1. Introduction
Glioblastoma (GBM) is a fatal primary brain tumor that affects men disproportionately compared to women (1.6:1), with data suggesting that males have more aggressive diseases and poorer outcomes [
1]. The biological reasons for these observations are as yet unclear, although molecular characterization reveals that females are more likely to have MGMT methylated disease, secondary GBMs, differences in tumor location, and molecular and metabolic mechanistic triggers [
2,
3,
4,
5,
6]. Sex differences in cancer as applied to serum proteomics and metabolomics are important given mounting data for the differential response to management and outcome differences between male and female patients. This is particularly the case in non-invasively collected biospecimens such as serum where molecular profiling aimed at sex differences is underexplored [
1,
6,
7,
8,
9,
10,
11]. Nonetheless, non-invasive specimens are easier and cheaper to acquire and provide the only means of obtaining data in real-time in tumors located in areas not amenable to repeat tissue sampling, such as the brain. Noninvasive biospecimens such as blood samples in the clinic are the mainstay of biomarker-directed clinical care in the real world and the most cost and time-effective means of obtaining information that can impact management in real-time. Blood-based biomarkers are not currently available for most cancers including GBM, limiting the ability to personalize management and curtailing access to precision medicine secondary to cost, particularly in resource-strained care environments [
12,
13]. Sex as a biological variable analysis (SABV) has been hampered by a paucity of data available with robust annotation for male and female as class labels and the prevalence of datasets where one sex is more frequently diagnosed with the primary tumor as compared to the other, as is the case for GBM [
14]. Most analyses in this space employ data as an aggregate of both men and women without separation of the samples for analysis, masking potential differences. Additional barriers in serum proteomic and metabolomic data include the biological complexity of sex differences comprised of hormonal influences, genetic and epigenetic factors, and immune response [
1]. Additional technical and methodological challenges contribute to the lack of conclusions in serum proteome and metabolome data, as both are highly complex, detecting both high- and low-abundance proteins and compounds, and there is difficulty in the attribution of differences to biological aspects compared to confounders including age and comorbidities [
15]. The identification of biomarkers that are equally effective in women and men remains an understudied aspect of serum omics data. Given existing data supporting differences in outcomes (both progression and survival) in GBM in men vs. women, addressing these issues requires integrative, multidisciplinary approaches combining advanced proteomic and metabolomic technologies, robust bioinformatics, and the consideration of biological sex in study design and analysis. We wanted to define serum proteomic and metabolomic signals in a cohort of women and men with pathologically proven GBM using serum-derived proteomic and metabolomic data, hypothesizing that identified signals may be representative of both known signals associated with sex differences, which can provide validation of the methodology, and possible additional signals, which may indicate a relationship with the underlying malignancy, with possible downstream mechanistic insights. We repeated the same analysis in publicly available data (CPTAC and TCGA) and compared the results, exploring the intersectionality between serum vs. tissue in an effort to guide future efforts aimed at identifying GBM biomarkers with clinical applicability.
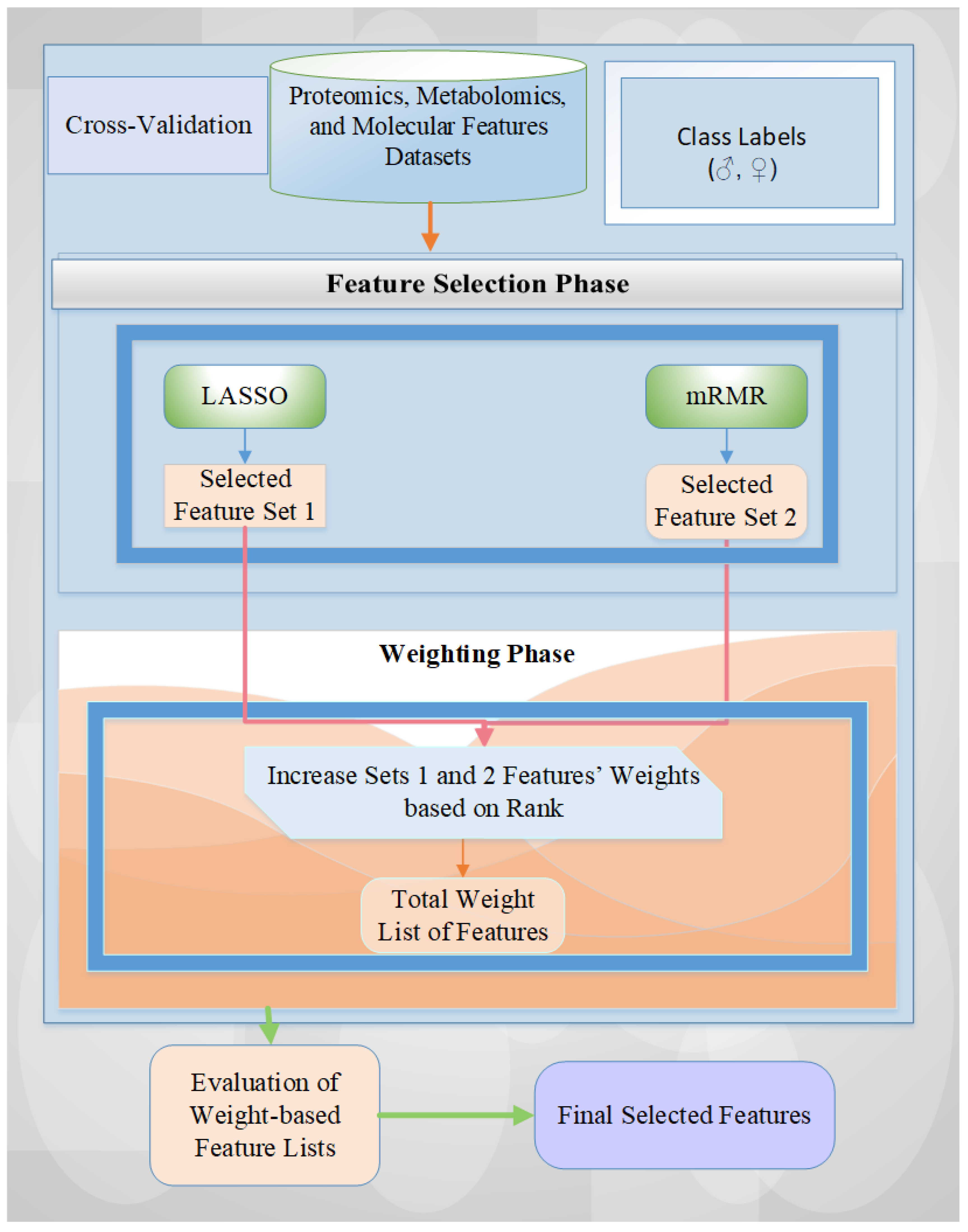
In this study, we proposed a hybrid feature selection method that combines LASSO (i.e., Least Absolute Shrinkage and Selection Operator) and mRMR (i.e., Minimum Redundancy Maximum Relevance). This approach leverages the advantages of sparse feature selection and reduced redundancy, effectively addressing the limitations of each method when applied individually. Through this integration, we sought to improve the reliability and interpretability of selected features, especially in the context of sex differences in glioblastoma. We also aimed to find the minimum number of selected features with the highest performance results in terms of accuracy rate.
The main contributions of this study, divided into technical and clinical aspects, are outlined below:
To the best of our knowledge, this is the first study that employs a combined feature selection and weighting methodology (i.e., GLIO-Select) employing female/male as a class label for classification tasks on different proteomic, metabolomic, and molecular datasets.
To increase the scope and motivation of this study, we apply and compare our approach to the five different case studies on -omics and molecular datasets for classification.
We adopted our previous MetaWise [
16] methodology to analyze different types of problems and omics data.
To address the effects of imbalanced class distribution in our datasets, we implemented stratified cross-validation, ensuring that each data fold retained the original class proportions.
We utilized a rank-based feature weighting methodology to identify the feature names despite potential variations across cross-validation folds.
We evaluated the effects of feature selection and weighting on six different machine learning models on proteomic, metabolomic, and molecular datasets to determine the optimal prediction model and minimal feature set for accurate classification.
We visualized and interpreted selected features by using clustergram (i.e., dendrogram and heatmap) plots according to the dataset’s male/female class labels.
We employed serum proteomic and metabolomic datasets applying feature selection operations to determine which serum signals distinguish samples obtained from females as compared to males. This is novel as serum proteomic and metabolomic profiles in GBM have not been described.
To determine whether there are overlapping or linked signals between tissue and serum, we carried out the same analysis in CPTAC proteomic and metabolomic data.
To link emerging signals and results derived using feature selection to transcriptomic data, we employed a TCGA glioma grading dataset we previously demonstrated as effective at glioma grading to identify mutations distinguishing tumor grade distribution between women and men.
The present approach, coupled with interpretable dimensionality reduction (i.e., feature selection), enabled the identification of biomarkers with high accuracy to differentiate males from females in patients with GBM.
Our methodology shows promising potential for future studies in several ways: (1) as a possible check for data accuracy in serum samples; (2) as a means of validating serum and tissue signals previously described using feature selection; (3) as a means of showcasing signals that have a distinct relationship to one sex vs. another, some of which represent novel signals; and (4) as a means of identifying signals that may help with further research into differential molecular and mechanistic aspects between females and males in GBM.
The remainder of this paper is structured as follows:
Section 2 outlines the experimental setup, performance indicators, and computational outcomes in detail.
Section 3 presents the results and discussion.
Section 4 provides a detailed description of the dataset, the employed feature selection and weighting methodologies, and the supervised learning models used for classification. Finally,
Section 5 summarizes the study’s findings and suggests potential directions for future research.
3. Discussion
While several studies have described sex differences in GBM employing transcriptomic or tissue-level proteomic data, noninvasive biospecimen-derived data and biological sex differences profiles in GBM serum proteomic and metabolomic data are not currently available [
1,
6,
9,
11,
23]. This paucity of data limits the ability to account for the impact of biological sex when interpreting emerging biomarkers in serum while also limiting our mechanistic understanding of observed outcome differences. In this study, we identified several hundred serum features associated with the male and female class labels in proteomic and metabolomic datasets. Using the local serum-based dataset, 17 features (100% ACC) and 16 features (92% ACC) were identified for the proteomic and metabolomic datasets, respectively, demonstrating that the serum in our cohort is a robust biospecimen of analysis for the identification of proteins associated with sex differences. Using the CPTAC tissue-based dataset (8828 proteomic and 59 metabolomic features), 5 features (99% ACC) and 13 features (80% ACC) were identified for the proteomic and metabolomic datasets, respectively. The proteomic data serum (local data) or tissue (CPTAC) achieved the highest accuracy rates (100% and 99%, respectively), followed by serum metabolome (92% and 80%) for the local set and CPTAC, respectively (
Table 6).
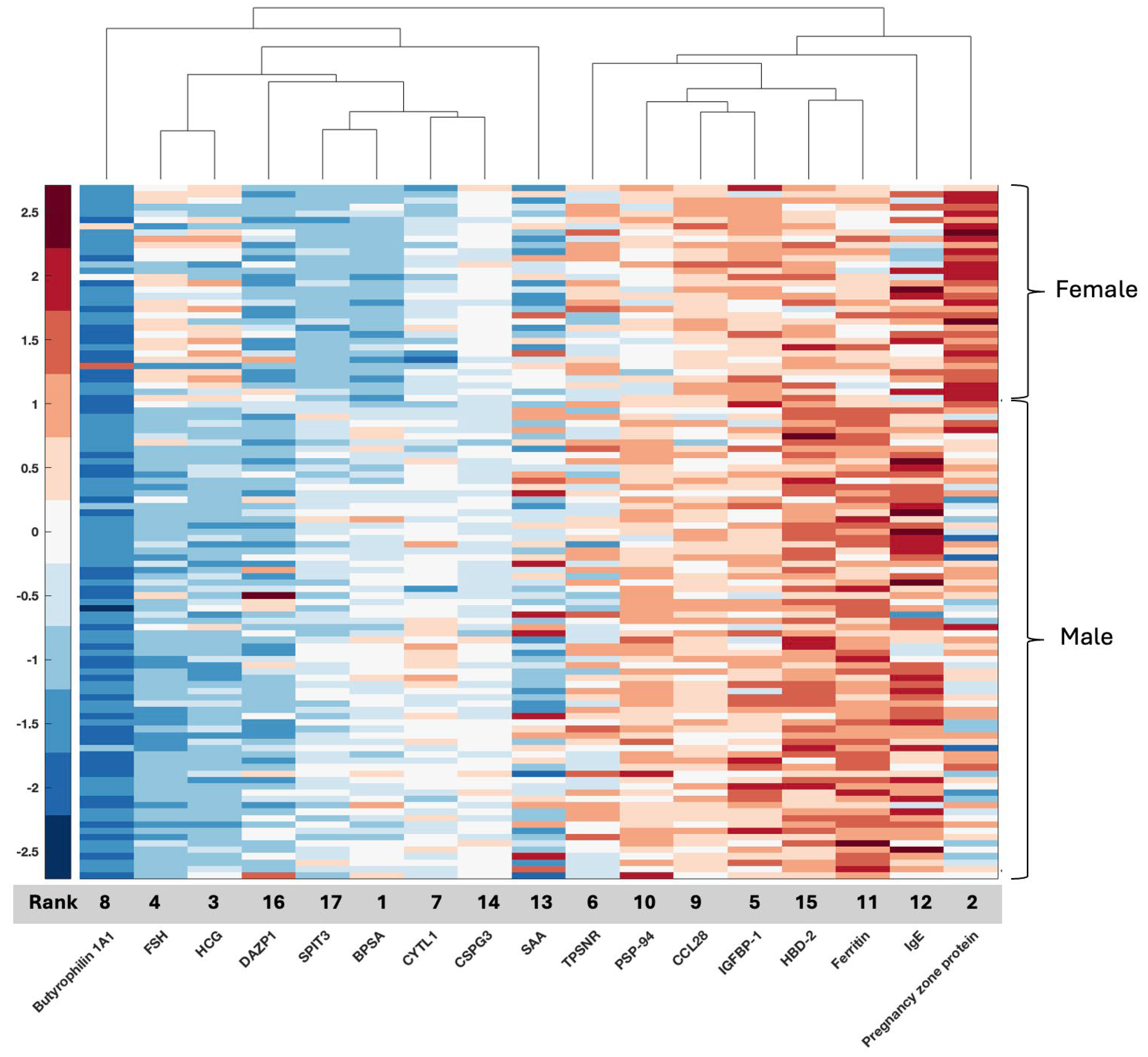
The current GLIO-Select method allowed for the selection of serum proteomic features that can differentiate a sample originating from a female GBM patient from a male GBM patient in serum with 100% accuracy for proteomic features and 92% accuracy for metabolomic features. The identified proteomic features offer both internal and methodological validation since several are already described as being associated with sex differences specifically in serum, including benign prostatic specific antigen (BPSA) [
24], pregnancy zone protein (PZP) [
25], PSP-94 [
26], and DAZP1 [
27]. Additional roles in glioma have also emerged for proteins known to have an association with one sex compared to the other, including HCG [
28], FSH [
29], PZP [
25,
30], and ferritin [
31] (
Table 7). However, while existing data support several of the identified features as being associated more with one sex than the other, for example, PZP with the female sex and PSA with the male sex, there are females with low PZP and males with PSA as low as that measured in most females. Thus, per the existing literature, no one serum protein or metabolite is 100% associated with either sex. For example, HCG may be measured in both men and women but may have physiological (pregnancy) or pathological (testicular cancer) implications. It should also be noted that just because certain serum proteins have a classical association with biological sex, such as HCG, they may have an additional role in GBM that is either unrecognized or subject to evolving data. HCG has been associated with GBM and may represent a possible biomarker [
32], being implicated in glioma cells’ redox homeostasis. Equally so, there is an emerging relationship between sex hormones and androgen levels and tumor aggressiveness in GBM, which may be reflected in the identification of PSA, FSH, and PSP-94 [
29,
33]. Some proteins, such as IGFBP-1 (associated with stemness and invasion) [
34] and BTN1A1 (novel immune checkpoint exclusive to PDL-1) [
35] may have a relationship with biological sex [
36], but this is evolving. In contrast, others are already associated with glioma (TPSNR (HLA1 antigen processing protein) [
37], IgE [
38], SAA1 (potential prognostic marker) [
39], NCAN (also known as CSPG3, glycosylated chondroitin sulfate proteoglycan implicated in the tumor microenvironment) [
40], and SPIT3 [
41], and, in our analysis, are also associated with biological sex (
Figure 6). Interestingly, SPIT3 clustered with BPSA in the serum, raising the hypothesis that there may be mechanistic features between the male sex and tumor behavior in males that may link to stemness and treatment resistance. DEFB4A (a component of the defensin family associated with several malignancies) [
42] was identified in serum, while DEFA3 was identified in tissue, with their roles in innate immunity and malignancy evolving. These proteins present an opportunity to identify potential mechanistic relationships that drive glioma proliferation and response differences between the sexes since they are, in some form, present and associated with sex in both biospecimens.
PZP was also identified in CPTAC tissue proteome in this analysis; however, it was not one of the five features that directly contributed to ACC of 99%.
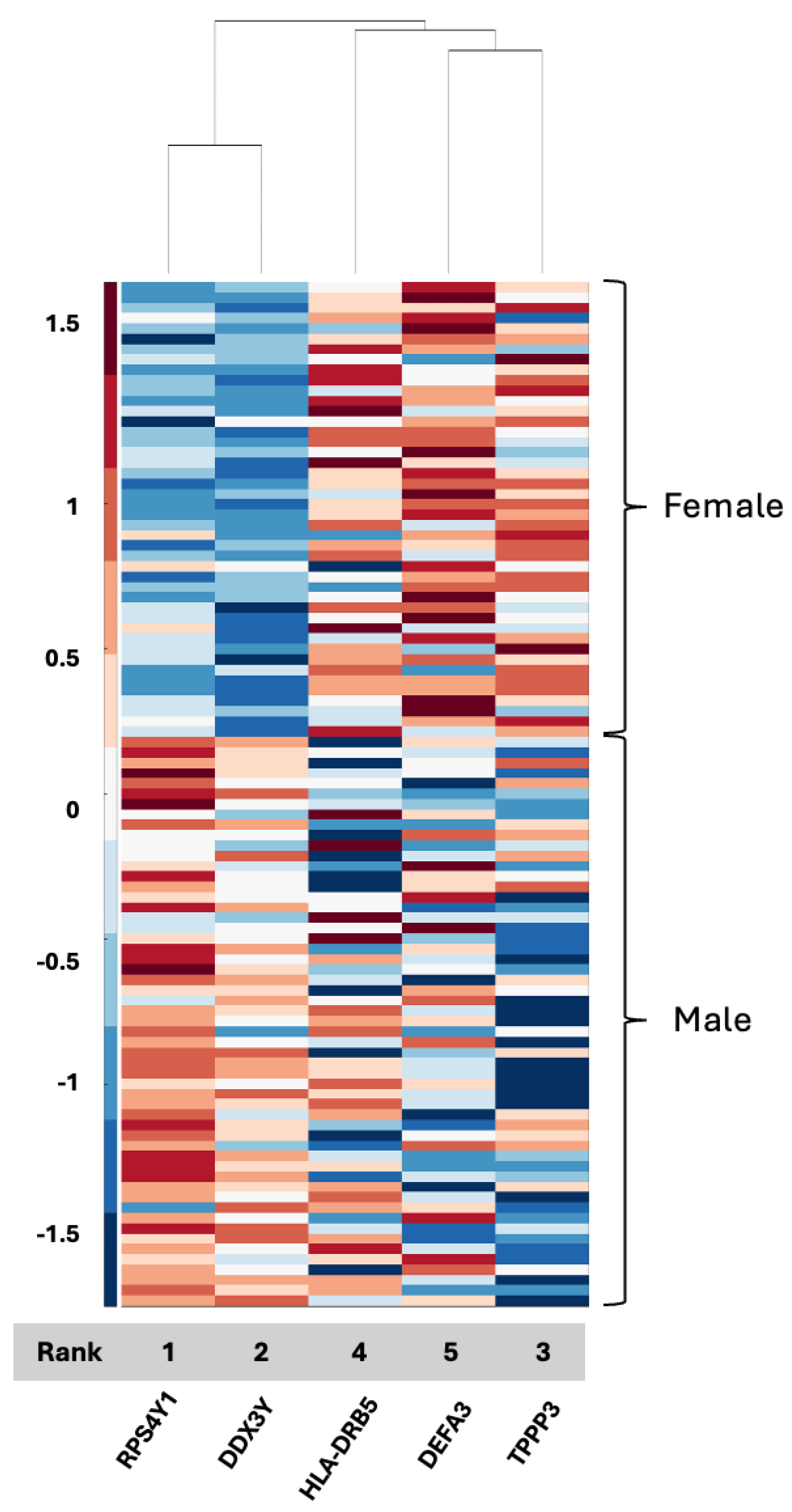
The CPTAC tissue proteome data achieved an accuracy rate of 99% resulting in five features: RPS4Y1, DDX3Y, HLA-DRB5, DEFA3, and TPPP3. Two of the features, RPS4Y1 and DDX3Y, are already Y chromosome-associated, and HLA-DRB5 and TPPP3 have been reported as being associated with GBM [
43,
44] with respect to prognosis and the epithelial–mesenchymal transition, respectively, albeit not in serum biospecimens as of yet. In contrast, DEFA3, a member of the defensin family (
Supplemental Table S1), has not been directly implicated in GBM or associated with sex differences specifically. Defensins, however, have been connected to cancer [
42,
45], and their identification in both serum (DEFB4A) and tissue (DEFA3) in association with GBM and biological sex could support previous data suggesting a relationship between innate immunity as a critical aspect of GBM propagation [
46] and its contribution to differential outcomes between men and women in GBM. Defensins captured in serum biospecimens thus merit additional research as a possible biomarker and a key to sex differences and differential responses to management. A connection between TPPP3, DEFA3, and HLA-DRB5, as evidenced by the clustering observed in the current analysis (
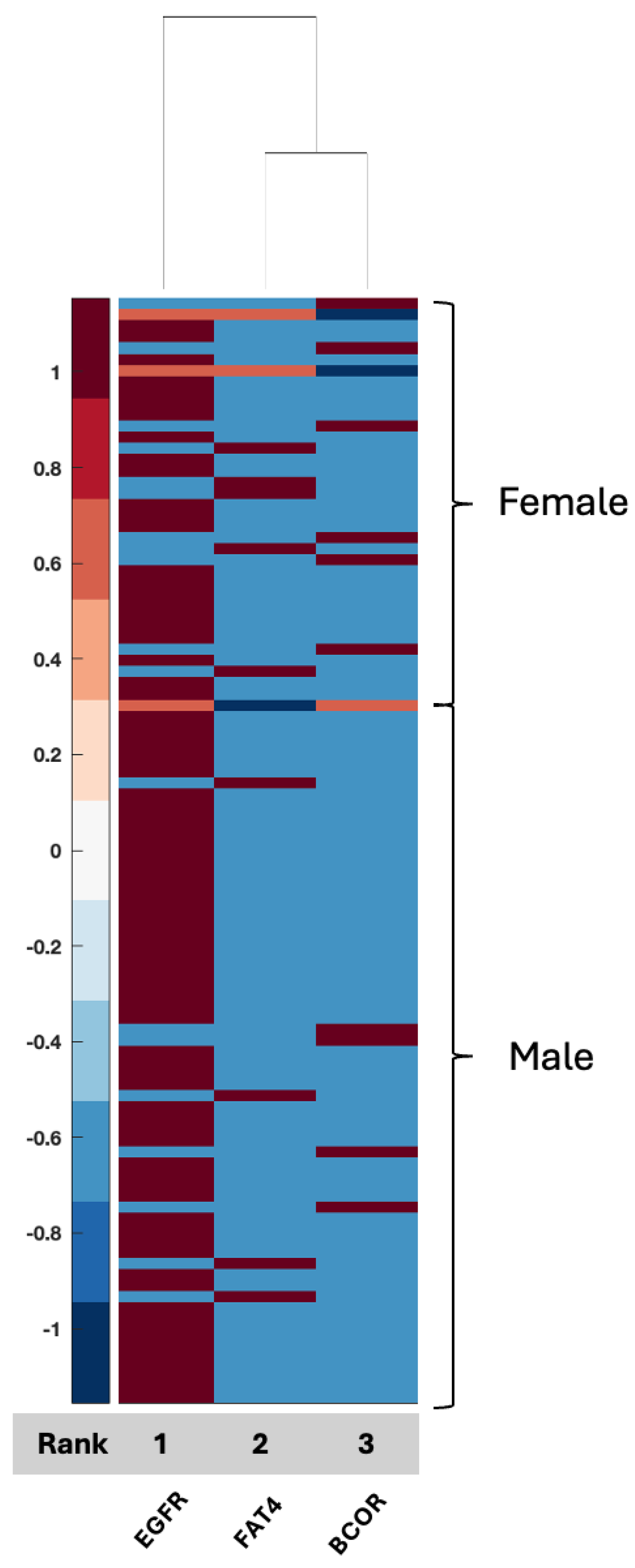
Figure 3), had not yet been described, and if it presents as a distinguishing signaling axis between men and women with GBM, this merits additional investigation as it links the epithelial–mesenchymal transition to tumor grade and prognosis and the innate immune system, with a clear link to biological sex in the GBM tissue proteome. We also observed limited overlap between proteins identified in serum and proteins identified in tissue, which may indicate that one biospecimen cannot serve as a validation of signals for the other, although far more data are needed in GBM using patient samples that have both serum and tissue for analysis.
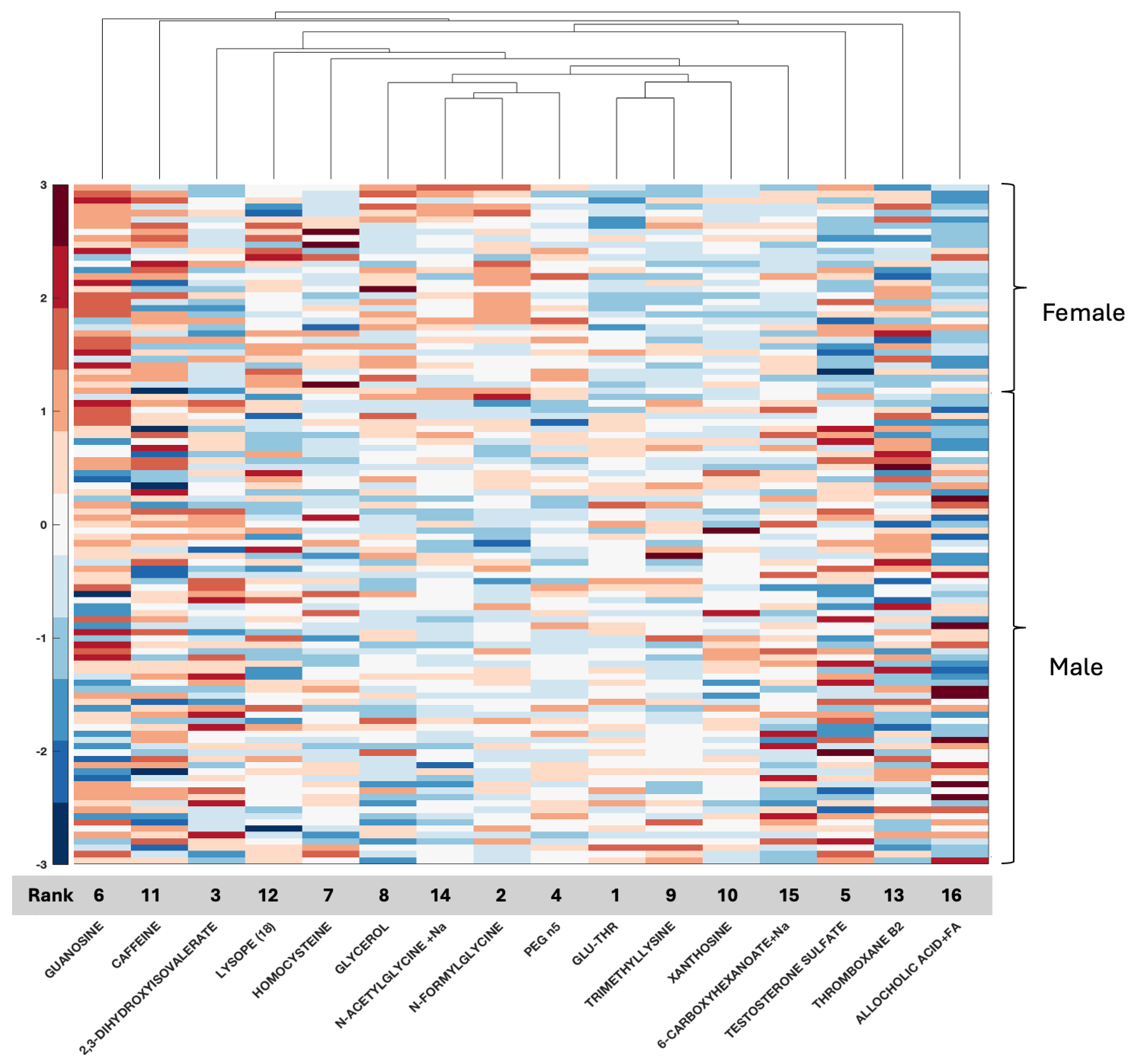
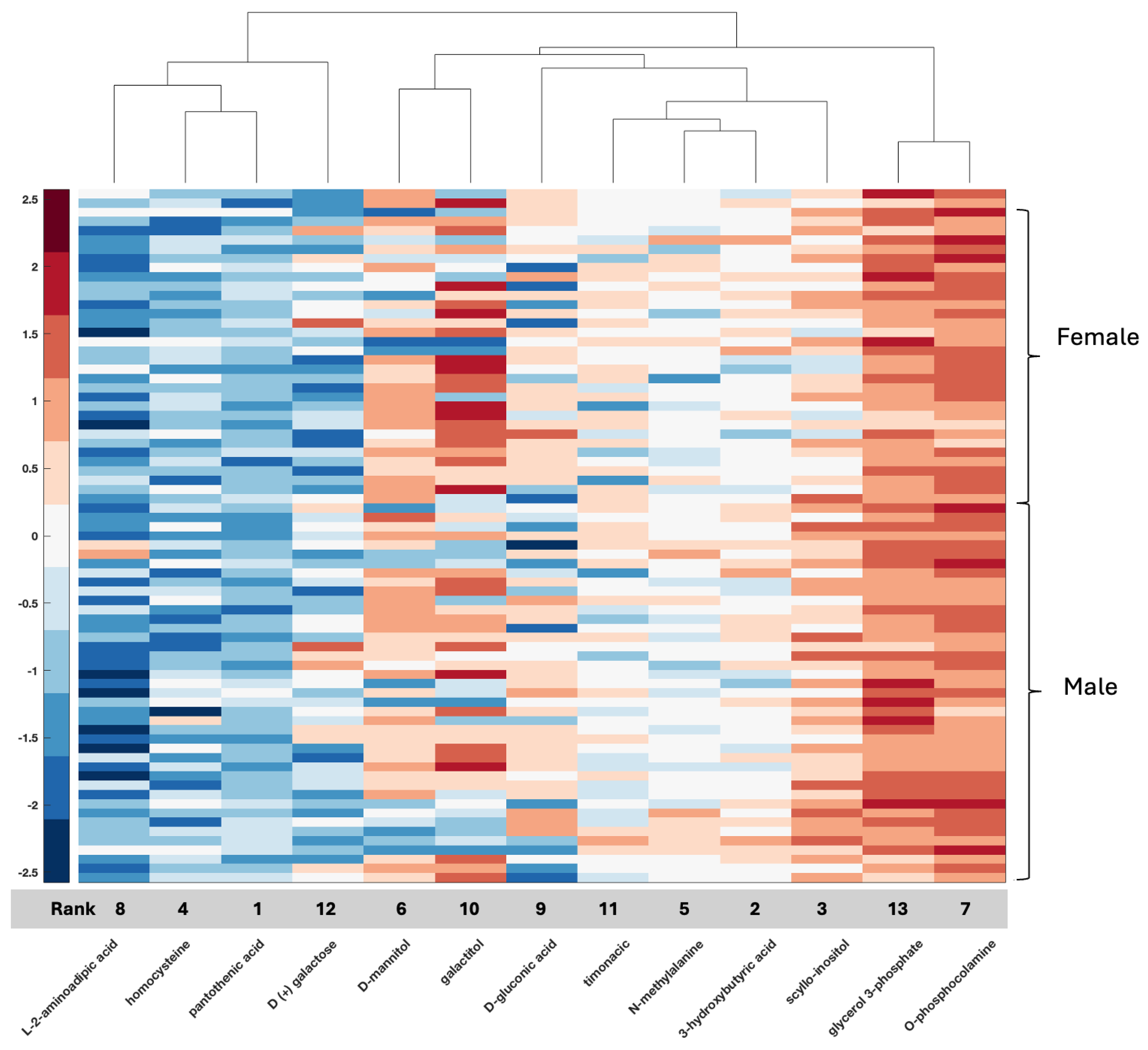
With respect to metabolomic data, the relationship between compounds in serum or tissue and biological sex is less well defined, although data are emerging. Compounds such as serum testosterone were identified in this study, again providing validation of the biospecimen of origin given clinical information with known clinical applicability for measurement in serum and supporting the accuracy of the feature selection method. Additional compounds such as GLU-THR (glutamine threonine), homocysteine, glycerol, xanthosine, and thromboxane B2 have also been associated with glioma and documented as differentially measured between men and women, while others such as allocholic acid are evolving (
Supplemental Table S7) [
1,
4,
47,
48,
49,
50,
51]. These compounds, as well as several others, relate to amino acid synthesis [
52] and purine metabolism, as well as fatty acid metabolism [
53]. Two metabolomic features shared between serum and tissue were homocysteine and pantothenic acid; however, overall, fewer compounds for analysis were present in CPTAC than in our dataset, with only 59 features to work with. Overall, both purine metabolism and fatty acid metabolism have been reported as highly significant in GBM, with emerging relationships to tumor resistance [
53,
54,
55], and the current analysis supports that, indeed, several compounds may be captured in the serum and tumor tissue of patients with GBM, leading to potential mechanistic relationships for further research. Blood homocysteine levels are related to primary brain tumors [
56] and homocysteine is associated with biological sex differences [
47]. In the present study, homocysteine ranked fourth in the CPTAC tissue data (ACC 80%) and seventh in the local serum (ACC 92%). Pantothenic acid or vitamin B5 has been associated with GBM, and in a recent study, it was one of several metabolites differentially expressed between core and edge tumor specimens in association with MGMT status [
57]. In the present study, it was ranked feature number 1 in tissue (CPTAC); however, it was ranked far lower in serum, not even reaching the 16 top identified features in serum. Since it emerged as a feature in both datasets, it warrants further study regarding its relationship with biological sex differences with respect to GBM.
EGFR, FAT4, and BCOR were the three features associated with 64% ACC using the TCGA-UCI glioma grading set aimed at tumor grading [
58]. The current analysis indicated that EGFR expression is differentially altered between men and women, with EGFR expression lower overall in women compared to men, which has been shown in other studies involving TCGA and other data [
23]. FAT4 and BCOR, however, were overall higher in women as compared to men. Neither has been explored in their relationship with biological sex differences; however, FAT4 acts as a tumor-suppressor gene with typically lower expression in tumors [
59], while BCOR (BCL6 corepressor gene) drives oncogenic transformation in neural cells [
60]. BCOR has also been linked to glioma via BCOR-altered gliomas, a newly identified glioma with a potential response to fluorinated pyrimidines [
61], indicating that glioma formation in women may occur via different pathways in women and men, resulting in differential tumor responses to chemotherapy. The relationship between the expression of these markers and sex differences is not well explored and the data are inconclusive. The current study illustrates potential relationships between well-known glioma drivers in males compared to females in GBM, some of which are better understood and studied; for example, the relationship between EGFR, BCOR, and FAT4 and connections to CD44 but with potential signaling via sex-related proteins such as HCG, as well as potential metabolic programming that may drive tumor behavior, e.g., via KLK3 (BPSA) and IGFBP1, which can connect to BCOR (
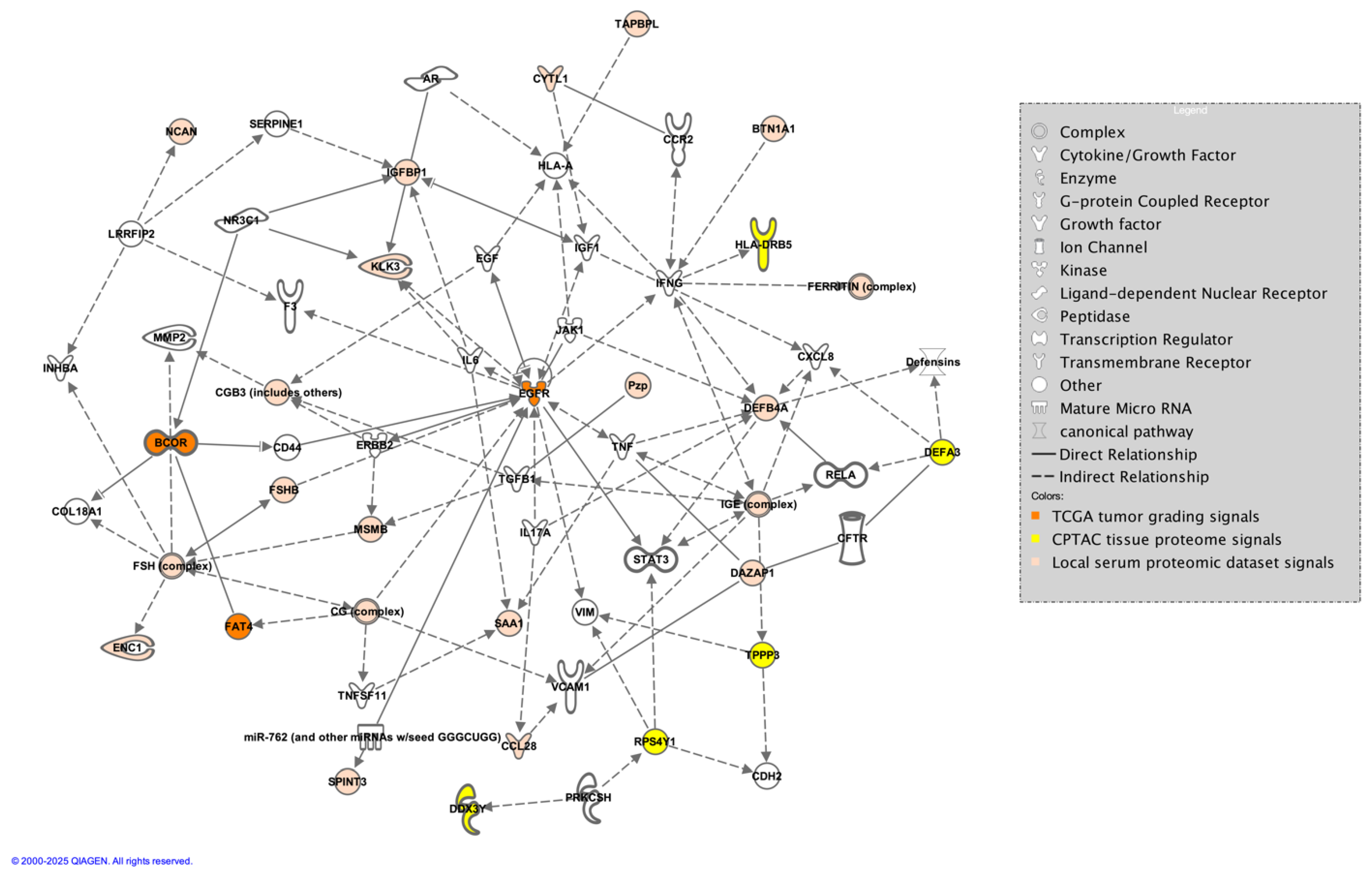
Figure 7).
Figure 6.
Proteins identified in serum have respective known relationships with biological sex and glioma, as well as novel signals with no direct or evolving relationships.
Figure 6.
Proteins identified in serum have respective known relationships with biological sex and glioma, as well as novel signals with no direct or evolving relationships.
Figure 7.
Direct and indirect connections between proteins identified in the TCGA tumor grading dataset (orange), the CPTAC tissue proteomic dataset (yellow), and the local serum dataset (tan) generated by the authors in IPA [
62].
Figure 7.
Direct and indirect connections between proteins identified in the TCGA tumor grading dataset (orange), the CPTAC tissue proteomic dataset (yellow), and the local serum dataset (tan) generated by the authors in IPA [
62].
Our analyses can suggest that integrating LASSO with mRMR offers notable benefits compared to existing hybrid methods, such as enhanced classification performance or robustness. This highlights the effectiveness of our hybrid feature selection and weighting approach in managing high-dimensional data efficiently. Future studies will focus on refining and validating the proposed method using larger datasets and alternative hybrid techniques.
Our analysis focused on traditional machine learning methods, including SVM, KNN, Logistic Regression, AdaBoost, and Random Forest. Although modern techniques, such as deep neural networks, autoencoders, and transformer-based models, may offer benefits, these methods can introduce additional complexity and computational demands. Traditional approaches were chosen for their established utility in high-dimensional data with limited sample sizes, as well as their ease of interpretation. Future studies will explore integrating advanced techniques to compare their potential advantages with our current framework.
Some limitations of our study can be expressed as follows: The datasets used in our study do not include unaffected controls, which limits our ability to distinguish GBM-related features from those that may arise due to general variability unrelated to GBM. Future studies incorporating unaffected control data will be crucial for enhancing the specificity of identified features. While our feature selection analysis was aimed at identifying sex differences in GBM, it was conducted on combined datasets rather than separately for male and female groups. As another limitation of this study, we utilized two independent proteomic (or metabolomic) datasets to identify features associated with male/female GBM classification and feature selection. Although both datasets provided meaningful insights, the shared features between them were limited. This presented a challenge for implementing cross-dataset classification, which relies on consistent features across datasets for reliable evaluation. To address this, we concentrated on feature selection within each dataset, aiming to extract biologically relevant patterns unique to each one. Nonetheless, we recognize that cross-dataset classification could offer a more comprehensive assessment of the generalizability and robustness of our findings. Moving forward, future research can prioritize the inclusion of datasets with a higher degree of feature overlap or incorporate cross-dataset validation to reinforce our conclusions. Another limitation of this study is the lack of independent external validation, which is critical for assessing the generalizability of the model. The predictive model in this study was validated using stratified cross-validation as external validation was not possible due to the absence of datasets with the same specifications. This validation strategy ensures our results’ robustness within the available data’s constraints. Future work will aim to address this limitation by identifying or generating additional datasets to validate the findings externally.
Since a large number of classifier models, different datasets, five-fold stratified cross-validation technique, and different feature weighting values were tried and applied in this study, the implementation of the SHAP values in tree-based models or deep learning-based feature attribution methods would be associated with a significant increase in both time and processing power; it is, however, our intention to test these methods in future studies. While these advanced methods are recognized for both their capacity to capture complex nonlinear interactions and offer robust frameworks for feature selection, LASSO and mRMR were chosen for their well-established interpretability, computational efficiency, and suitability for high-dimensional omics datasets. Future work will address this limitation by comparing our approach with additional state-of-the-art techniques.



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}