

Interactions of CFTR and Arylsulfatase B (ARSB; N-acetylgalactosamine-4-sulfatase) in Prostate Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
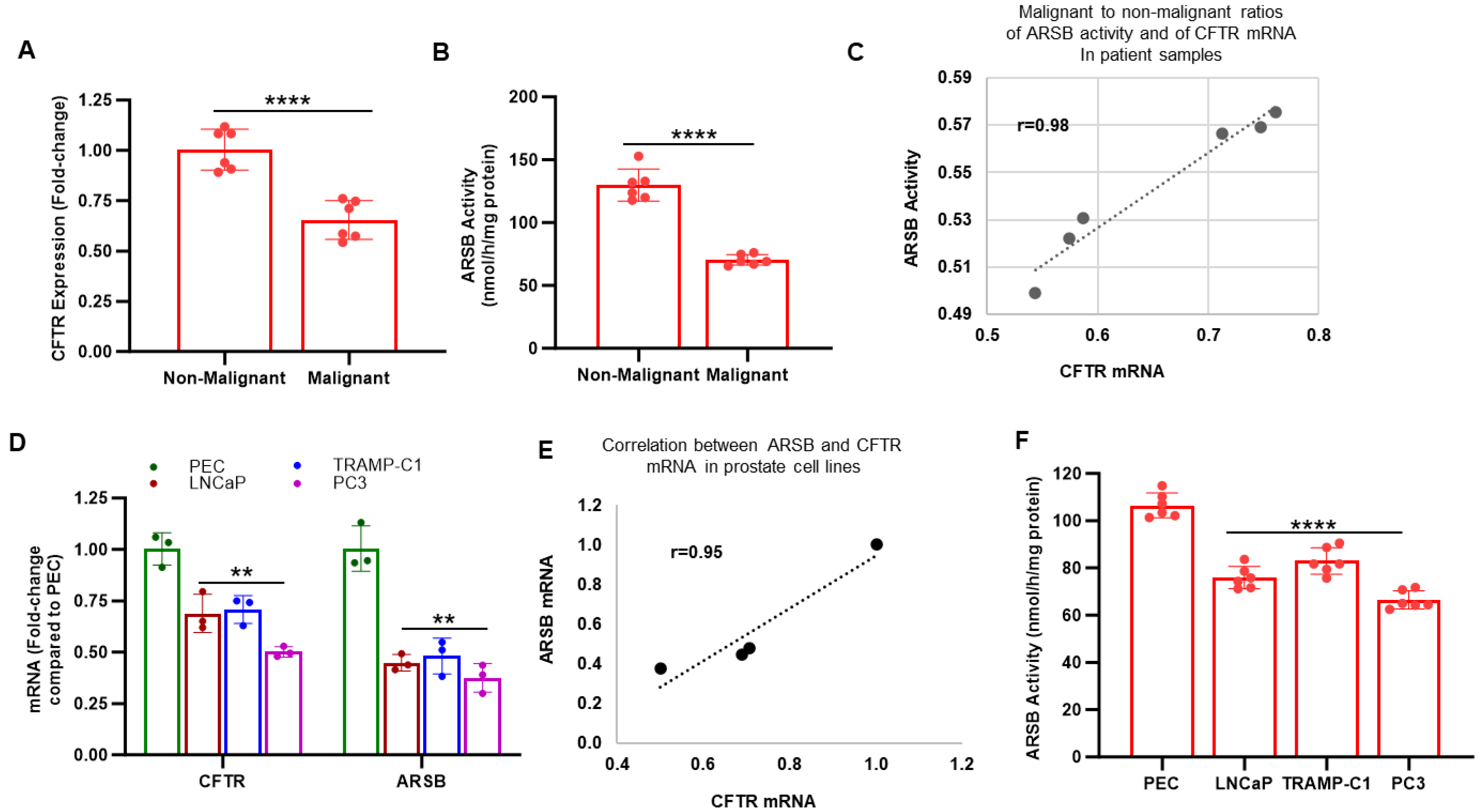
2.1. CFTR and ARSB in Normal and Malignant Human Prostate Tissue and Cells
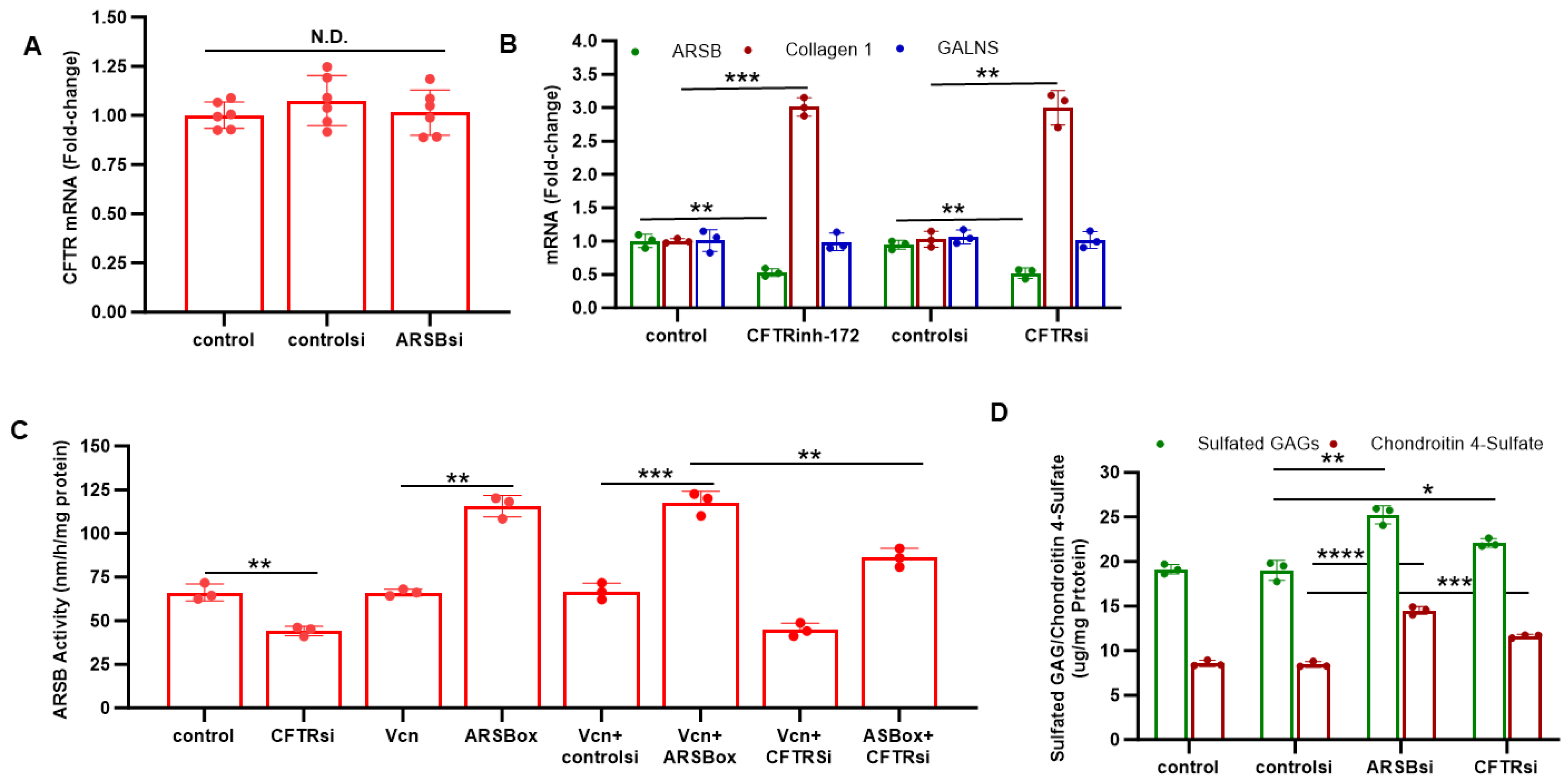
2.2. Effects of ARSB and CFTR Inhibition
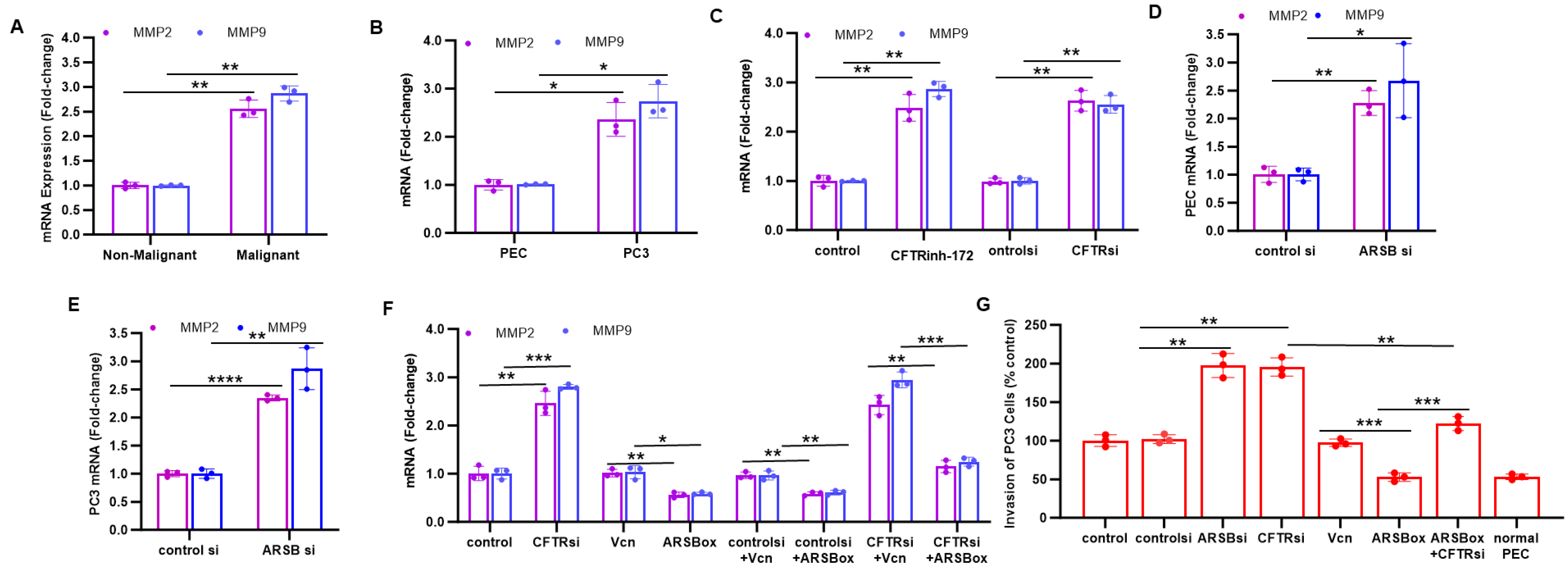
2.3. Inhibition of CFTR Replicates Effects of ARSB Knockdown on MMP2, MMP9 and Invasiveness
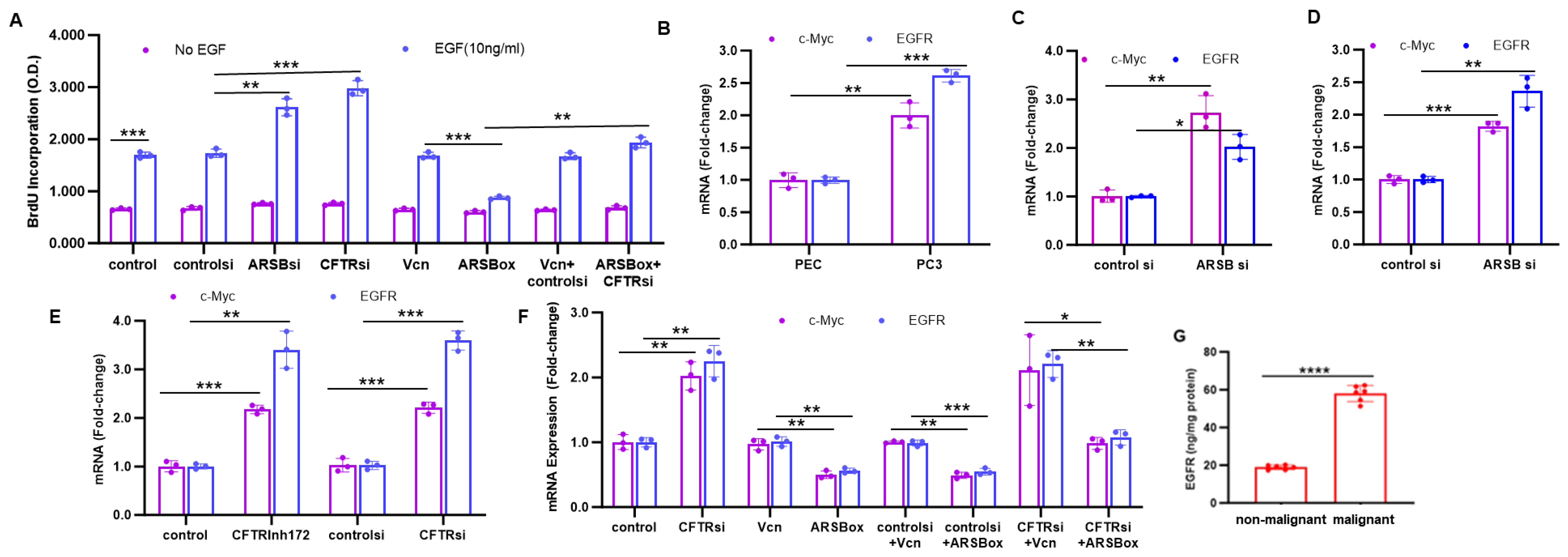
2.4. Effects of ARSB and CFTR on c-Myc, EGFR, and BrdU Incorporation
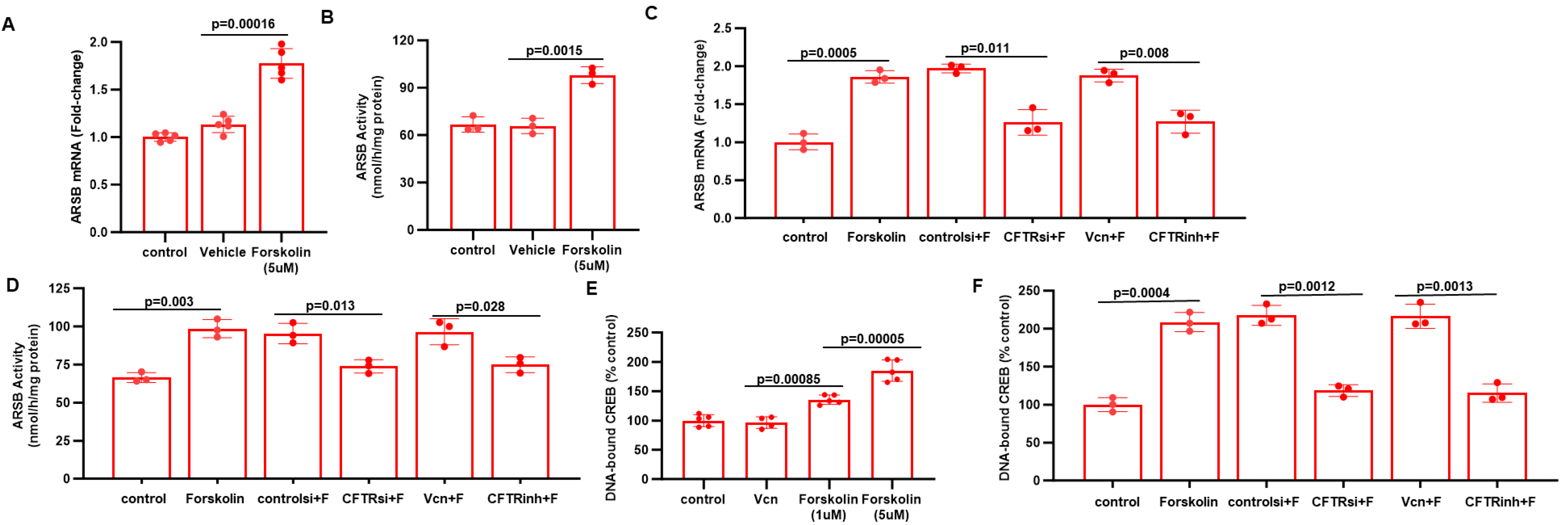
2.5. Impact of CFTR, Forskolin, and CREB on ARSB Expression
3. Discussion
4. Materials and Methods
4.1. Prostate Cell Lines and Tissue
4.2. Laser Capture Microdissection
4.3. ARSB and CFTR Silencing by siRNA
4.4. ARSB Overexpression
4.5. ARSB Activity
4.6. Total Sulfated Glycosaminoglycan GAG and Chondroitin 4-Sulfate Assays
4.7. QRT-PCR for CFTR, EGFR, GATA-3, Collagen 1, ARSB, MMP2, MMP9, Cyclin-D1, c-Myc
4.8. Total EGFR Assay
4.9. Nuclear Phospho-CREB Assay
4.10. BrdU Incorporation Assay
4.11. Invasion Assay
4.12. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What role does CFTR play in development, differentiation, regeneration and cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef] [PubMed]
- Parisi, G.F.; Papale, M.; Pecora, G.; Rotolo, N.; Manti, S.; Russo, G.; Leonardi, S. Cystic Fibrosis and cancer: Unraveling the complex role of CFTR gene in cancer susceptibility. Cancers 2023, 15, 4244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Jiang, X.; Chan, H.C. Cystic fibrosis transmembrane conductance regulator-emerging regulator of cancer. Cell. Mol. Life Sci. 2018, 75, 1737–1756. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jiang, X.H.; Zhang, J.T.; Sun, T.T.; Dong, J.D.; Sanders, A.J.; Diao, R.Y.; Wang, Y.; Fok, K.L.; Tsang, L.L.; et al. CFTR suppresses tumor progression through miR-193b targeting urokinase plasminogen activator (uPA) in prostate cancer. Oncogene 2013, 32, 2282–2291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jia, S.; Zheng, J.; Xiang, R.; Cui, Y.; Zhang, J.; Xu, Y.; Zhao, M. Analysis of CFTR gene expression as an immunological and prognostic biomarker in pan-cancers. Comput. Biol. Med. 2022, 146, 105614. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Burke, J.; Bhattacharyya, S.; Sharma, N.; Katyal, S.; Park, R.L.; Tobacman, J. Reduced Arylsulfatase B activity in leukocytes from cystic fibrosis patients. Pediatr. Pulmonol. 2013, 48, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Tobacman, J.K.; Bhattacharyya, S. Profound impact of decline in N-acetylgalactosamine-4-sulfatase (Arylsulfatase B) on molecular pathophysiology and human diseases. Int. J. Mol. Sci. 2022, 23, 13146. [Google Scholar] [CrossRef] [PubMed]
- Roeser, D.; Schmidt, B.; Preusser-Kunze, A.; Rudolph, M.G. Probing the oxygen-binding site of the human formylglycine-generating enzyme using halide ions. Acta Crystallogr. D Biol. Crystallogr. 2007, 63 Pt 5, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Keitany, G.J.; Jenkins, B.J.; Obiakor, H.T.; Daniel, S.; Muehlenbachs, A.; Semblat, J.P.; Gamain, B.; ADoritchamou, J.Y.; Desai, S.A.; MacDonald, N.J.; et al. An invariant protein that colocalizes with VAR2CSA on Plasmodium falciparum-Infected red cells binds to chondroitin sulfate A. J. Infect. Dis. 2022, 225, 2011–2022. [Google Scholar] [CrossRef] [PubMed]
- Achur, R.N.; Valiyaveettil, M.; Gowda, D.C. The low sulfated chondroitin sulfate proteoglycans of human placenta have sulfate group-clustered domains that can efficiently bind Plasmodium falciparum-infected erythrocytes. J. Biol. Chem. 2003, 278, 11705–11713. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, A.; Achur, R.N.; Bhavanandan, V.P.; Fouda, G.G.; Taylor, D.W.; Gowda, D.C. Plasmodium falciparum-infected erythrocytes adhere both in the intervillous space and on the villous surface of human placenta by binding to the low-sulfated chondroitin sulfate proteoglycan receptor. Am. J. Pathol. 2004, 164, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Feferman, L.; Tobacman, J.K. Dihydrotestosterone inhibits arylsulfatase B and Dickkopf Wnt signaling pathway inhibitor (DKK)-3 leading to enhanced Wnt signaling in prostate epithelium in response to stromal Wnt3A. Prostate 2019, 79, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ou, C.; Zhu, X.; Tan, C.; Xiang, X.; He, Y. Potential role of CFTR in bisphenol A-induced malignant transformation of prostate cells via mitochondrial apoptosis. Toxicol. Ind. Health 2020, 36, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Gao, L.; Yan, C.; He, Y. A novel role and mechanism of cystic fibrosis transmembrane conductance regulator in bisphenol A-induced prostate cancer. J. Cell. Biochem. 2019, 120, 8689–8695. [Google Scholar] [CrossRef] [PubMed]
- Salamanca-Fernández, E.; Rodríguez-Barranco, M.; Amiano, P.; Delfrade, J.; Chirlaque, M.D.; Colorado, S.; Guevara, M.; Jimenez, A.; Arrebola, J.P.; Vela, F.; et al. Bisphenol-A exposure and risk of breast and prostate cancer in the Spanish European Prospective Investigation into Cancer and Nutrition study. Environ. Health 2021, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, K.; Han, T.; Zhang, X.; Xu, X.; Liu, Z.; Ren, X.; Zhang, X.; Lu, Z.; Qin, C. Bisphenol A promote the cell proliferation and invasion ability of prostate cancer cells via regulating the androgen receptor. Ecotoxicol. Environ. Saf. 2024, 269, 115818. [Google Scholar] [CrossRef] [PubMed]
- Hengstler, J.G.; Foth, H.; Gebel, T.; Kramer, P.J.; Lilienblum, W.; Schweinfurth, H.; Völkel, W.; Wollin, K.M.; Gundert-Remy, U. Critical evaluation of key evidence on the human health hazards of exposure to bisphenol A. Crit. Rev. Toxicol. 2011, 41, 263–291. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Hector, A.; Bratcher, P.E.; Mall, M.A.; Griese, M.; Hartl, D. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir J. 2011, 38, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Devereux, G.; Steele, S.; Jagelman, T.; Fielding, S.; Muirhead, R.; Brady, J.; Grierson, C.; Brooker, R.; Winter, J.; Fardon, T.; et al. An observational study of matrix metalloproteinase (MMP)-9 in cystic fibrosis. J. Cyst. Fibros. 2014, 13, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Capraro, M.; Pedrazzi, M.; De Tullio, R.; Manfredi, M.; Cresta, F.; Castellani, C.; Averna, M. Modulation of Plasmatic Matrix Metalloprotease 9: A promising new tool for understanding the variable clinical responses of patients with Cystic Fibrosis to cystic fibrosis transmembrane conductance regulator modulators. Int. J. Mol. Sci. 2023, 24, 13384. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharyya, S.; Tobacman, J.K. Interactions of CFTR and Arylsulfatase B (ARSB; N-acetylgalactosamine-4-sulfatase) in Prostate Carcinoma. Int. J. Mol. Sci. 2025, 26, 4350. https://doi.org/10.3390/ijms26094350
Bhattacharyya S, Tobacman JK. Interactions of CFTR and Arylsulfatase B (ARSB; N-acetylgalactosamine-4-sulfatase) in Prostate Carcinoma. International Journal of Molecular Sciences. 2025; 26(9):4350. https://doi.org/10.3390/ijms26094350
Chicago/Turabian StyleBhattacharyya, Sumit, and Joanne K. Tobacman. 2025. "Interactions of CFTR and Arylsulfatase B (ARSB; N-acetylgalactosamine-4-sulfatase) in Prostate Carcinoma" International Journal of Molecular Sciences 26, no. 9: 4350. https://doi.org/10.3390/ijms26094350
APA StyleBhattacharyya, S., & Tobacman, J. K. (2025). Interactions of CFTR and Arylsulfatase B (ARSB; N-acetylgalactosamine-4-sulfatase) in Prostate Carcinoma. International Journal of Molecular Sciences, 26(9), 4350. https://doi.org/10.3390/ijms26094350