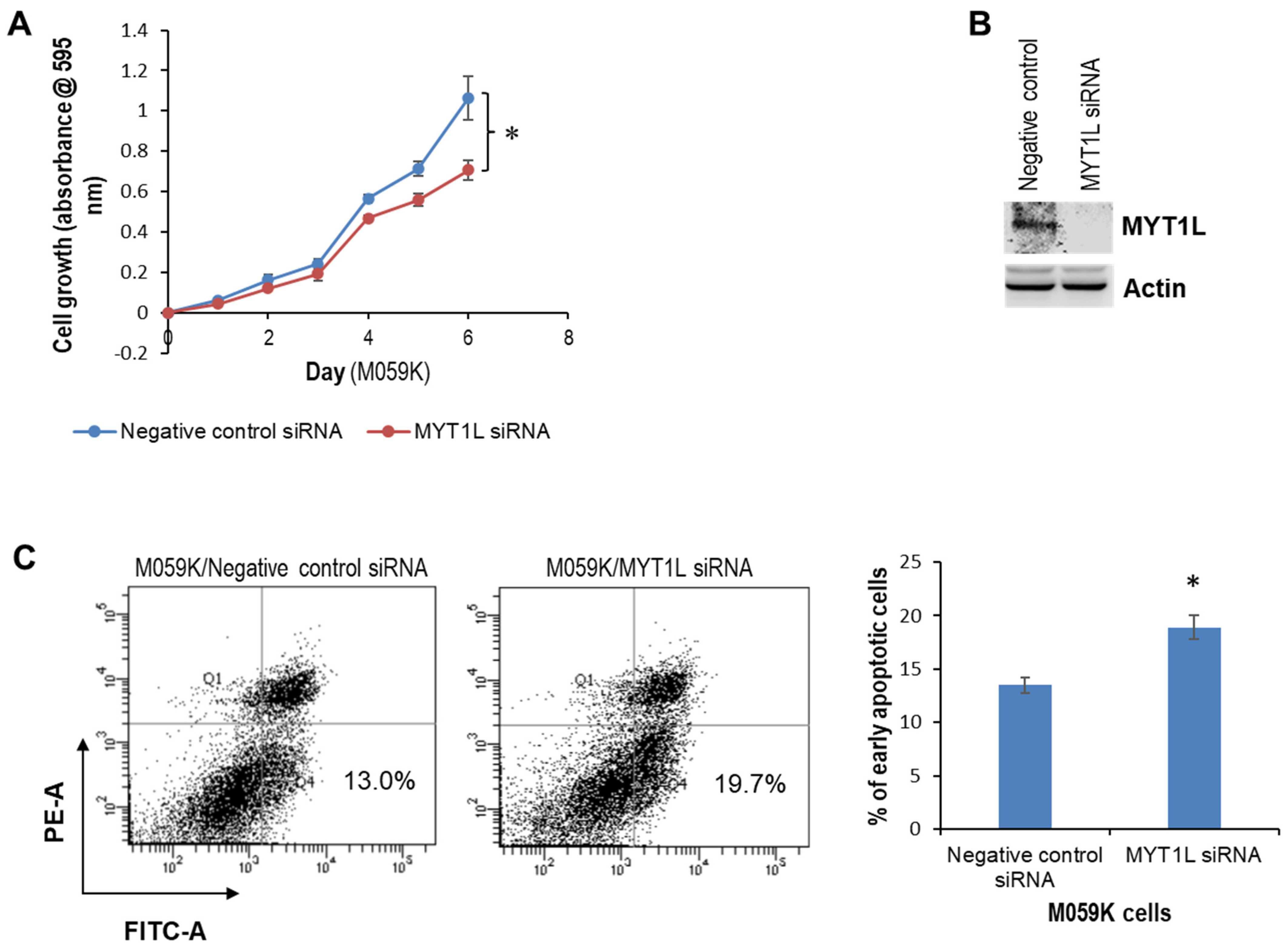
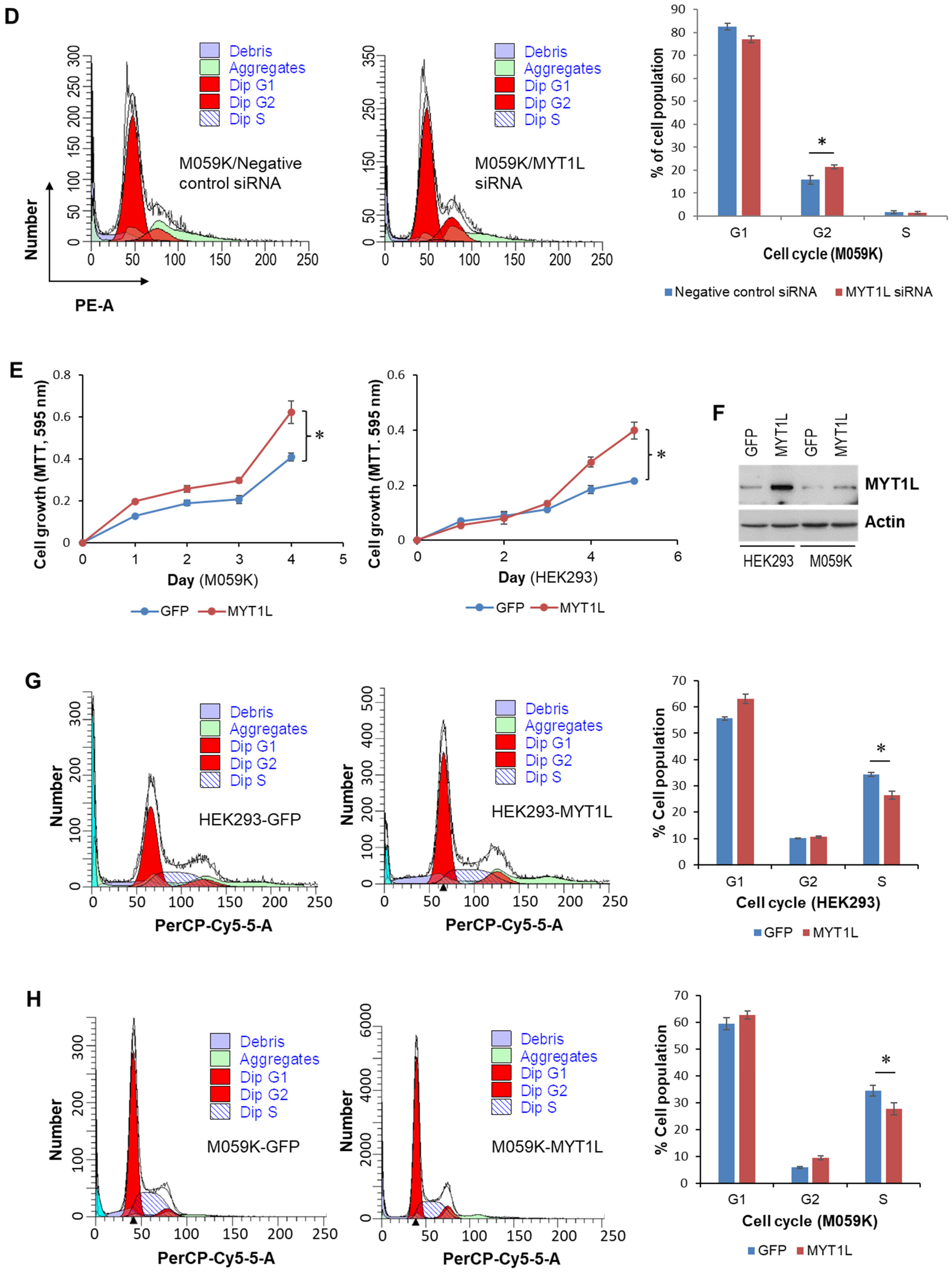
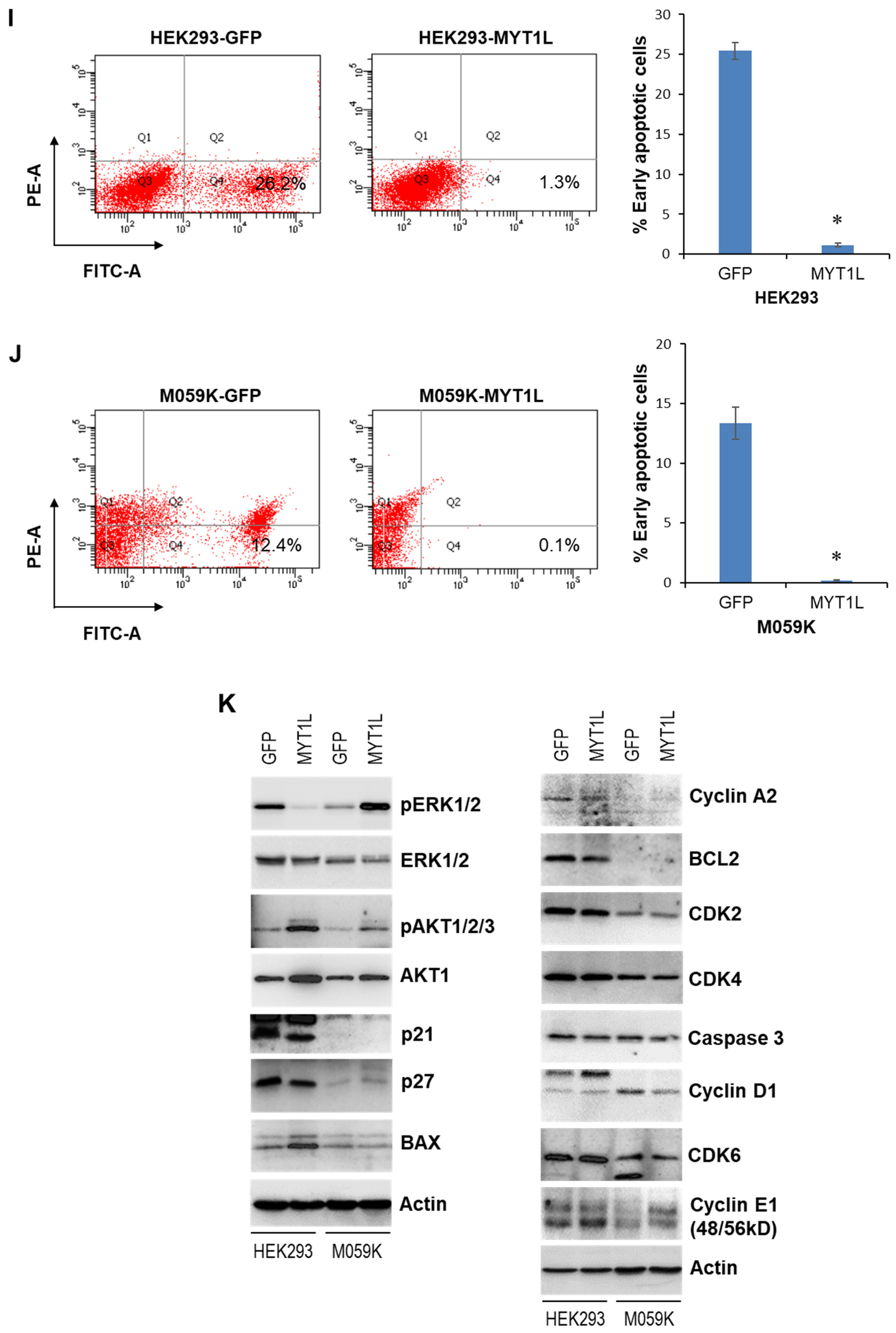
2.1. MYT1L Acts as an Oncogene in Glioblastoma Cells with Normal DNA-PK Activity
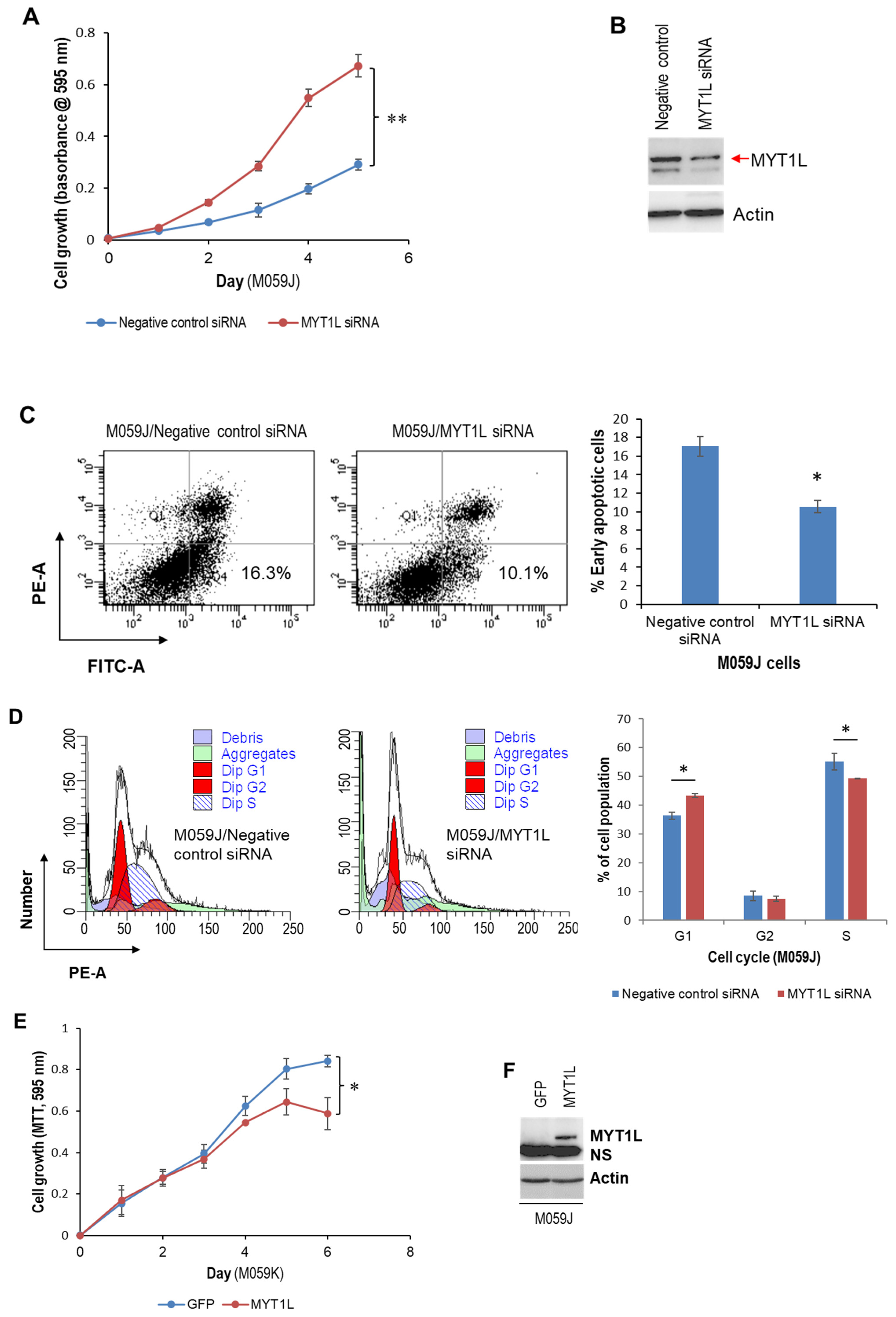
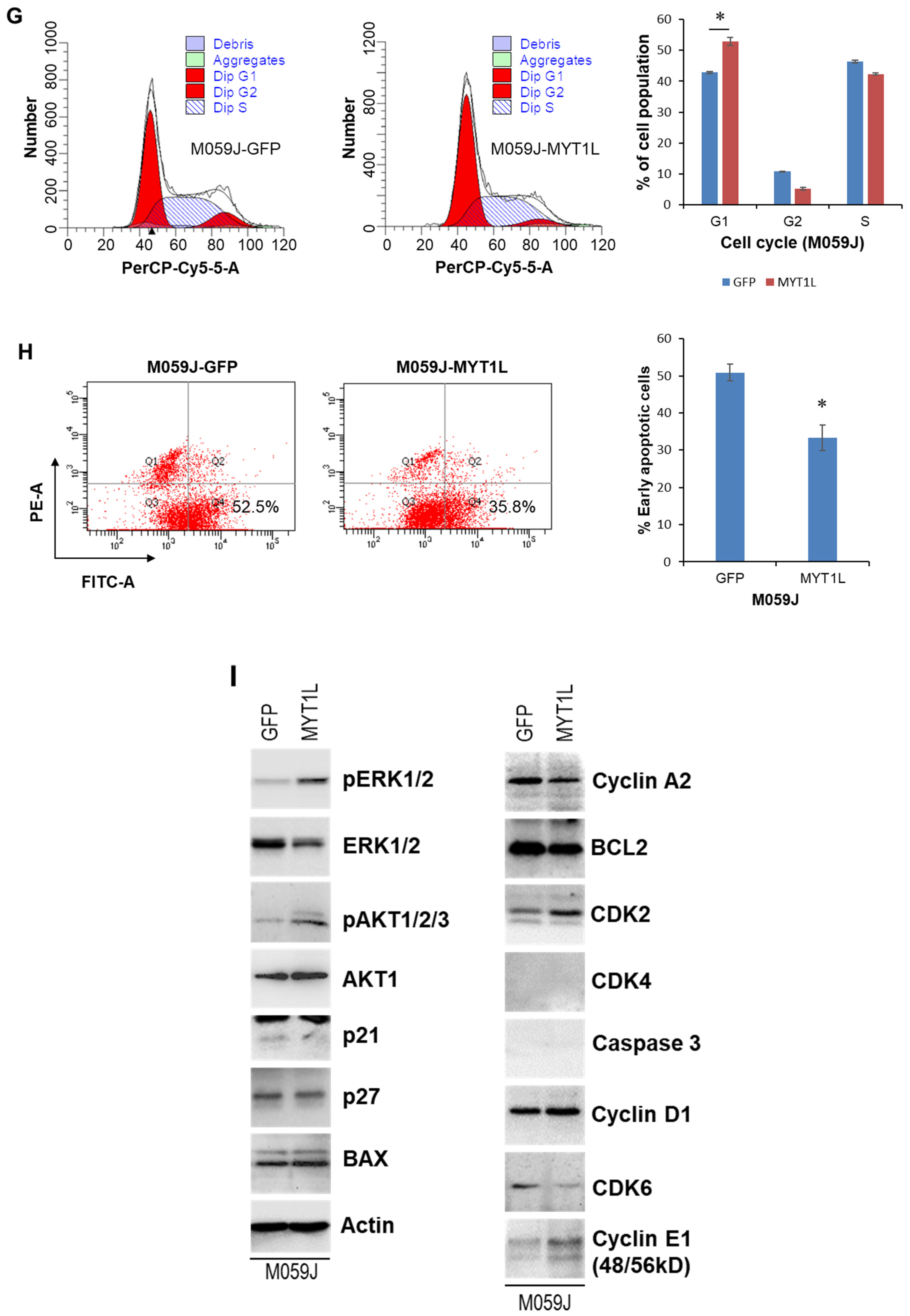
2.2. MYT1L Inhibited Proliferation and Induced Apoptosis in Glioblastoma Cells with Loss of DNA-PK Function
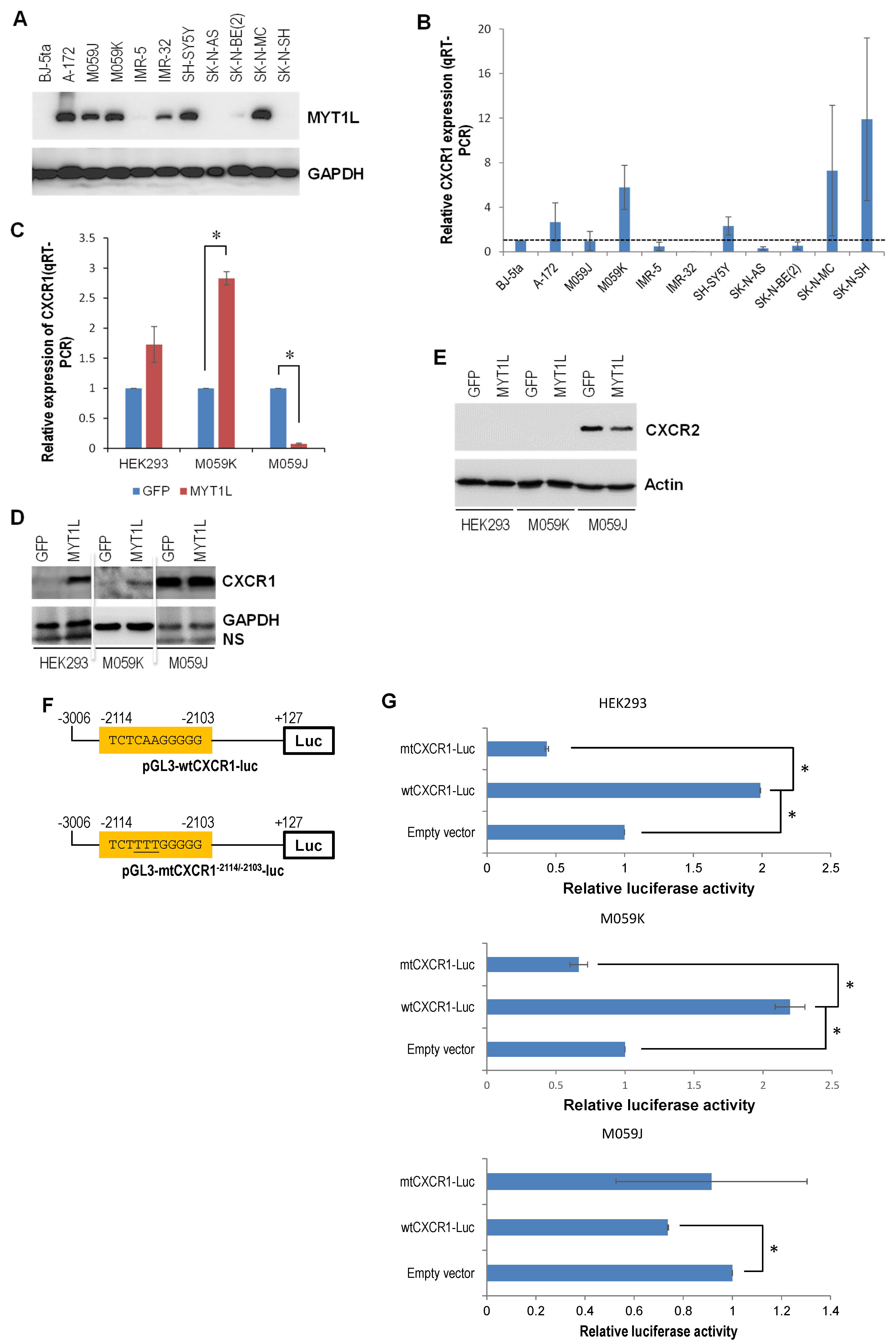
2.3. DNA-PK and MYT1L Are Both Involved in CXCR1 Transcription
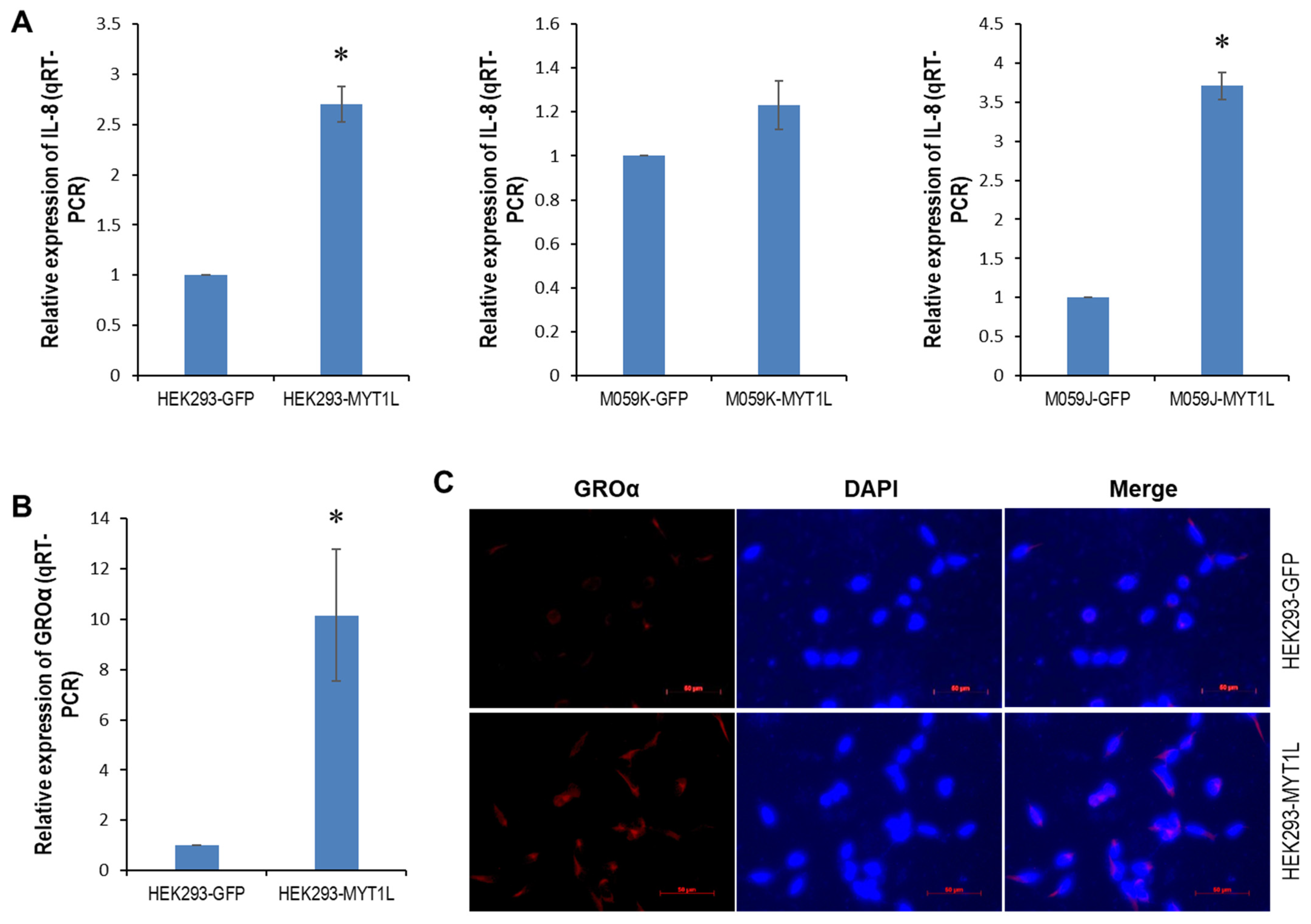
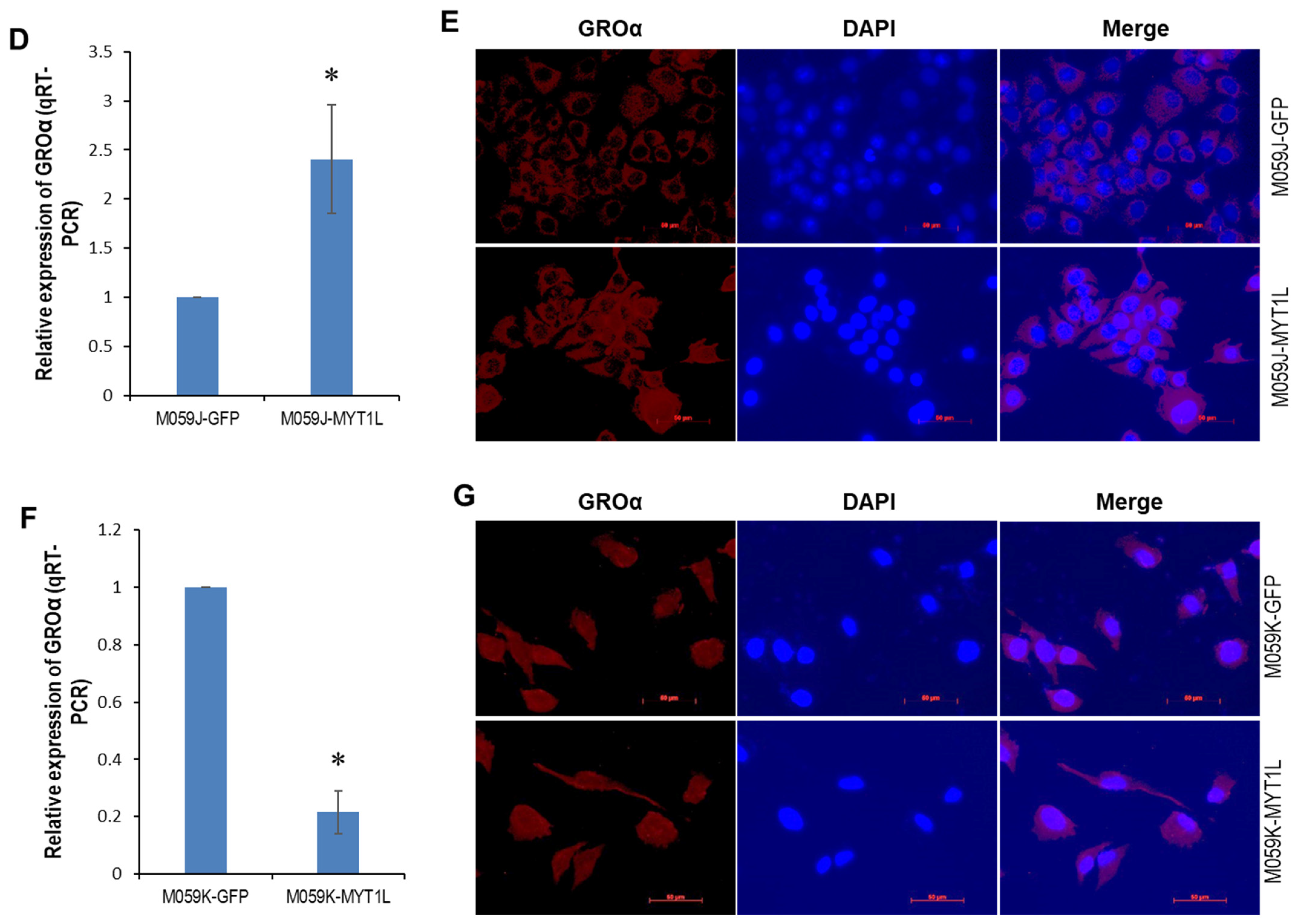
2.4. IL-8- and GROα-CXCR1/ERK1/2 Proliferative Signaling in Glioblastoma Cells
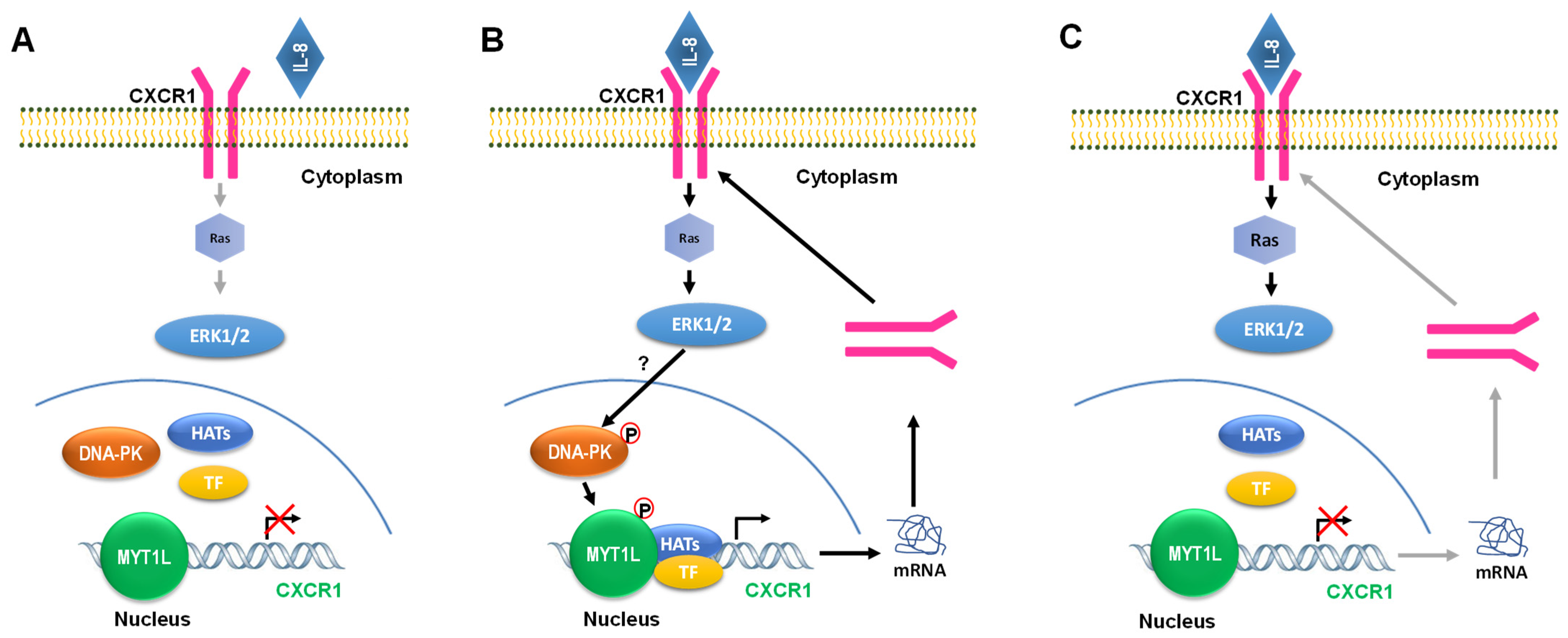
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MTT Assay
4.3. Apoptosis and Cell Cycle Analyses
4.4. Generation of Stable Cell Lines Expressing MYT1L
4.5. Western Blot Analysis
4.6. Whole-Genome Gene Expression Profiling
4.7. Quantitative Real-Time RT-PCR (qRT-PCR)
4.8. Immunofluorescence
4.9. Bioinformatics Analysis
4.10. Site-Directed Mutagenesis
4.11. Transient Transfection and Luciferase Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.P.; Harrison, R.A.; Lapointe, S.; Lim-Fat, M.J.; MacNeil, M.V.; Mathieu, D.; Perry, J.R.; Pitz, M.W.; Roberge, D.; Tsang, D.S.; et al. Canadian expert consensus recommendations for the diagnosis and management of glioblastoma: Results of a Delphi study. Curr. Oncol. 2025, 32, 207. [Google Scholar] [CrossRef] [PubMed]
- Noonan, J.; Zarrer, J.; Murphy, B.M. Targeting autophagy in glioblastoma. Crit. Rev. Oncog. 2016, 21, 241–252. [Google Scholar] [CrossRef]
- Stieber, D.; Abdul Rahim, S.A.; Niclou, S.P. Novel ways to target brain tumour metabolism. Expert Opin. Ther. Targets 2011, 15, 1227–1239. [Google Scholar] [CrossRef]
- Taylor, T.E.; Furnari, F.B.; Cavenee, W.K. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef]
- Rao, R.D.; Uhm, J.H.; Krishnan, S.; James, C.D. Genetic and signaling pathway alterations in glioblastoma: Relevance to novel targeted therapies. Front. Biosci. 2003, 8, e270–e280. [Google Scholar]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nistér, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar]
- Lokker, N.A.; Sullivan, C.M.; Hollenbach, S.J.; Israel, M.A.; Giese, N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002, 62, 3729–3735. [Google Scholar] [PubMed]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef]
- Arrillaga-Romany, I.; Reardon, D.A.; Wen, R.Y. Current status of anti-angiogenic therapies for glioblastoma. Expert Opin. Investig. Drugs 2014, 23, 199–210. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastoma. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Saxena, A.; Clark, W.C.; Robertson, J.T.; Oldfield, E.H.; Aaronson, S.A.; Ali, I.U. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992, 52, 4550–4553. [Google Scholar]
- Martinho, O.; Longatto-Filho, A.; Lambros, M.B.K.; Martins, A.; Pinheiro, C.; Silva, A.; Pardal, F.; Amorim, J.; Mackay, A.; Milanezi, F.; et al. Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. Br. J. Cancer 2009, 101, 973–982. [Google Scholar] [CrossRef]
- Hermanson, M.; Funa, K.; Koopmann, J.; Maintz, D.; Waha, A.; Westermark, B.; Heldin, C.H.; Wiestler, O.D.; Louis, D.N.; Von Deimling, A.; et al. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor α receptor expression in human malignant gliomas. Cancer Res. 1996, 56, 164–171. [Google Scholar] [PubMed]
- Plate, K.H.; Breier, G.; Farrell, C.L.; Risau, W. Platelet-derived growth factor receptor-β is induced during tumor development and up-regulated during tumor progression in endothelial cells in human gliomas. Lab. Investig. 1992, 67, 529–534. [Google Scholar]
- Kim, Y.; Kim, E.; Wu, Q.; Guryanova, O.; Hitomi, M.; Lathia, J.D.; Serwanski, D.; Sloan, A.E.; Weil, R.J.; Lee, J.; et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev. 2012, 26, 1247–1262. [Google Scholar] [CrossRef]
- Jiang, Y.; Boije, M.; Westermark, B.; Uhrbom, L. PDGF-B can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia 2011, 13, 492–503. [Google Scholar] [CrossRef]
- Uhrbom, L.; Hesselager, G.; Nistér, M.; Westermark, B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998, 58, 5275–5279. [Google Scholar] [PubMed]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human glioma in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef]
- Sonoda, Y.; Kanamori, M.; Deen, D.F.; Cheng, S.Y.; Berger, M.S.; Pieper, R.O. Overexpression of vascular endothelial growth factor isoforms drives oxygenation and growth but not progression to glioblastoma multiforme in a human model of gliomagenesis. Cancer Res. 2003, 63, 1962–1968. [Google Scholar] [PubMed]
- Huang, H.; Held-Feindt, J.; Buhl, R.; Mehdorn, H.M.; Mentlein, R. Expression of VEGF and its receptors in different brain tumors. Neurol. Res. 2005, 27, 371–377. [Google Scholar] [CrossRef]
- Wong, M.L.; Prawira, A.; Kaye, A.H.; Hovens, C.M. Tumour angiogenesis: Its mechanism and therapeutic implications in malignant gliomas. J. Clin. Neurosci. 2009, 16, 1119–1130. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Sharma, I.; Singh, A.; Siraj, F.; Saxena, S. IL-8/CXCR1/2 signalling promotes tumor cell proliferation, invasion and vascular mimicry in glioblastoma. J. Biomed. Sci. 2018, 25, 62. [Google Scholar] [CrossRef]
- Wang, B.; Li, D.; Yao, Y.; Heyns, M.; Kovalchuk, A.; Ilnytskyy, Y.; Rodriguez-Juarez, R.; Bronson, R.T.; Metz, G.A.; Kovalchuk, O.; et al. The crucial role of DNA-dependent protein kinase and myelin transcription factor 1-like protein in the miR-141 tumor suppressor network. Cell Cycle 2019, 18, 2876–2892. [Google Scholar] [CrossRef]
- Mi, J.; Dziegielewski, J.; Bolesta, E.; Brautigan, D.L.; Larner, J.M. Activation of DNA-PK by ionizing radiation is mediated by protein phosphatase 6. PLoS ONE 2009, 4, e4395. [Google Scholar] [CrossRef]
- Farber-Katz, S.E.; Dippold, H.C.; Buschman, M.D.; Peterman, M.C.; Xing, M.; Noakes, C.J.; Tat, J.; Ng, M.M.; Rahajeng, J.; Cowan, D.M.; et al. DNA damage triggers golgi dispersal via DNA-PK and GOLPH3. Cell 2014, 156, 413–427. [Google Scholar] [CrossRef] [PubMed]
- De Rocker, N.; Vergult, S.; Koolen, D.; Jacobs, E.; Hoischen, A.; Zeesman, S.; Bang, B.; Béna, F.; Bockaert, N.; Bongers, E.M.; et al. Refinement of the critical 2p25.3 deletion region: The role of MYT1L in intellectual disability and obesity. Genet. Med. 2015, 17, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Pfisterer, U.; Kirkeby, A.; Torper, O.; Wood, J.; Nelander, J.; Dufour, A.; Björklund, A.; Lindvall, O.; Jakobsson, J.; Parmar, M. Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 10343–10348. [Google Scholar] [CrossRef]
- Ambasudhan, R.; Talantova, M.; Coleman, R.; Yuan, X.; Zhu, S.; Lipton, S.A.; Ding, S. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 2011, 9, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, J.A.; Sauer, K.; Jones, L.; Richarddson, H.; Saint, R.; Lehner, C.F. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell 1994, 77, 107–120. [Google Scholar] [CrossRef]
- Sauer, K.; Knoblich, J.A.; Richardson, H.; Lehner, C.F. Distinct modes of cyclin E/cdc2c kinase regulation and S-phase control in mitotic and endoreduplication cycles of Drosophila embryogenesis. Genes Dev. 1995, 9, 1327–1339. [Google Scholar] [CrossRef]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef]
- Mohapatra, P.; Preet, R.; Das, D.; Satapathy, S.R.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Quinacrine-mediated autophagy and apoptosis in colon cancer cells is through a p53- and p21-dependent mechanism. Oncol. Res. 2012, 20, 81–91. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Wang, J.; Ye, Z.; Li, J.C. Oridonin up-regulates expression of p21 and induces autophagy and apoptosis in human prostate cancer cells. Int. J. Biol. Sci. 2012, 8, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Knudsen, K.E. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef]
- Jackson, S.P.; MacDonald, J.J.; Lees-Miller, S.; Tjian, R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell 1990, 63, 155–165. [Google Scholar] [CrossRef]
- Xu, P.; LaVallee, P.A.; Lin, J.J.; Hoidal, J.R. Characterization of proteins binding to E-box/Ku86 sites and function of Ku86 in transcriptional regulation of the human xanthine oxidoreductase gene. J. Biol. Chem. 2004, 279, 16057–16063. [Google Scholar] [CrossRef]
- Mayeur, G.L.; Kung, W.J.; Martinez, A.; Izumiya, C.; Chen, D.J.; Kung, H.J. Ku is a novel transcriptional recycling coactivator of the androgen receptor in prostate cancer cells. J. Biol. Chem. 2005, 280, 10827–10833. [Google Scholar] [CrossRef]
- Ebnet, K.; Vestweber, D. Molecular mechanisms that control leukocyte extravasation: The selectins and chemokines. Histochem. Cell Biol. 1999, 112, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. Chemokines in pathology and medicine. J. Intern. Med. 2001, 250, 91–104. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Shattuck-Brandt, R.; Haghnegahdar, H.; Owen, J.D.; Strieter, R.; Burdick, M.; Nirodi, C.; Beauchamp, D.; Johnson, K.N.; Richmond, A. Mechanism and biological significance of constitutive expression of MGSA/GRO chemokines in malignant melanoma tumor progression. J. Leukoc. Biol. 1997, 62, 588–597. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Zeelenberg, I.S.; Ruuls-Van Stalle, L.; Roos, E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastasis. Cancer Res. 2003, 63, 3833–3839. [Google Scholar] [PubMed]
- Wang, B.; Hendricks, D.T.; Wamunyokoli, F.; Parker, M.I. A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer. Cancer Res. 2006, 66, 3071–3077. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Arenberg, D.A.; Stoy, K.; Morgan, T.; Addison, C.L.; Morris, S.B.; Glass, M.; Wilke, C.; Xue, Y.Y.; Sitterding, S.; et al. Distinct CXC chemokines mediate tumorigenicity of prostate cancer cells. Am. J. Pathol. 1999, 154, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.M.; Webster, S.J.; Flower, D.; Woll, P.J. Interleukin-8/CXCL8 is a growth factor for human lung cancer cells. Br. J. Cancer 2004, 91, 1970–1976. [Google Scholar] [CrossRef]
- Lowman, H.B.; Slagle, P.H.; DeForge, L.E.; Wirth, C.M.; Gillece-Castro, B.L.; Bourell, J.H.; Fairbrother, W.J. Exchanging interleukin-8 and melanoma growth-stimulating activity receptor binding specificities. J. Biol. Chem. 1996, 271, 14344–14352. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radio-resistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Migliozzi, S.; Oh, Y.T.; Hasanain, M.; Garofano, L.; D’angelo, F.; Najac, R.D.; Picca, A.; Bielle, F.; Di Stefano, A.L.; Lerond, J.; et al. Integrative multi-omics networks identify PKCδ and DNA-PK as master kinases of glioblastoma subtypes and guide targeted cancer therapy. Nat. Cancer 2023, 4, 181–202. [Google Scholar] [CrossRef]
- Infanger, D.W.; Cho, Y.; Lopez, B.S.; Mohanan, S.; Liu, S.C.; Gursel, D.; Boockvar, J.A.; Fischbach, C. Glioblastoma stem cells are regulated by interleukin-8 signaling in a tumoral perivascular niche. Cancer Res. 2013, 73, 7079–7089. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, L.; Ouyang, Q.; Xu, L.; Cui, H.; Xu, M. Neurotensin signaling regulates stem-like traits of glioblastoma stem cells though activation of IL-8/CXCR1/STAT3 pathway. Cell Signal. 2014, 26, 2896–2902. [Google Scholar] [CrossRef]
- Looyenga, B.D.; Resau, J.; MacKeigan, J.P. Cytokine receptor-like factor 1 (CRLF1) protects against 6-hydroxydopamine toxicity independent of the gp130/JAK signaling pathway. PLoS ONE 2013, 8, e66548. [Google Scholar] [CrossRef]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8/CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef]
- McClelland, S.; Maxwell, P.J.; Branco, C.; Barry, S.T.; Eberlain, C.; LaBonte, M.J. Targeting IL-8 and its receptors in prostate cancer: Inflammation, stress response, and treatment resistance. Cancer 2024, 16, 2797. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, C.; Teijeira, A.; Oñate, C.; Pérez, G.; Sanmamed, M.F.; Andueza, M.P.; Alignani, D.; Labiano, S.; Azpilikueta, A.; Rodriguez-Paulete, A.; et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin. Cancer Res. 2016, 22, 3924–3936. [Google Scholar] [CrossRef]
- Molczyk, C.; Singh, R.K. CXCR1: A cancer stem cell marker and therapeutic target in solid tumors. Biomedicines 2023, 11, 576. [Google Scholar] [CrossRef] [PubMed]
- Gulhati, P.; Schalck, A.; Jiang, S.; Shang, X.; Wu, C.J.; Hou, P.; Ruiz, S.H.; Soto, L.S.; Parra, E.; Ying, H.; et al. Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer. Nat. Cancer 2023, 4, 62–80. [Google Scholar] [CrossRef] [PubMed]
- iu, H.; Zhao, Q.; Tan, L.; Wu, X.; Huang, R.; Zuo, Y.; Chen, L.; Yang, J.; Zhang, Z.-X.; Ruan, W.; et al. Neutralizing IL-8 potentiates immune checkpoint blockade efficacy for glioma. Cancer Cell 2023, 41, 693–710. [Google Scholar]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Christensen, C.; Guldberg, P. Growth factors rescue cutaneous melanoma cells from apoptosis induced by knockdown of mutated (V600E) B-RAF. Oncogene 2005, 24, 6292–6302. [Google Scholar] [CrossRef]
- Shin, M.; Franks, C.E.; Hsu, K.L. Isoform-selective activity-based profiling of ERK signaling. Chem. Sci. 2018, 9, 2419–2431. [Google Scholar] [CrossRef]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef]
- Zielińska, K.A.; Katanaev, V.L. Information theory: New look at oncogenic signaling pathways. Trends Cell Biol. 2019, 29, 862–875. [Google Scholar] [CrossRef] [PubMed]
- Zerbib, J.; Ippolito, M.R.; Eliezer, Y.; De Feudis, G.; Reuveni, E.; Kadmon, A.S.; Martin, S.; Viganò, S.; Leor, G.; Berstler, J.; et al. Human aneuploidy cells depend on the RAF/MEK/ERK pathway for overcoming increased DNA damage. Nat. Commun. 2024, 15, 7772. [Google Scholar] [CrossRef] [PubMed]
- Tislevoll, B.S.; Hellesøy, M.; Fagerholt, O.H.E.; Gullaksen, S.-E.; Srivastava, A.; Birkeland, E.; Kleftogiannis, D.; Ayuda-Durán, P.; Piechaczyk, L.; Tadele, D.S.; et al. Early response evaluation by single cell signaling profiling in acute myeloid leukemia. Nat. Commun. 2023, 14, 115. [Google Scholar] [CrossRef]
- Grogan, L.; Shapiro, P. Chapter Five—Progress in the development of ERK1/2 inhibitors for treating cancer s and other diseases. Adv. Pharmacol. 2024, 100, 181–207. [Google Scholar]
- Fu, L.; Chen, S.; He, G.; Chen, Y.; Liu, B. Targeting extracellular signal-regulated protein kinase 1/2 (ERK1/2) in cancer: An update on pharmacological small-molecule inhibitors. J. Med. Chem. 2022, 65, 13561–13573. [Google Scholar] [CrossRef]
- Xiong, Q.; Liu, A.; Ren, Q.; Xue, Y.; Yu, X.; Ying, Y.; Gao, H.; Tan, H.; Zhang, Z.; Li, W.; et al. Cuprous oxide nanoparticles trigger reactive oxygen species-induced apoptosis through activation of erk-dependent autophagy in bladder cancer. Cell Death Dis. 2020, 11, 366. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Tran, E.; Nguyen, T.H.; Do, P.T.; Huynh, T.H.; Huynh, H. The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis 2004, 25, 647–659. [Google Scholar] [CrossRef]
- Thamkachy, R.; Kumar, B.; Rajasekharan, K.N.; Sengupta, S. ERK mediated upregulation of death receptor 5 overcome the lack of p53 functionality in the diaminothiazole DAT1 induced apoptosis in colon cancer model: Efficiency of DAT1 in Ras-Raf mutated cells. Mol. Cancer 2016, 15, 22. [Google Scholar] [CrossRef]
- Pettersson, F.; Couture, M.C.; Hanna, N.; Miller, W.H. Enhanced retinoid-induced apoptosis of MDA-MB-231 breast cancer cells by PKC inhibitors involves activation of ERK. Oncogene 2004, 23, 7053–7066. [Google Scholar] [CrossRef]
- Yu, W.; Liao, Q.Y.; Hantash, F.M.; Sanders, B.G.; Kline, K. Activation of extracellular signal-regulated kinase and c-Jun-NH(2)-terminal kinase but not p38 mitogen-activated protein kinases is required for RRR-alpha-tocopheryl succinate-induced apoptosis of human breast cancer cells. Cancer Res. 2001, 61, 6569–6576. [Google Scholar]
- Schweyer, S.; Soruri, A.; Meschter, O.; Heintze, A.; Zschunke, F.; Miosge, N.; Thelen, P.; Schlott, T.; Radzun, H.J.; Fayyazi, A. Cisplatin-induced apoptosis in human malignant testicular germ cell lines depends on MEK/ERK activation. Br. J. Cancer 2004, 91, 589–598. [Google Scholar] [CrossRef]
- Kim, H.J.; Chakravarti, N.; Oridate, N.; Choe, C.; Claret, F.X.; Lotan, R. N-(4-hydroxyphenyl)retinamide-induced apoptosis triggered by reactive oxygen species is mediated by activation of MAPKs in head and neck squamous carcinoma cells. Oncogene 2006, 25, 2785–2794. [Google Scholar] [CrossRef]
- An, J.; Li, L.; Zhang, X. Curcusone C induces apoptosis in endometrial cancer cells via mitochondria-dependent apoptosis and ERK pathway. Biotechnol. Lett. 2021, 43, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Tuergong, G.; Zhu, H.; Wang, X.; Weng, G.; Ren, Y. Norcantharidin induces G2/M arrest and apoptosis via activation of ERK and JNK, but not p38 signaling in human renal cell carcinoma ACHN cells. Acta Pharm. 2021, 71, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Sengelen, A.; Önay-Ucar, E. Rosmarinic acid attenuates glioblastoma cells and spheroids’ growth and EMT/stem-like state by PTEN/PI3K/AKT downregulation and ERK-induced apoptosis. Phytomedicine 2024, 135, 156060. [Google Scholar] [CrossRef]
- Petroni, G.; Buqué, A.; Coussens, L.M.; Galluzzi, L. Targeting oncogene and non-oncogene addiction to inflame the tumour microenvironment. Nat. Rev. Drug Discov. 2022, 21, 440–462. [Google Scholar] [CrossRef] [PubMed]
- Negarini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Riabinska, A.; Daheim, M.; Herter-Sprie, G.S.; Winkler, J.; Fritz, C.; Hallek, M.; Thomas, R.K.; Kreuzer, K.-A.; Frenzel, L.P.; Monfared, P.; et al. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci. Transl. Med. 2013, 5, 189ra78. [Google Scholar] [CrossRef] [PubMed]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK inhibitor VX-984 enhances the radiosensitivity of glioblastoma cells gown in vitro and orthotopic xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacological inhibitor of DNA-PK, M3814, potentiates radiotherapy and regresses human tumors in mouse models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, D.; Rodriguez-Juarez, R.; Farfus, A.; Storozynsky, Q.; Malach, M.; Carpenter, E.; Filkowski, J.; Lykkesfeldt, A.E.; Kovalchuk, O. A suppressive role of guanine nucleotide-binding protein subunit beta-4 inhibited by DNA methylation in the growth of anti-estrogen resistant breast cancer cells. BMC Cancer 2018, 18, 817. [Google Scholar] [CrossRef]
- Wang, B.; Li, D.; Filkowski, J.; Rodriguez-Juarez, R.; Storozynsky, Q.; Malach, M.; Carpenter, E.; Kovalchuk, O. A dual role of miR-22 modulated by RelA/p65 in resensitizing fulvestrant-resistant breast cancer cells to fulvestrant by targeting FOXP1 and HDAC4 and constitutive acetylation of p53 at Lys382. Oncogenesis 2018, 7, 54. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Li, D.; Ilnytskyy, Y.; Khachigian, L.M.; Zhong, N.; Rodriguez-Juarez, R.; Kovalchuk, I.; Kovalchuk, O. A Positive Feedback DNA-PK/MYT1L-CXCR1-ERK1/2 Proliferative Signaling Loop in Glioblastoma. Int. J. Mol. Sci. 2025, 26, 4398. https://doi.org/10.3390/ijms26094398
Wang B, Li D, Ilnytskyy Y, Khachigian LM, Zhong N, Rodriguez-Juarez R, Kovalchuk I, Kovalchuk O. A Positive Feedback DNA-PK/MYT1L-CXCR1-ERK1/2 Proliferative Signaling Loop in Glioblastoma. International Journal of Molecular Sciences. 2025; 26(9):4398. https://doi.org/10.3390/ijms26094398
Chicago/Turabian StyleWang, Bo, Dongping Li, Yaroslav Ilnytskyy, Levon M. Khachigian, Nuanying Zhong, Rocio Rodriguez-Juarez, Igor Kovalchuk, and Olga Kovalchuk. 2025. "A Positive Feedback DNA-PK/MYT1L-CXCR1-ERK1/2 Proliferative Signaling Loop in Glioblastoma" International Journal of Molecular Sciences 26, no. 9: 4398. https://doi.org/10.3390/ijms26094398
APA StyleWang, B., Li, D., Ilnytskyy, Y., Khachigian, L. M., Zhong, N., Rodriguez-Juarez, R., Kovalchuk, I., & Kovalchuk, O. (2025). A Positive Feedback DNA-PK/MYT1L-CXCR1-ERK1/2 Proliferative Signaling Loop in Glioblastoma. International Journal of Molecular Sciences, 26(9), 4398. https://doi.org/10.3390/ijms26094398