
Harnessing Azelaic Acid for Acute Myeloid Leukemia Treatment: A Novel Approach to Overcoming Chemoresistance and Improving Outcomes

 ,
,  ,
, 


Abstract
1. Introduction
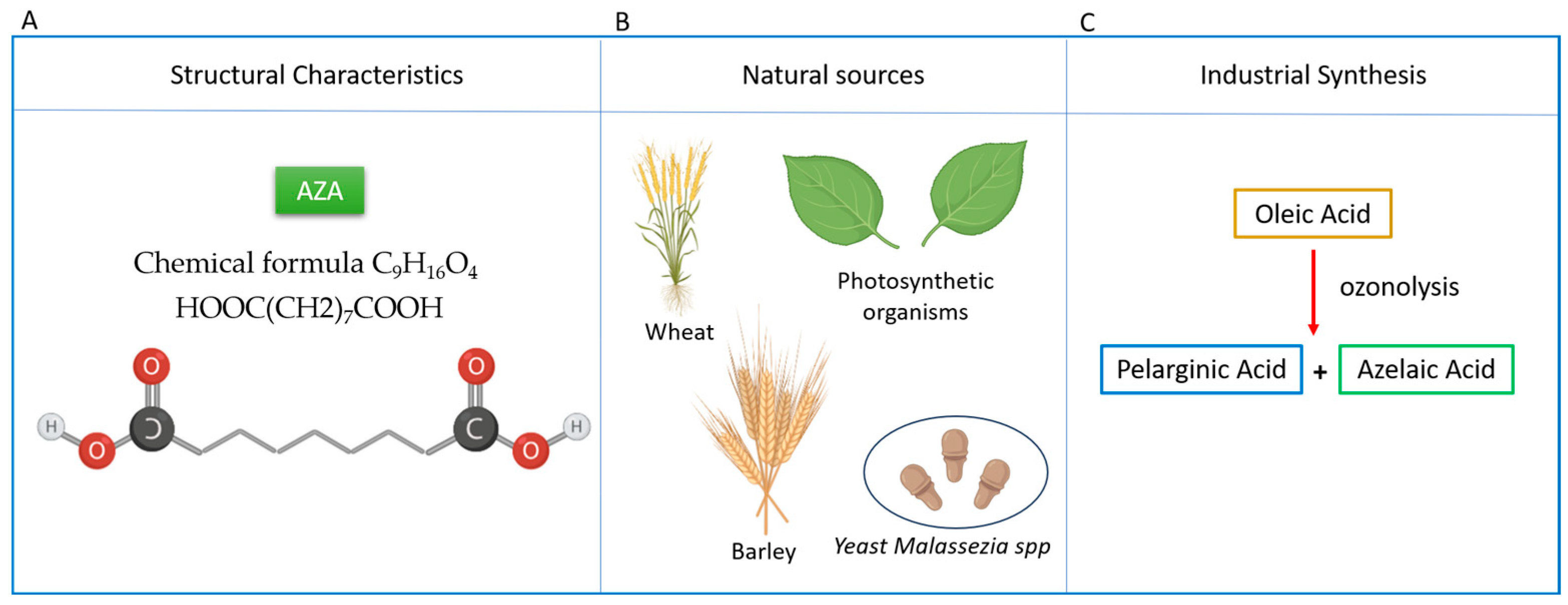
2. Exploring the Structure and Molecular Mechanisms of Azelaic Acid
2.1. Structure, Pharmacological Benefits, and Dietary Sources
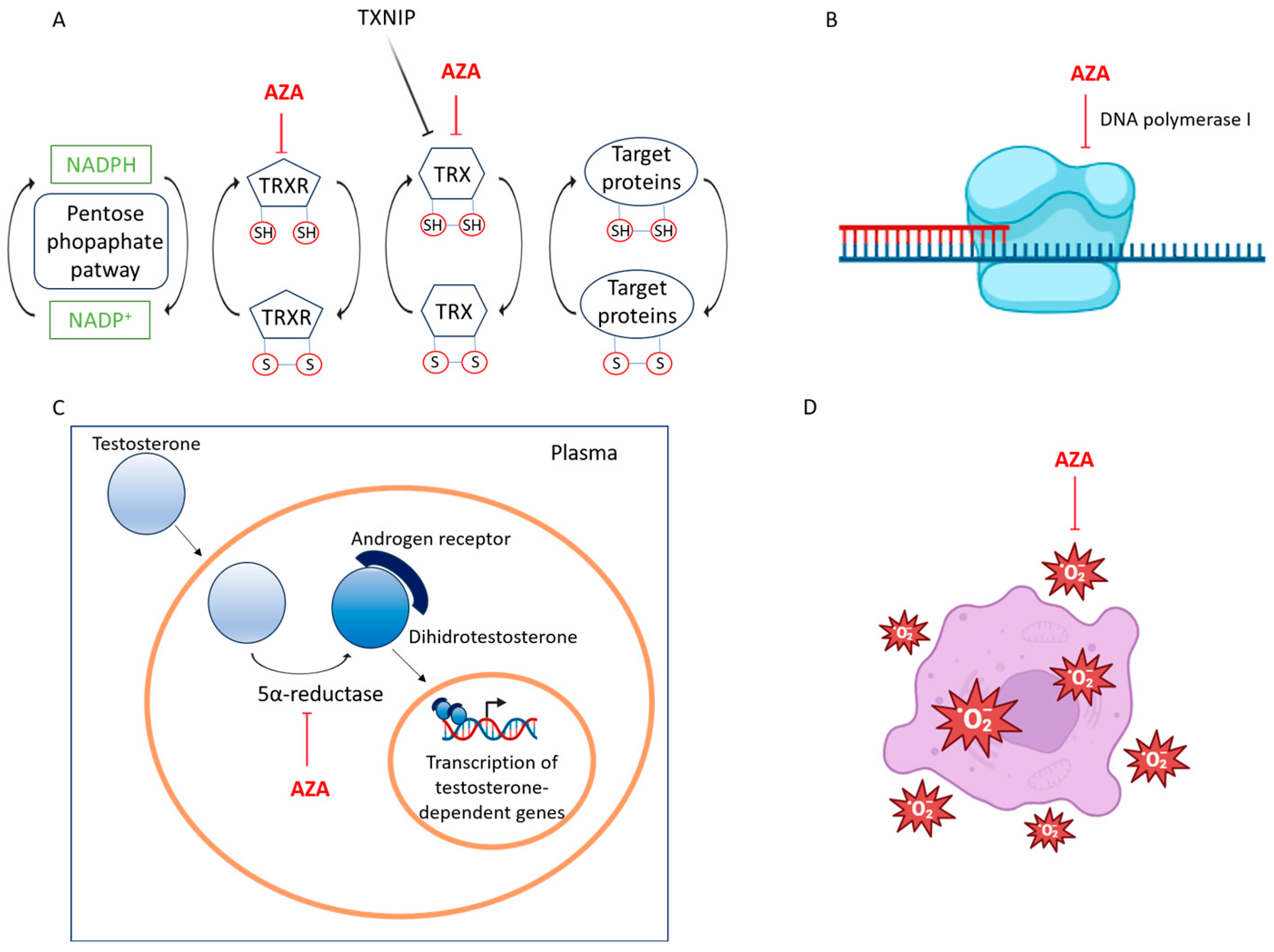
2.2. Physiological Functions and Metabolism
2.3. Effects on Tyrosinase and Melanogenesis
2.4. Inhibition of DNA Synthesis and Mitochondrial Function
2.5. Targeting Steroid Metabolism and Energy Production via 5α-Reductase and Glycolytic Pathway Suppression
2.6. Regulation of ROS Signaling and Antioxidant Defense
2.7. Anti-Inflammatory and Immunomodulatory Effects
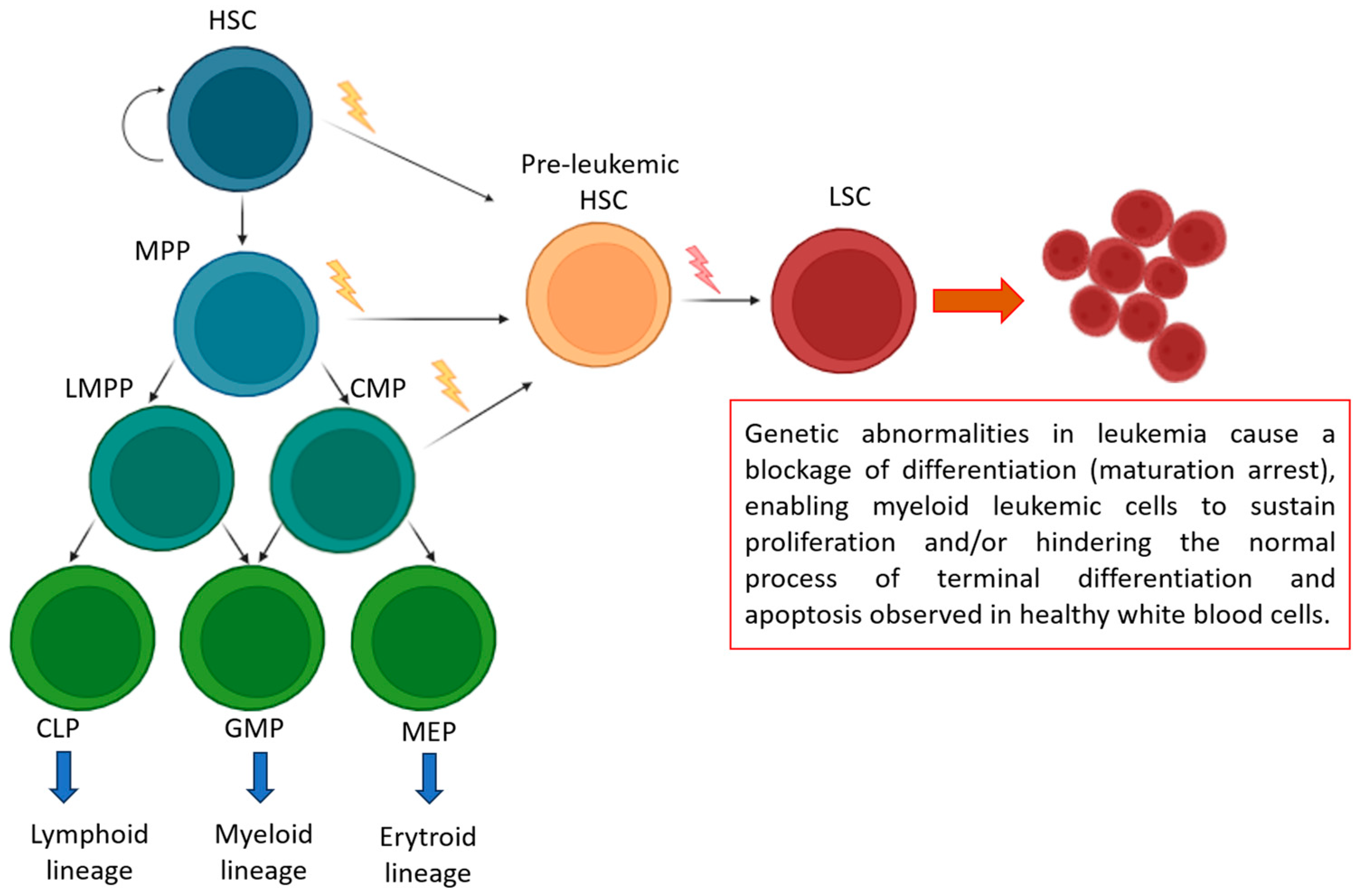
3. Advancements and Challenges in Acute Myeloid Leukemia: From Genetic Insights to Therapeutic Strategies
4. Antileukemic Potential of AZA in Acute Myeloid Leukemia: Cellular Mechanisms and Immunomodulatory Effects
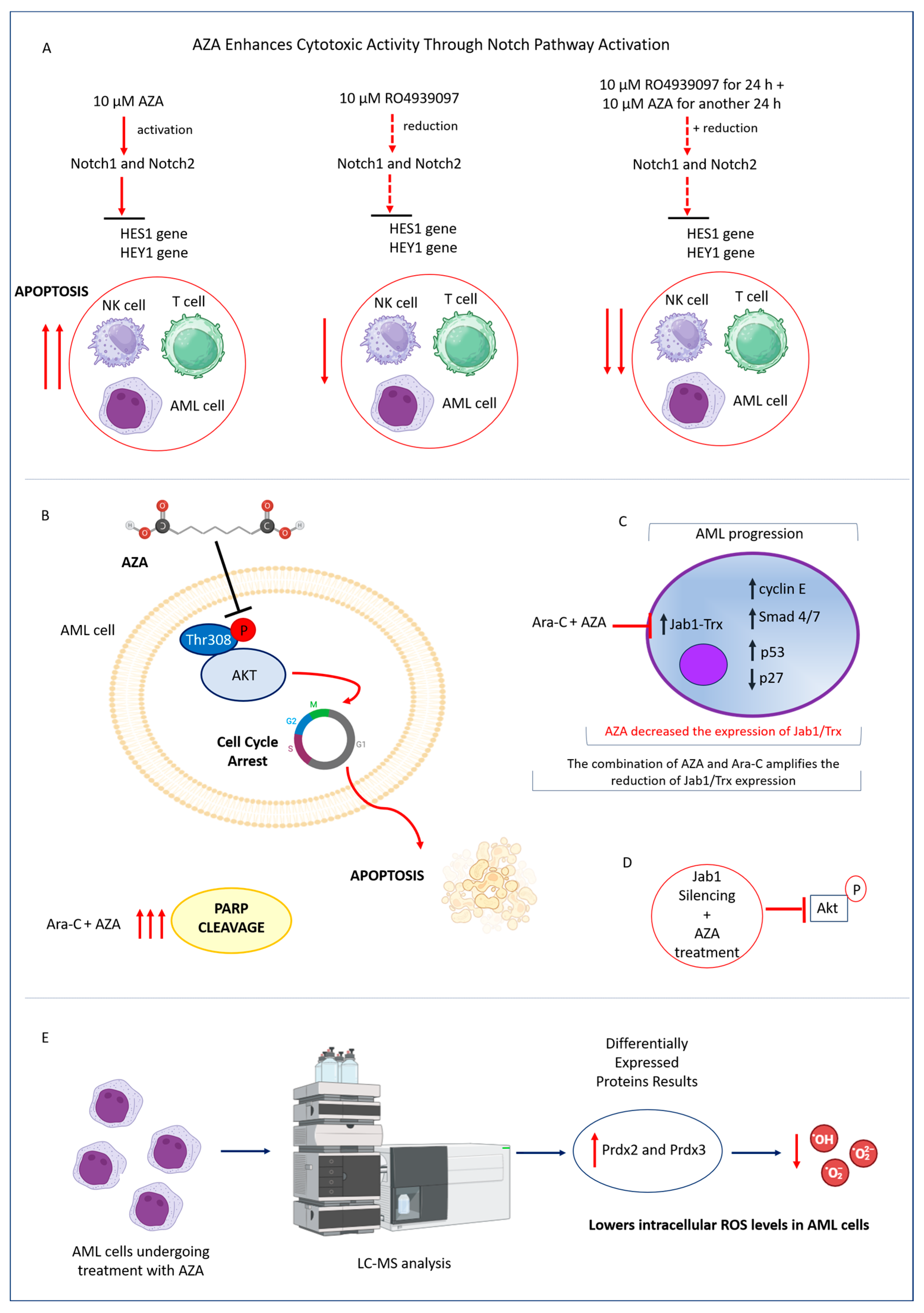
4.1. In Vitro Therapeutic Effects of Azelaic Acid on Acute Myeloid Leukemia Cells
4.2. Mechanisms of AZA-Mediated Cytotoxicity in AML: Modulation of Notch Signaling, PI3K/Akt Pathway, and Redox Balance
4.3. Enhancing AML Treatment: AZA as a Sensitizer to Cytarabine’s Antileukemic Activity
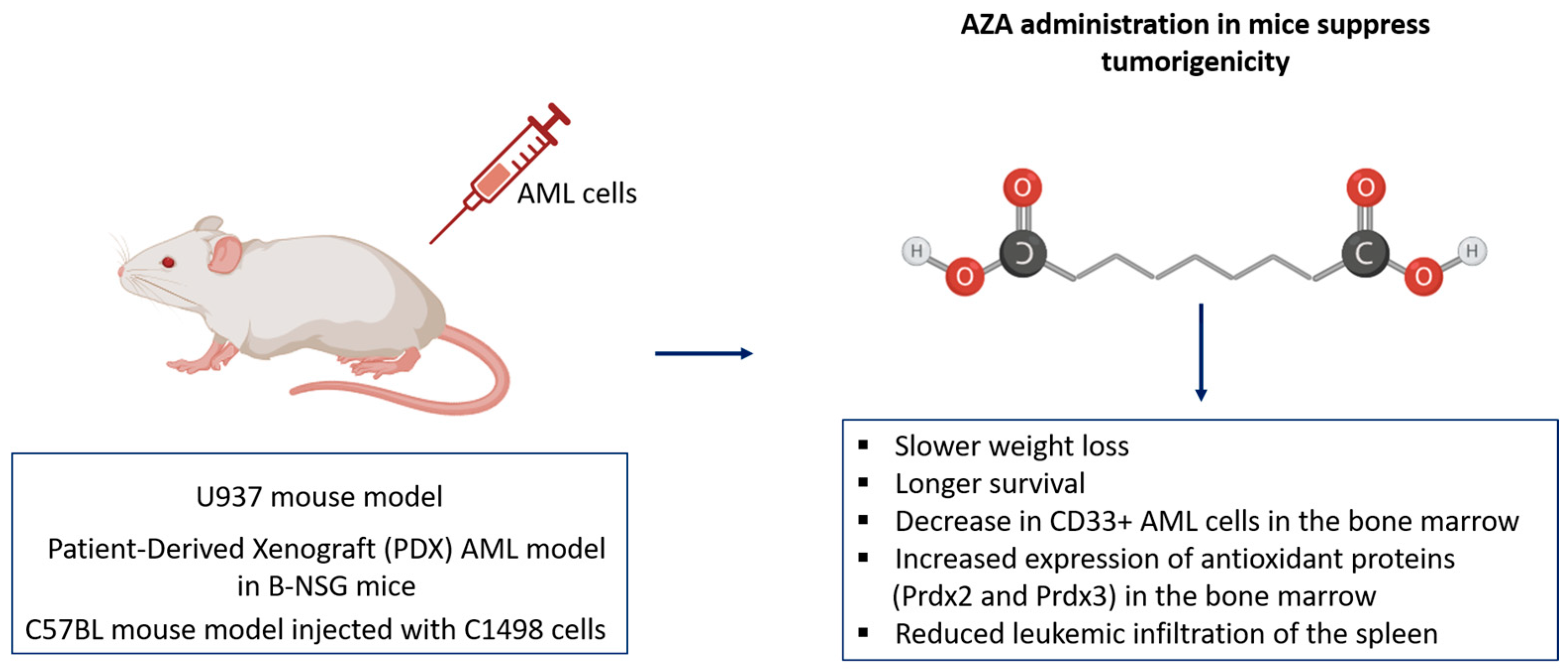
4.4. Effective In Vivo Validation of AZA in Suppressing AML Tumorigenicity and Enhancing Immunologic Response Across Mouse Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Term |
| AML | Acute Myeloid Leukemia |
| AZA | Azelaic Acid |
| ROS | Reactive Oxygen Species |
| PBMC | Peripheral Blood Mononuclear Cell |
| HSC | Hematopoietic Stem Cell |
| LSC | Leukemic Stem Cell |
| CMP | Common Myeloid Progenitor |
| CLP | Common Lymphoid Progenitor |
| GMP | Granulocyte-Macrophage Progenitor |
| MEP | Megakaryocyte-Erythroid Progenitor |
| PDX | Patient-Derived Xenograft |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| PI3K | Phosphatidylinositol 3-Kinase |
| Akt | Protein Kinase B |
| PARP | Poly(ADP-ribose) Polymerase |
| Trx | Thioredoxin |
| TrxR | Thioredoxin Reductase |
| TXNIP | Thioredoxin-Interacting Protein |
| RNR | Ribonucleotide Reductase |
| dNTP | Deoxyribonucleotide Triphosphate |
| SOD | Superoxide Dismutase |
| GSH | Glutathione |
| Prdx | Peroxiredoxin |
| CAT | Catalase |
| MMP | Mitochondrial Membrane Potential |
| NK | Natural Killer (cells) |
| TGF-β | Transforming Growth Factor Beta |
| IFN-γ | Interferon Gamma |
| TNF-α | Tumor Necrosis Factor Alpha |
| ELN | European LeukemiaNet |
| WHO | World Health Organization |
| ICC | International Consensus Classification |
| MRD | Minimal Residual Disease |
| FLT3–ITD | FMS-like Tyrosine Kinase 3–Internal Tandem Duplication |
| IDH | Isocitrate Dehydrogenase |
| BCL-2 | B-cell Lymphoma 2 |
| HMA | Hypomethylating Agent |
| DAs | Dicarboxylic Acids |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| PPP | Pentose Phosphate Pathway |
| TF | Transcription Factor |
References
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Erba, H.P.; Freeman, S.D.; Wei, A.H. Acute Myeloid Leukaemia. Lancet 2023, 401, 2073–2086. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Millman, S.E.; Zhang, L. Metabolism in Acute Myeloid Leukemia: Mechanistic Insights and Therapeutic Targets. Blood 2023, 141, 1119. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, D.; Wei, Y.; Su, D.; Lu, C.; Hu, Y.; Zhou, F. Azelaic Acid Exerts Antileukemic Activity in Acute Myeloid Leukemia. Front. Pharmacol. 2017, 8, 359. [Google Scholar] [CrossRef]
- Zhang, D.; Luo, Z.; Jin, Y.; Chen, Y.; Yang, T.; Yang, Q.; Wu, B.; Shang, Y.; Liu, X.; Wei, Y.; et al. Azelaic Acid Exerts Antileukemia Effects against Acute Myeloid Leukemia by Regulating the Prdxs/ROS Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 1295984. [Google Scholar] [CrossRef]
- Dongdong, Z.; Jin, Y.; Yang, T.; Yang, Q.; Wu, B.; Chen, Y.; Luo, Z.; Liang, L.; Liu, Y.; Xu, A.; et al. Antiproliferative and Immunoregulatory Effects of Azelaic Acid against Acute Myeloid Leukemia via the Activation of Notch Signaling Pathway. Front. Pharmacol. 2019, 10, 1396. [Google Scholar] [CrossRef]
- Radtke, F.; MacDonald, H.R.; Tacchini-Cottier, F. Regulation of Innate and Adaptive Immunity by Notch. Nat. Rev. Immunol. 2013, 13, 427–437. [Google Scholar] [CrossRef]
- Breathnach, A.S. Azelaic Acid: Potential as a General Antitumoural Agent. Med. Hypotheses 1999, 52, 221–226. [Google Scholar] [CrossRef]
- Rau, A.; Keri, J.; Murase, J.E. Management of Acne in Pregnancy. Am. J. Clin. Dermatol. 2024, 25, 465–471. [Google Scholar] [CrossRef]
- Hung, W.-H.; Chen, P.-K.; Fang, C.-W.; Lin, Y.-C.; Wu, P.-C. Preparation and Evaluation of Azelaic Acid Topical Microemulsion Formulation: In Vitro and In Vivo Study. Pharmaceutics 2021, 13, 410. [Google Scholar] [CrossRef]
- Parveen, N.; Sheikh, A.; Molugulu, N.; Annadurai, S.; Wahab, S.; Kesharwani, P. Drug Permeation Enhancement, Efficacy, and Safety Assessment of Azelaic Acid Loaded SNEDDS Hydrogel to Overcome the Treatment Barriers of Atopic Dermatitis. Environ. Res. 2023, 236, 116850. [Google Scholar] [CrossRef] [PubMed]
- Grego, A.V.; Mingrone, G. Dicarboxylic Acids, an Alternate Fuel Substrate in Parenteral Nutrition: An Update. Clin. Nutr. 1995, 14, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Gover, M.D. Azelaic Acid (15% Gel) in the Treatment of Acne Rosacea. Int. J. Dermatol. 2007, 46, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Uyama, H. Tyrosinase Inhibitors from Natural and Synthetic Sources: Structure, Inhibition Mechanism and Perspective for the Future. Cell. Mol. Life Sci. 2005, 62, 1707–1723. [Google Scholar] [CrossRef]
- Ferreira, A.M.; de Souza, A.A.; Koga, R.d.C.R.; Sena, I.d.S.; Matos, M.d.J.S.; Tomazi, R.; Ferreira, I.M.; Carvalho, J.C.T. Anti-Melanogenic Potential of Natural and Synthetic Substances: Application in Zebrafish Model. Molecules 2023, 28, 1053. [Google Scholar] [CrossRef]
- Lemic-Stojcevic, L.; Nias, A.H.W.; Breathnach, A.S. Effect of Azelaic Acid on Melanoma Cells in Culture. Exp. Dermatol. 1995, 4, 79–81. [Google Scholar] [CrossRef]
- Sauer, N.; Oślizło, M.; Brzostek, M.; Wolska, J.; Lubaszka, K.; Karłowicz-Bodalska, K. The Multiple Uses of Azelaic Acid in Dermatology: Mechanism of Action, Preparations, and Potential Therapeutic Applications. Adv. Dermatol. Allergol./Postȩpy Dermatol. I Alergologii 2023, 40, 716. [Google Scholar] [CrossRef]
- Varela, M.T.; de Castro Levatti, E.V.; Tempone, A.G.; Fernandes, J.P.S. Investigation of Structure-Activity Relationships for Benzoyl and Cinnamoyl Piperazine/Piperidine Amides as Tyrosinase Inhibitors. ACS Omega 2023, 8, 44265–44275. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Hassan Khan, M.T.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A Comprehensive Review on Tyrosinase Inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef]
- Shawon, J.; Khan, A.M.; Rahman, A.; Hoque, M.M.; Khan, M.A.K.; Sarwar, M.G.; Halim, M.A. Molecular Recognition of Azelaic Acid and Related Molecules with DNA Polymerase I Investigated by Molecular Modeling Calculations. Interdiscip. Sci. 2018, 10, 525–537. [Google Scholar] [CrossRef]
- Nazzaro-Porro, M. Azelaic Acid. J. Am. Acad. Dermatol. 1987, 17, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.; Hassan, M.H.A.; Mohamed, E.I.A.; Azmy, A.F.; Moawad, A.; Mohammed, R.; Zaki, M.A. New Insights into the Anti-Inflammatory and Anti-Melanoma Mechanisms of Action of Azelaic Acid and Other Fusarium Solani Metabolites via in Vitro and in Silico Studies. Sci. Rep. 2024, 14, 14370. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Li, L.; Deng, X.; Tian, M.; Wang, Z.; Yang, L. Fully Automated Chip-Based Nanoelectrospray Ionization-Mass Spectrometry as an Effective Tool for Rapid and High-Throughput Screening of 5α-Reductase Inhibitors. Anal. Bioanal. Chem. 2020, 412, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Charnock, C.; Brudeli, B.; Klaveness, J. Evaluation of the Antibacterial Efficacy of Diesters of Azelaic Acid. Eur. J. Pharm. Sci. 2004, 21, 589–596. [Google Scholar] [CrossRef]
- Akamatsu, H.; Komura, J.; Asada, Y.; Miyachi, Y.; Niwa, Y. Inhibitory Effect of Azelaic Acid on Neutrophil Functions: A Possible Cause for Its Efficacy in Treating Pathogenetically Unrelated Diseases. Arch. Dermatol. Res. 1991, 283, 162–166. [Google Scholar] [CrossRef]
- Coda, A.B.; Hata, T.; Miller, J.; Audish, D.; Kotol, P.; Two, A.; Shafiq, F.; Yamasaki, K.; Harper, J.C.; Del Rosso, J.Q.; et al. Cathelicidin, Kallikrein 5, and Serine Protease Activity Is Inhibited during Treatment of Rosacea with Azelaic Acid 15% Gel. J. Am. Acad. Dermatol. 2013, 69, 570–577. [Google Scholar] [CrossRef]
- Mastrofrancesco, A.; Ottaviani, M.; Aspite, N.; Cardinali, G.; Izzo, E.; Graupe, K.; Zouboulis., C.C.; Camera, E.; Picardo, M. Azelaic acid modulates the inflammatory response in normal human keratinocytes through PPARgamma activation. Exp. Dermatol. 2010, 9, 813–820. [Google Scholar] [CrossRef]
- Briganti, S.; Flori, E.; Mastrofrancesco, A.; Kovacs, D.; Camera, E.; Ludovici, M.; Cardinali, G.; Picardo, M. Azelaic Acid Reduced Senescence-like Phenotype in Photo-Irradiated Human Dermal Fibroblasts: Possible Implication of PPARγ. Exp. Dermatol. 2013, 22, 41–47. [Google Scholar] [CrossRef]
- Passi, S.; Picardo, M.; Mingrone, G.; Breathnach, A.S.; Nazzaro-Porro, M. Azelaic Acid—Biochemistry and Metabolism. Acta Derm. Venereol. Suppl. 1989, 143, 8–13. [Google Scholar] [CrossRef]
- Long, N.A.; Golla, U.; Sharma, A.; Claxton, D.F. Acute Myeloid Leukemia Stem Cells: Origin, Characteristics, and Clinical Implications. Stem Cell Rev. Rep. 2022, 18, 1211. [Google Scholar] [CrossRef]
- Magliulo, D.; Simoni, M.; Caserta, C.; Fracassi, C.; Belluschi, S.; Giannetti, K.; Pini, R.; Zapparoli, E.; Beretta, S.; Uggè, M.; et al. The Transcription Factor HIF2α Partakes in the Differentiation Block of Acute Myeloid Leukemia. EMBO Mol. Med. 2023, 15, e17810. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, E.; Carrá, G.; Scicchitano, S.; Codispoti, B.; Mega, T.; Lupia, M.; Pelaggi, D.; Marafioti, M.G.; Aloisio, A.; Giordano, M.; et al. UMG Lenti: Novel Lentiviral Vectors for Efficient Transgene- and Reporter Gene Expression in Human Early Hematopoietic Progenitors. PLoS ONE 2014, 9, e114795. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Liesveld, J.L. NPM 1 Mutations in AML-The Landscape in 2023. Cancers 2023, 15, 1177. [Google Scholar] [CrossRef]
- Chiarella, E.; Aloisio, A.; Scicchitano, S.; Bond, H.M.; Mesuraca, M. Regulatory Role of MicroRNAs Targeting the Transcription Co-Factor ZNF521 in Normal Tissues and Cancers. Int. J. Mol. Sci. 2021, 22, 8461. [Google Scholar] [CrossRef]
- Codispoti, B.; Rinaldo, N.; Chiarella, E.; Lupia, M.; Spoleti, C.B.; Marafioti, M.G.; Aloisio, A.; Scicchitano, S.; Giordano, M.; Nappo, G.; et al. Recombinant TAT-BMI-1 Fusion Protein Induces Ex Vivo Expansion of Human Umbilical Cord Blood-Derived Hematopoietic Stem Cells. Oncotarget 2017, 8, 43782–43798. [Google Scholar] [CrossRef]
- Sasaki, K.; Ravandi, F.; Kadia, T.M.; DiNardo, C.D.; Short, N.J.; Borthakur, G.; Jabbour, E.; Kantarjian, H.M. De Novo Acute Myeloid Leukemia: A Population-Based Study of Outcome in the United States Based on the Surveillance, Epidemiology, and End Results (SEER) Database, 1980 to 2017. Cancer 2021, 127, 2049–2061. [Google Scholar] [CrossRef]
- Shimony, S.; Stahl, M.; Stone, R.M. Acute Myeloid Leukemia: 2023 Update on Diagnosis, Risk-Stratification, and Management. Am. J. Hematol. 2023, 98, 502–526. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V.; Gómez-Guijosa, M.Á.; Cortes-Penagos, C. Acute Myeloid Leukemia—Genetic Alterations and Their Clinical Prognosis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 328. [Google Scholar]
- Nisticò, C.; Chiarella, E. An Overview on Lipid Droplets Accumulation as Novel Target for Acute Myeloid Leukemia Therapy. Biomedicines 2023, 11, 3186. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Panyosak, T. Selective Effects of Azelaic Acid in Nanovesicles on Cell Lines. In Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–7. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch Signaling Pathway: Architecture, Disease, and Therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Gallay, N.; Dos Santos, C.; Cuzin, L.; Bousquet, M.; Gouy, V.S.; Chaussade, C.; Attal, M.; Payrastre, B.; Demur, C.; Récher, C. The Level of AKT Phosphorylation on Threonine 308 but Not on Serine 473 Is Associated with High-Risk Cytogenetics and Predicts Poor Overall Survival in Acute Myeloid Leukaemia. Leukemia 2009, 23, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Pan, Y.; Wei, Y.; Zhang, R.; Bai, G.; Shen, Q.; Meng, S.; Le, X.F.; Andreeff, M.; Claret, F.X. Jab1/Csn5-Thioredoxin Signaling in Relapsed Acute Monocytic Leukemia under Oxidative Stress. Clin. Cancer Res. 2017, 23, 4450–4461. [Google Scholar] [CrossRef]
- Dennis, M.; Hills, R.K.; Russell, N.H.; Copland, M.; Thomas, I.; McMullin, M.F.F.; Ali, S.; Burnett, A.K. An Evaluation of 17 Years of Low Dose Cytarabine As Therapy for AML Patients Not Fit for Intensive Treatment, Including Patients with Adverse Cytogenetics, Shows Improving Survival, Potential Underutilisation and Highlights the Need for New Therapy. Blood 2017, 130, 3874. [Google Scholar] [CrossRef]
- Ishii, H.; Yano, S. New Therapeutic Strategies for Adult Acute Myeloid Leukemia. Cancers 2022, 14, 2806. [Google Scholar] [CrossRef]
- Heo, S.K.; Noh, E.K.; Yu, H.M.; Kim, D.K.; Seo, H.J.; Lee, Y.J.; Cheon, J.; Koh, S.J.; Min, Y.J.; Choi, Y.; et al. Radotinib Enhances Cytarabine (Ara-C)-Induced Acute Myeloid Leukemia Cell Death. BMC Cancer 2020, 20, 1193. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Hours of AZA Treatment | AZA Concentration | Biological Effects | Ref. |
|---|---|---|---|---|
U937 THP-1 KG-1 NB-4 HL-60 | 72 | IC50: 1.4 mM 1.2 mM 1.7 mM 1.7 mM 1.9 mM | Reduction in cell viability, Induction of apoptosis | [4] |
| U937 THP-1 KG-1 NB-4 HL-60 | 72 | 3.0 mM | Reduction in colony formation | [4] |
| U937 HL-60 Molm-13 human AML cells | 48 | 5.0 mM | Reduction in cell viability, Induction of apoptosis | [6] |
| THP-1 U937 human AML cells | 6 | Co-culture with 10 µM AZA-treated NK and T cell supernatants | Reduction in cell viability, Enhanced expression of cytolysis-related receptors | [6] |
| Healthy PBMCs | 72 | 5–10 mM | No cytolytic effects observed | [4] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Agostino, S.; Di Vito, A.; Aloisio, A.; Piazzetta, G.L.; Lobello, N.; Bria, J.; Chiarella, E. Harnessing Azelaic Acid for Acute Myeloid Leukemia Treatment: A Novel Approach to Overcoming Chemoresistance and Improving Outcomes. Int. J. Mol. Sci. 2025, 26, 4362. https://doi.org/10.3390/ijms26094362
Di Agostino S, Di Vito A, Aloisio A, Piazzetta GL, Lobello N, Bria J, Chiarella E. Harnessing Azelaic Acid for Acute Myeloid Leukemia Treatment: A Novel Approach to Overcoming Chemoresistance and Improving Outcomes. International Journal of Molecular Sciences. 2025; 26(9):4362. https://doi.org/10.3390/ijms26094362
Chicago/Turabian StyleDi Agostino, Silvia, Anna Di Vito, Annamaria Aloisio, Giovanna Lucia Piazzetta, Nadia Lobello, Jessica Bria, and Emanuela Chiarella. 2025. "Harnessing Azelaic Acid for Acute Myeloid Leukemia Treatment: A Novel Approach to Overcoming Chemoresistance and Improving Outcomes" International Journal of Molecular Sciences 26, no. 9: 4362. https://doi.org/10.3390/ijms26094362
APA StyleDi Agostino, S., Di Vito, A., Aloisio, A., Piazzetta, G. L., Lobello, N., Bria, J., & Chiarella, E. (2025). Harnessing Azelaic Acid for Acute Myeloid Leukemia Treatment: A Novel Approach to Overcoming Chemoresistance and Improving Outcomes. International Journal of Molecular Sciences, 26(9), 4362. https://doi.org/10.3390/ijms26094362