
Transcriptome Insights into Protective Mechanisms of Ferroptosis Inhibition in Aortic Dissection

 ,
,
Abstract
1. Introduction
2. Results
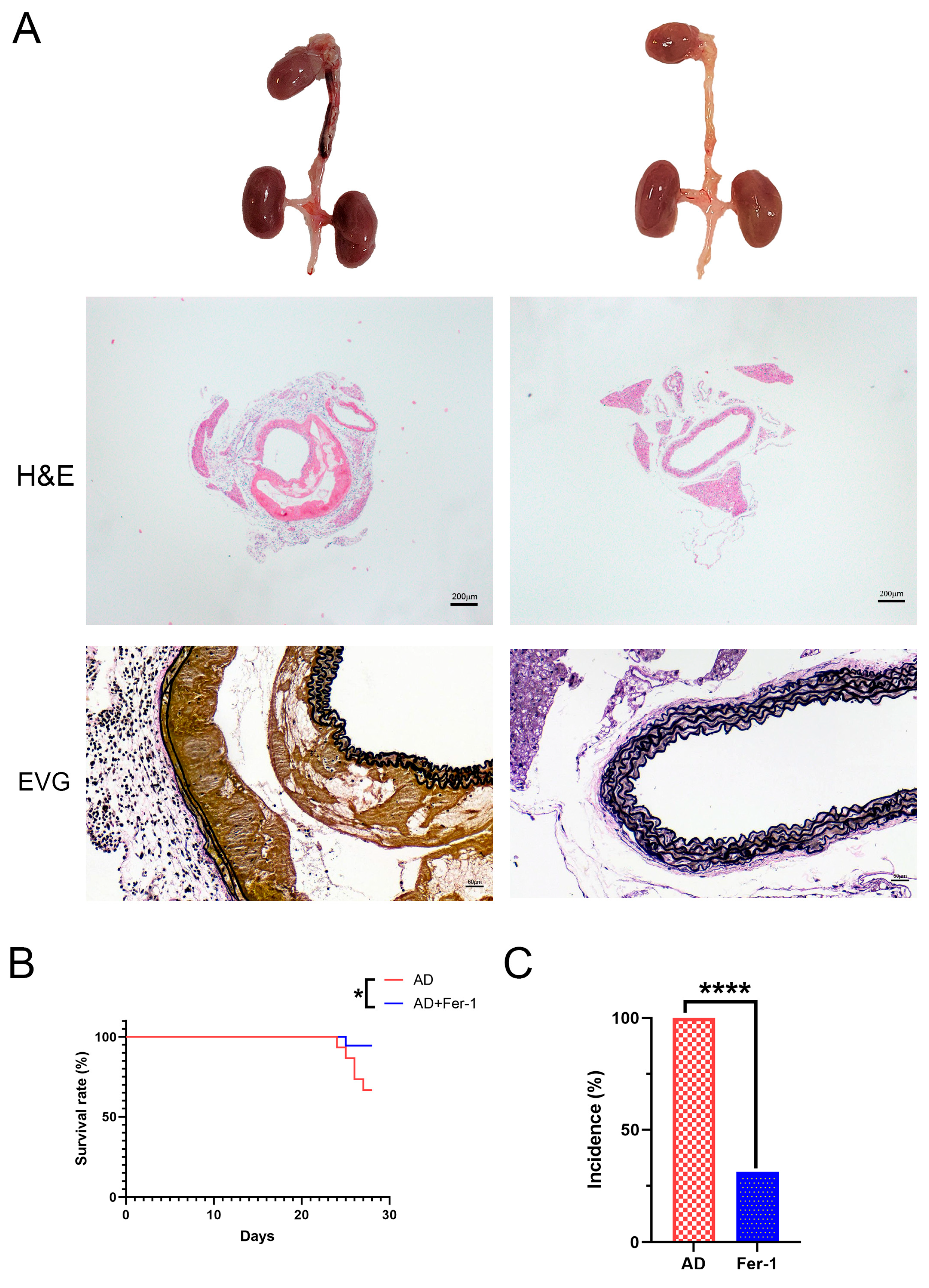
2.1. Treatment with Fer-1 Attenuated AD Development in Mice
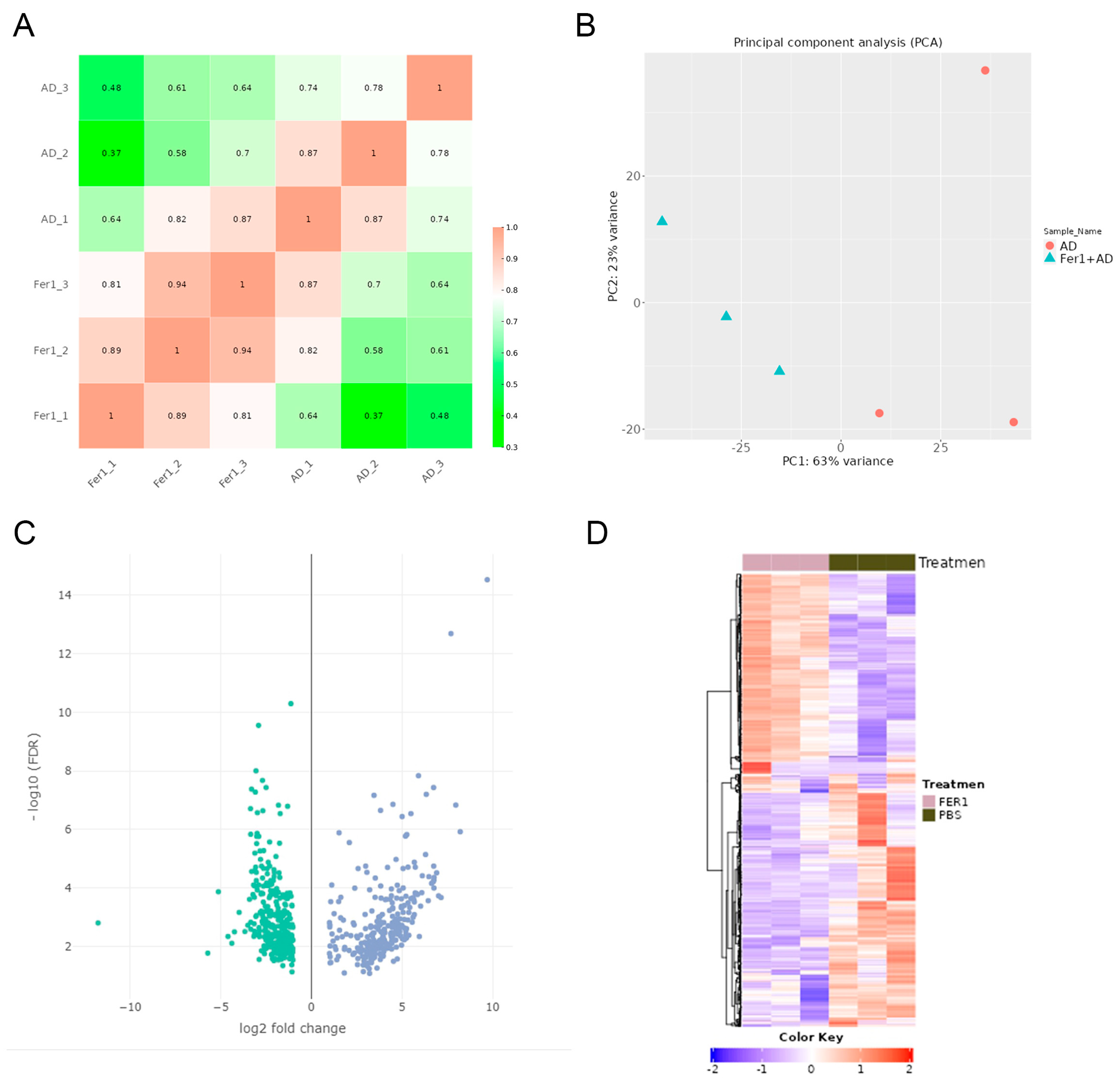
2.2. Identification of Differentially Expressed Genes (DEGs) After Fer-1 Treatment in AD Mice
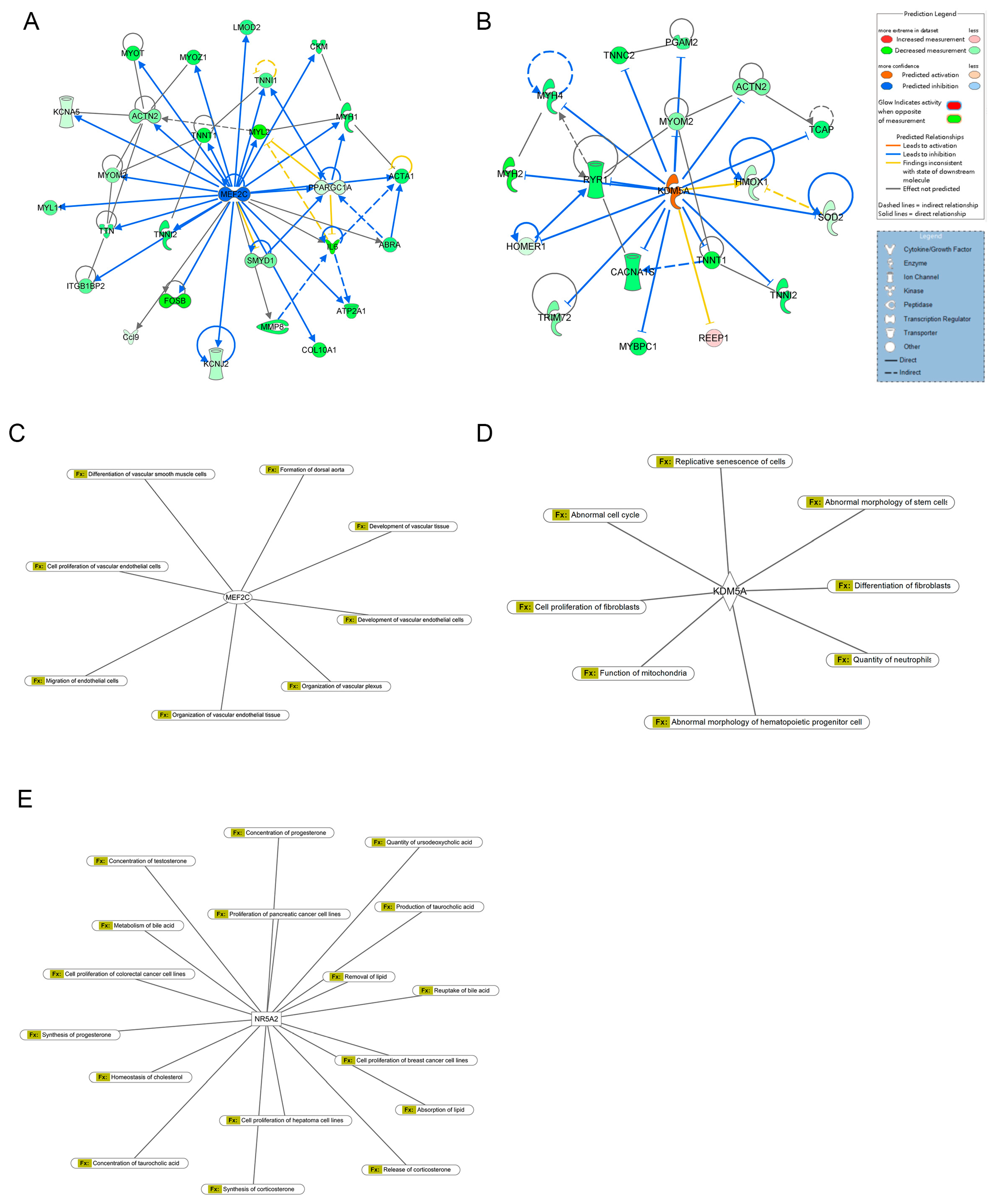
2.3. Upstream Regulators of Differential Gene Expression
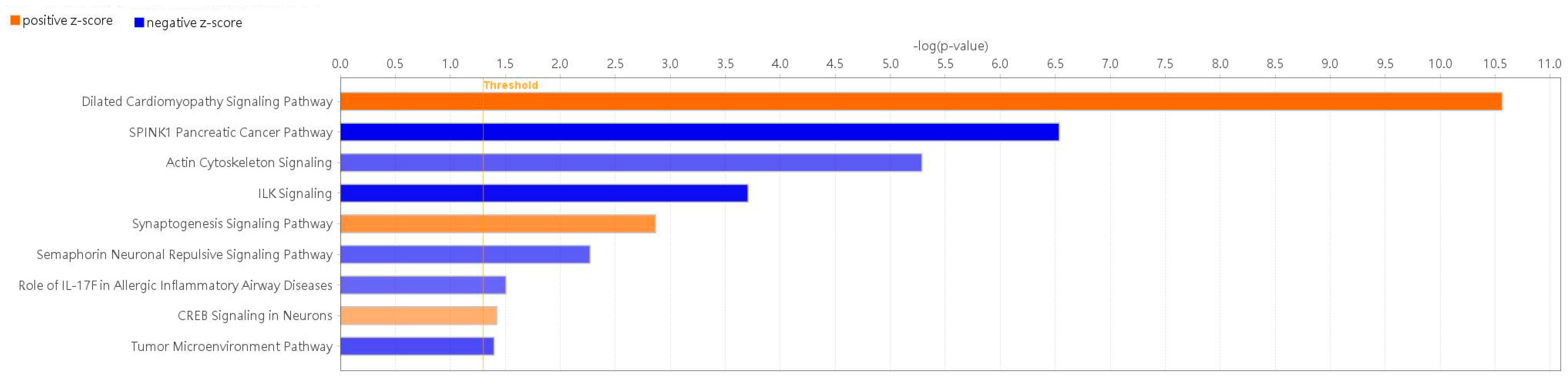
2.4. Functional Prediction and Enriched Canonical Pathways
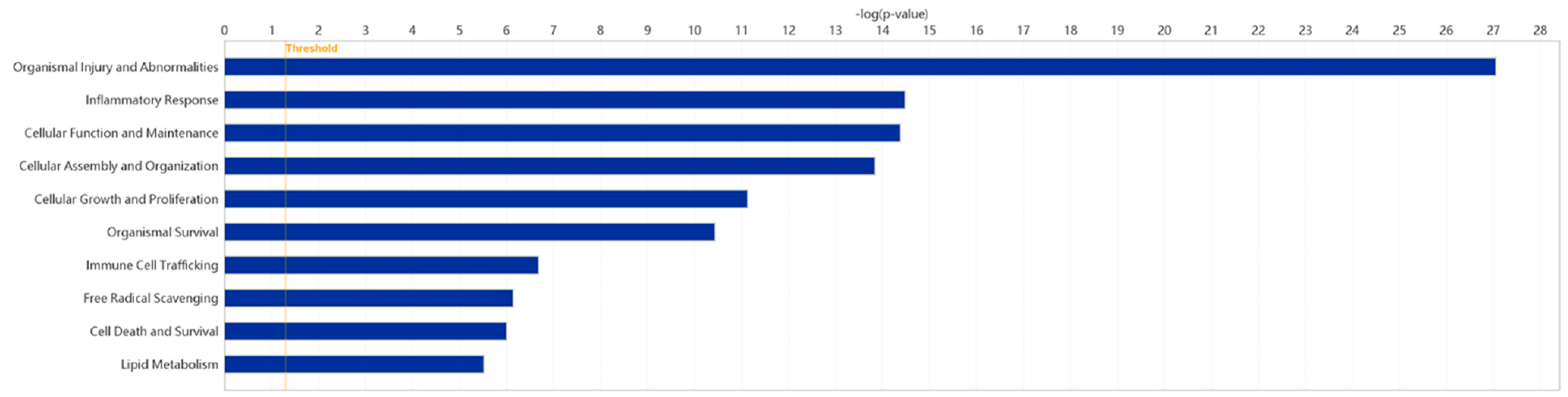
2.5. Disease and Biofunction Analysis
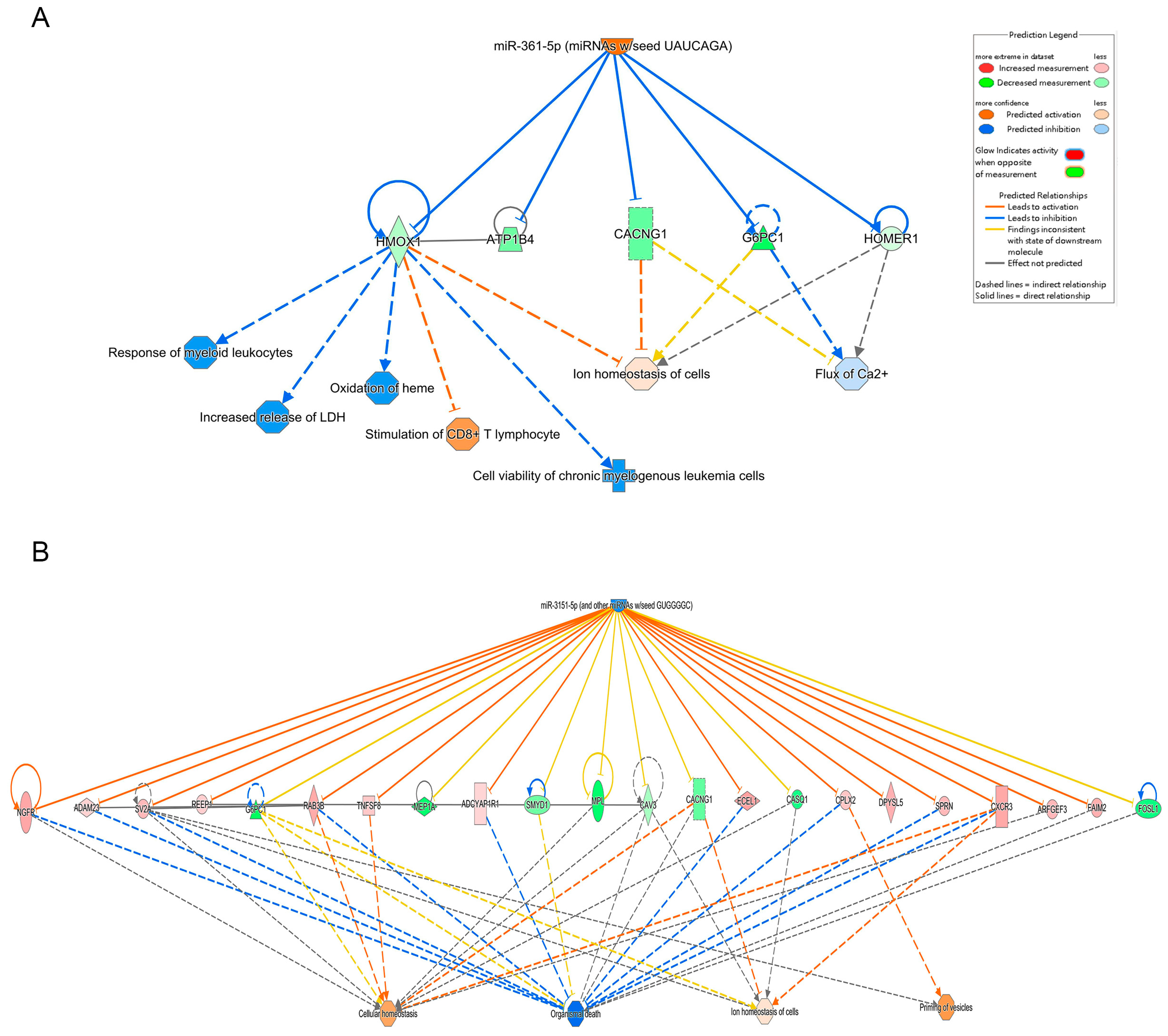
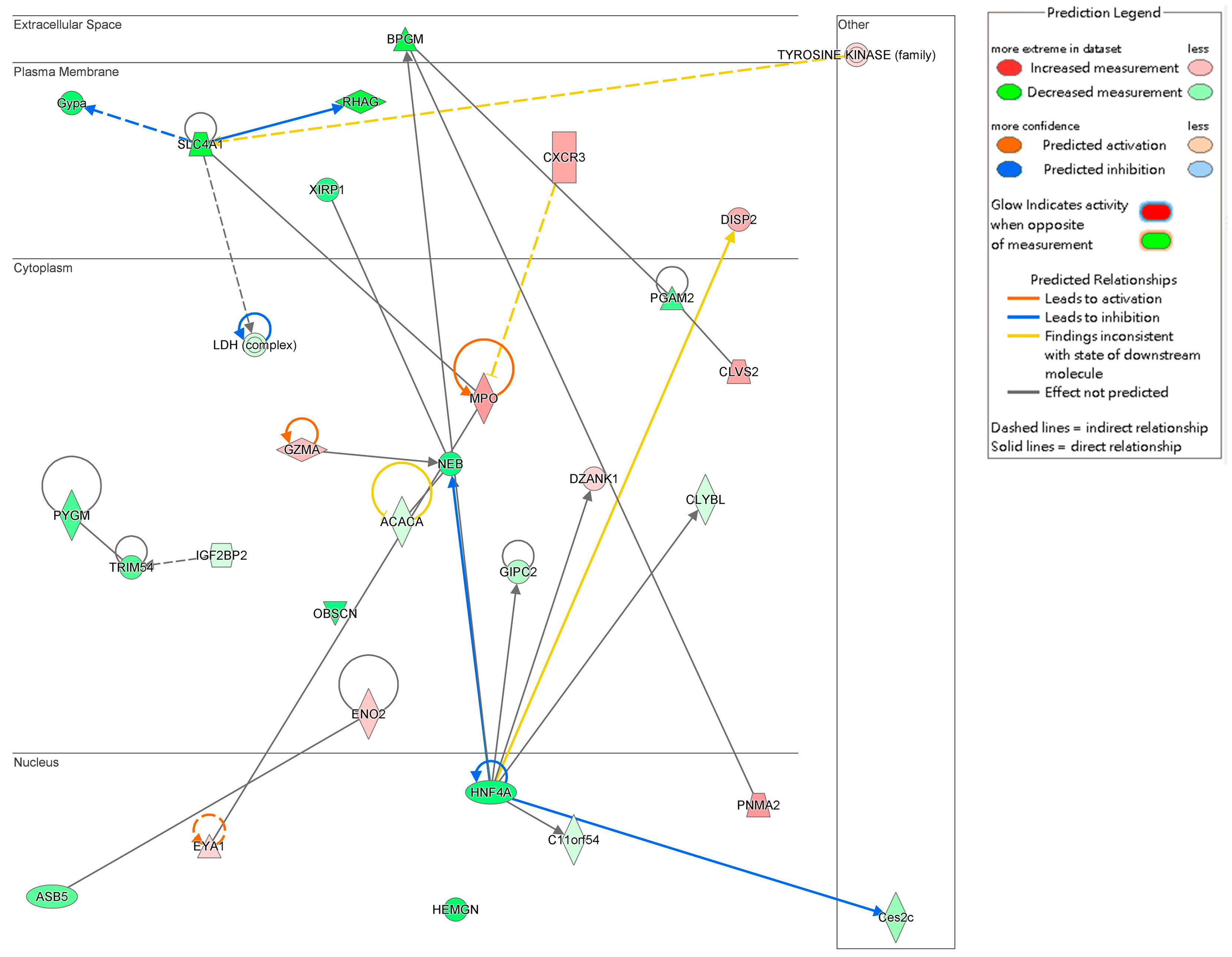
2.6. Interaction Network Analysis
3. Discussion
4. Materials and Methods
4.1. Model Establishment and Interventions
4.2. Histological Analysis
4.3. RNA Sequencing
4.4. Bioinformatics and Network Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Aortic Dissection |
| Fer-1 | Ferrostatin-1 |
| SMC | Smooth Muscle Cell |
| ECM | Extracellular Matrix |
| BAPN | β-aminopropionitrile |
| Ang-II | Angiotensin II |
| SPF | Specific Pathogen-free |
| IPA | Ingenuity Pathway Analysis |
| PCA | Principal Component Analysis |
| DEGs | Differentially Expressed Genes |
| FC | Fold Change |
References
- Nienaber, C.A.; Clough, R.E.; Sakalihasan, N.; Suzuki, T.; Gibbs, R.; Mussa, F.; Jenkins, M.P.; Thompson, M.M.; Evangelista, A.; Yeh, J.S.; et al. Aortic dissection. Nat. Rev. Dis. Primers 2016, 2, 16053. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.; Isselbacher, E.M.; Bossone, E.; Gleason, T.G.; Eusanio, M.D.; Sechtem, U.; Ehrlich, M.P.; Trimarchi, S.; Braverman, A.C.; Myrmel, T.; et al. Insights from the International Registry of Acute Aortic Dissection: A 20-Year Experience of Collaborative Clinical Research. Circulation 2018, 137, 1846–1860. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; LeMaire, S.A.; Webb, N.R.; Cassis, L.A.; Daugherty, A.; Lu, H.S. Aortic Aneurysms and Dissections Series. Arter. Thromb. Vasc. Biol. 2020, 40, e37–e46. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.D.; Narracott, A.; von Tengg-Kobligk, H.; Silva Soto, D.A.; Hsiao, S.; Lungu, A.; Evans, P.; Bressloff, N.W.; Lawford, P.V.; Hose, D.R.; et al. Computational fluid dynamics modelling in cardiovascular medicine. Heart 2016, 102, 18–28. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, Y.; Fan, J. Stanford type A aortic dissection presenting as acute inferior myocardial infarction. Br. J. Hosp. Med. 2022, 83, 1–3. [Google Scholar] [CrossRef]
- Han, L.; Dai, L.; Zhao, Y.F.; Li, H.Y.; Liu, O.; Lan, F.; Jiang, W.J.; Zhang, H.J. CD40L promotes development of acute aortic dissection via induction of inflammation and impairment of endothelial cell function. Aging 2018, 10, 371–385. [Google Scholar] [CrossRef]
- Kusters, P.J.H.; Seijkens, T.T.P.; Beckers, L.; Lievens, D.; Winkels, H.; de Waard, V.; Duijvestijn, A.; Lindquist Liljeqvist, M.; Roy, J.; Daugherty, A.; et al. CD40L Deficiency Protects Against Aneurysm Formation. Arter. Thromb. Vasc. Biol. 2018, 38, 1076–1085. [Google Scholar] [CrossRef]
- He, R.; Guo, D.C.; Estrera, A.L.; Safi, H.J.; Huynh, T.T.; Yin, Z.; Cao, S.N.; Lin, J.; Kurian, T.; Buja, L.M.; et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J. Thorac. Cardiovasc. Surg. 2006, 131, 671–678. [Google Scholar] [CrossRef]
- Li, N.; Yi, X.; He, Y.; Huo, B.; Chen, Y.; Zhang, Z.; Wang, Q.; Li, Y.; Zhong, X.; Li, R.; et al. Targeting Ferroptosis as a Novel Approach to Alleviate Aortic Dissection. Int. J. Biol. Sci. 2022, 18, 4118–4134. [Google Scholar] [CrossRef]
- Qi, Z.; Wang, Q.G.; Huang, M.X.; Zeng, Y.F.; Li, J.Y.; Duan, Z.C.; Tan, L.; Tang, H. Dual functions of silibinin in attenuating aortic dissection via regulating iron homeostasis and endoplasmic reticulum stress against ferroptosis. Cell Death Dis. 2024, 15, 900. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Yi, X.; Zhu, X.H.; Wei, X.; Jiang, D.S. Regulated cell death in myocardial ischemia-reperfusion injury. Trends Endocrinol. Metab. 2024, 35, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Tian, X.M.; Li, W.; Hao, L.Y. Ferroptosis in cardiac hypertrophy and heart failure. Biomed. Pharmacother. 2023, 168, 115765. [Google Scholar] [CrossRef]
- Chen, Y.; Yi, X.; Huo, B.; He, Y.; Guo, X.; Zhang, Z.; Zhong, X.; Feng, X.; Fang, Z.M.; Zhu, X.H.; et al. BRD4770 functions as a novel ferroptosis inhibitor to protect against aortic dissection. Pharmacol. Res. 2022, 177, 106122. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef]
- Zhang, S.; Bei, Y.; Huang, Y.; Huang, Y.; Hou, L.; Zheng, X.L.; Xu, Y.; Wu, S.; Dai, X. Induction of ferroptosis promotes vascular smooth muscle cell phenotypic switching and aggravates neointimal hyperplasia in mice. Mol. Med. 2022, 28, 121. [Google Scholar] [CrossRef]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef]
- Song, W.; Chen, Y.; Qin, L.; Xu, X.; Sun, Y.; Zhong, M.; Lu, Y.; Hu, K.; Wei, L.; Chen, J. Oxidative stress drives vascular smooth muscle cell damage in acute Stanford type A aortic dissection through HIF-1alpha/HO-1 mediated ferroptosis. Heliyon 2023, 9, e22857. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vuckovic, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Hassannia, B.; Vanden Berghe, T.; Augustyns, K. Beyond ferrostatin-1: A comprehensive review of ferroptosis inhibitors. Trends Pharmacol. Sci. 2023, 44, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Krebs, J.R.; Bellotti, P.; Valisno, J.A.C.; Su, G.; Sharma, S.; Kollareth, D.J.M.; Hartman, J.B.; Adithan, A.; Spinosa, M.; Kamat, M.; et al. Pharmacologic Inhibition of Ferroptosis Attenuates Experimental Abdominal Aortic Aneurysm Formation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lu, Y.W.; Lowery, A.M.; Sun, L.Y.; Singer, H.A.; Dai, G.; Adam, A.P.; Vincent, P.A.; Schwarz, J.J. Endothelial Myocyte Enhancer Factor 2c Inhibits Migration of Smooth Muscle Cells Through Fenestrations in the Internal Elastic Lamina. Arter. Thromb. Vasc. Biol. 2017, 37, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, Y.; Bao, M.; Kato, Y.; Han, J.; Eaton, J.W. Vascular smooth muscle cell proliferation requires both p38 and BMK1 MAP kinases. Arch. Biochem. Biophys. 2002, 400, 199–207. [Google Scholar] [CrossRef]
- Firulli, A.B.; Miano, J.M.; Bi, W.; Johnson, A.D.; Casscells, W.; Olson, E.N.; Schwarz, J.J. Myocyte enhancer binding factor-2 expression and activity in vascular smooth muscle cells. Association with the activated phenotype. Circ. Res. 1996, 78, 196–204. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kita, S.; Okuzaki, D.; Fujishima, Y.; Otsuki, M.; Kato, H.; Nishizawa, Y.; Miyashita, K.; Yokoyama, C.; Fukuhara, A.; et al. Favine/CCDC3 deficiency accelerated atherosclerosis and thrombus formation is associated with decreased MEF2C-KLF2 pathway. iScience 2022, 25, 105252. [Google Scholar] [CrossRef]
- Chen, Z.; Di, X.; Chen, H.; Song, S.; Chen, R.; Kou, L.; Chu, M. MEF2C mitigates coronary artery lesions in Kawasaki disease by enhancing endothelial barrier function through KLF2 regulation. Int. Immunopharmacol. 2025, 148, 114030. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, T.; Ma, L.; Liu, Z.; Xin, Y.; Jia, Z.; Chen, Y.; Li, C.; Sun, R. Prothrombotic effects of high uric acid in mice via activation of MEF2C-dependent NF-kappaB pathway by upregulating let-7c. Aging 2020, 12, 17976–17989. [Google Scholar] [CrossRef]
- Wang, R.M.; Zhang, Q.G.; Li, J.; Yang, L.C.; Yang, F.; Brann, D.W. The ERK5-MEF2C transcription factor pathway contributes to anti-apoptotic effect of cerebral ischemia preconditioning in the hippocampal CA1 region of rats. Brain Res. 2009, 1255, 32–41. [Google Scholar] [CrossRef]
- Xu, Z.; Yoshida, T.; Wu, L.; Maiti, D.; Cebotaru, L.; Duh, E.J. Transcription factor MEF2C suppresses endothelial cell inflammation via regulation of NF-kappaB and KLF2. J. Cell Physiol. 2015, 230, 1310–1320. [Google Scholar] [CrossRef]
- Wang, S.; Wu, Z.; Bu, X.; Peng, X.; Zhou, Q.; Song, W.; Gao, W.; Wang, W.; Xia, Z. MEF2C Alleviates Postoperative Cognitive Dysfunction by Repressing Ferroptosis. CNS Neurosci. Ther. 2024, 30, e70066. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Jablonowski, C.; Cheng, P.H.; AlTahan, A.; Li, C.; Wang, Y.; Palmer, L.; Lan, C.; Sun, B.; Abu-Zaid, A.; et al. KDM5A Regulates a Translational Program that Controls p53 Protein Expression. iScience 2018, 9, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lin, J.; Zhou, W.; Moses, R.; Dai, Z.; Kossenkov, A.V.; Drapkin, R.; Bitler, B.G.; Karakashev, S.; Zhang, R. KDM5A Inhibits Antitumor Immune Responses Through Downregulation of the Antigen-Presentation Pathway in Ovarian Cancer. Cancer Immunol. Res. 2022, 10, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.A.; Naya, F.J. The Function of the MEF2 Family of Transcription Factors in Cardiac Development, Cardiogenomics, and Direct Reprogramming. J. Cardiovasc. Dev. Dis. 2016, 3, 26. [Google Scholar] [CrossRef]
- Stein, S.; Schoonjans, K. Molecular basis for the regulation of the nuclear receptor LRH-1. Curr. Opin. Cell Biol. 2015, 33, 26–34. [Google Scholar] [CrossRef]
- Mueller, M.; Cima, I.; Noti, M.; Fuhrer, A.; Jakob, S.; Dubuquoy, L.; Schoonjans, K.; Brunner, T. The nuclear receptor LRH-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J. Exp. Med. 2006, 203, 2057–2062. [Google Scholar] [CrossRef]
- Coste, A.; Dubuquoy, L.; Barnouin, R.; Annicotte, J.S.; Magnier, B.; Notti, M.; Corazza, N.; Antal, M.C.; Metzger, D.; Desreumaux, P.; et al. LRH-1-mediated glucocorticoid synthesis in enterocytes protects against inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2007, 104, 13098–13103. [Google Scholar] [CrossRef]
- Yang, X.; Song, Y.; Sun, Y.; Wang, M.; Xiang, Y. Down-regulation of miR-361-5p promotes the viability, migration and tube formation of endothelial progenitor cells via targeting FGF1. Biosci. Rep. 2020, 40, BSR20200557. [Google Scholar] [CrossRef]
- Zhang, W.; Chang, G.; Cao, L.; Ding, G. Dysregulation of serum miR-361-5p serves as a biomarker to predict disease onset and short-term prognosis in acute coronary syndrome patients. BMC Cardiovasc. Disord. 2021, 21, 74. [Google Scholar] [CrossRef]
- Wang, M.; Li, C.; Zhang, Y.; Zhou, X.; Liu, Y.; Lu, C. LncRNA MEG3-derived miR-361-5p regulate vascular smooth muscle cells proliferation and apoptosis by targeting ABCA1. Am. J. Transl. Res. 2019, 11, 3600–3609. [Google Scholar]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, Q.; Wang, Y.; Jin, M.; Tao, Z.; Wan, J. Identification of HMOX1 as a Critical Ferroptosis-Related Gene in Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 833642. [Google Scholar] [CrossRef] [PubMed]
- Khoukaz, H.B.; Vadali, M.; Schoenherr, A.; Ramirez-Perez, F.I.; Morales-Quinones, M.; Sun, Z.; Fujie, S.; Foote, C.A.; Lyu, Z.; Zeng, S.; et al. PAI-1 Regulates the Cytoskeleton and Intrinsic Stiffness of Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2024, 44, 2191–2203. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wu, C. ILK: A pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr. Opin. Cell Biol. 2012, 24, 607–613. [Google Scholar] [CrossRef]
- Imai, Y.; Clemmons, D.R. Roles of phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways in stimulation of vascular smooth muscle cell migration and deoxyriboncleic acid synthesis by insulin-like growth factor-I. Endocrinology 1999, 140, 4228–4235. [Google Scholar] [CrossRef]
- Troussard, A.A.; Costello, P.; Yoganathan, T.N.; Kumagai, S.; Roskelley, C.D.; Dedhar, S. The integrin linked kinase (ILK) induces an invasive phenotype via AP-1 transcription factor-dependent upregulation of matrix metalloproteinase 9 (MMP-9). Oncogene 2000, 19, 5444–5452. [Google Scholar] [CrossRef]
- Alique, M.; Civantos, E.; Sanchez-Lopez, E.; Lavoz, C.; Rayego-Mateos, S.; Rodrigues-Diez, R.; Garcia-Redondo, A.B.; Egido, J.; Ortiz, A.; Rodriguez-Puyol, D.; et al. Integrin-linked kinase plays a key role in the regulation of angiotensin II-induced renal inflammation. Clin. Sci. 2014, 127, 19–31. [Google Scholar] [CrossRef]
- Rui, H.; Zhao, F.; Yuhua, L.; Hong, J. Suppression of SMOC2 alleviates myocardial fibrosis via the ILK/p38 pathway. Front. Cardiovasc. Med. 2022, 9, 951704. [Google Scholar] [CrossRef]
- Ju, X.; Ijaz, T.; Sun, H.; Ray, S.; Lejeune, W.; Lee, C.; Recinos, A., 3rd; Guo, D.C.; Milewicz, D.M.; Tilton, R.G.; et al. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arter. Thromb. Vasc. Biol. 2013, 33, 1612–1621. [Google Scholar] [CrossRef]
- Nishida, N.; Aoki, H.; Ohno-Urabe, S.; Nishihara, M.; Furusho, A.; Hirakata, S.; Hayashi, M.; Ito, S.; Yamada, H.; Hirata, Y.; et al. High Salt Intake Worsens Aortic Dissection in Mice: Involvement of IL (Interleukin)-17A-Dependent ECM (Extracellular Matrix) Metabolism. Arter. Thromb. Vasc. Biol. 2020, 40, 189–205. [Google Scholar] [CrossRef]
- Chang, S.L.; Hsiao, Y.W.; Tsai, Y.N.; Lin, S.F.; Liu, S.H.; Lin, Y.J.; Lo, L.W.; Chung, F.P.; Chao, T.F.; Hu, Y.F.; et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J. Mol. Cell Cardiol. 2018, 122, 69–79. [Google Scholar] [CrossRef]
- Mora-Ruiz, M.D.; Blanco-Favela, F.; Chavez Rueda, A.K.; Legorreta-Haquet, M.V.; Chavez-Sanchez, L. Role of interleukin-17 in acute myocardial infarction. Mol. Immunol. 2019, 107, 71–78. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fer-1/PBS | |||||||
|---|---|---|---|---|---|---|---|
| Symbol | Entrez Gene Name | Function | Fold Change | Symbol | Entrez Gene Name | Function | Fold Change |
| Ctrl | Chymotrypsin-like | Hydrolase, Protease, Serine protease | 6.160 | Bpifa1 | BPI fold containing family A member 1 | Antibiotic, Antimicrobial | −9.438 |
| Pnlip | Pancreatic lipase | Hydrolase | 5.944 | Myl2 | Myosin light chain 2 | Motor protein, Muscle protein, Myosin | −8.398 |
| Rnase1 | Ribonuclease, RNase A family, 1 | Endonuclease, Hydrolase, Lyase, Nuclease | 5.754 | Myh7 | Myosin heavy chain 7 | Actin-binding, Calmodulin-binding, Motor protein, Muscle protein, Myosin | −8.184 |
| Ctrb1 | Chymo-trypsinogen B1 | Hydrolase, Protease, Serine protease | 5.483 | Scgb1a1 | Secretoglobin family 1A member 1 | Phospholipase A2 inhibitor | −7.8139 |
| Try4 | Serine protease 3 | Hydrolase, Protease, Serine protease | 5.395 | Cxcl2 | C-X-C motif chemokine ligand 2 | Cytokine | −7.309 |
| Cela2a | Chymotrypsin like elastase 2A | Hydrolase, Protease, Serine protease | 5.379 | Csf3 | Colony stimulating factor 3 | Cytokine, Growth factor | −7.091 |
| Cel | Carboxyl ester lipase | Hydrolase, Serine esterase | 4.822 | Mstn | Myostatin | Cytokine, Growth factor, Heparin-binding | −6.867 |
| Cpa2 | Carboxypeptidase A2 | Carboxypeptidase, Hydrolase, Metalloprotease, Protease | 4.789 | Alas2 | 5′-aminolevulinate synthase 2 | Acyltransferase, Transferase | −6.860 |
| Cd40lg | CD40 ligand | Cytokine | 4.0546 | Il6 | Interleukin 6 | Cytokine, Growth factor | −6.843 |
| Klhl14 | Deleted in malignant brain tumors 1 | Developmental protein | 3.869 | Fosb | FosB proto-oncogene, AP-1 transcription factor subunit | DNA-binding | −6.702 |
| Upstream Regulator | Molecule Type | Predicted Activation State | Activation z-Score | p-Value of Overlap | Target Molecules in Dataset |
|---|---|---|---|---|---|
| MEF2C | Transcription regulator | Inhibited | −4.358 | 3.06 × 10−18 | ABRA, ACTA1, ACTN2, ATP2A1, Ccl9, CKM, COL10A1, CXCL2, CXCL6, FOSB, IL6, ITGB1BP2, KCNA5, KCNJ2, LMOD2, MMP8, MYH1, MYH7, MYL11, MYL2, MYOM2, MYOT, MYOZ1, PPARGC1A, SMYD1, TNNI1, TNNI2, TNNT1, TTN |
| LHX1 | Transcription regulator | Inhibited | −4.199 | 4.66 × 10−10 | ACSM2A, ALDOB, CLDN2, Cyp2j5, FUT9, GSTA5, HNF4A, Kap, Keg1, LRP2, LRRC19, MEP1A, MMP8, PAH, SLC34A1, SLC47A1, SLC5A8, Sult1d1, UMOD |
| GATA1 | Transcription regulator | Inhibited | −3.8 | 6.01 × 10−7 | ALAS2, ALOX12, CCL5, CXCR3, EPB42, FECH, FOSB, GATA3, GP1BA, GP6, GP9, Gypa, HBA1/HBA2, HBB, Hbb-b1, Hbb-b2, HDAC11, ITGAX, MPL, NEFH, NFE2, SLC4A1, SNCA, SPTA1, TUBB1 |
| ERK (family) | Group | Inhibited | −3.774 | 0.00357 | ARG2, Ccl7, CSF2, CXCL2, CXCL6, DIO2, FOSL1, GDF15, HAS1, HMOX1, HOMER1, IL1B, IL6, LIF, SERPINB2 |
| F2 | Peptidase | Inhibited | −3.741 | 0.00927 | Ccl9, CXCL3, CYP2B6, DHRS9, EPB41, FOSB, FOSL1, GP6, HMOX1, IL1B, IL6, OSM, SELE, SOD2, TFPI2 |
| Hydrogen peroxide | Chemical–endogenous mammalian | Inhibited | −3.721 | 0.0384 | AKR1C1/AKR1C2, CKMT2, CSF3, CXCL3, CXCL6, CXCR3, FOSL1, GDF15, GSTA5, HDC, HMOX1, IL1B, IL6, MB, MIOX, MMP8, NOS1, PAH, PPARGC1A, SOD2, SQLE, TCF3 |
| TGFB1 | Growth factor | Inhibited | −3.686 | 8.56 × 10−5 | ACTA1, ADAMTS4, AKR1C1/AKR1C2, ALDOB, ALOX12, ALOX15, AMY2B, BPIFB1, CCL5, Ccl7, CD40, CDHR1, Ces2c, CHRNB4, CKM, COL10A1, CRHR2, CRMP1, CSF2, CXCL2, CXCL3, CXCR3, DISP2, EGF, ENO1, ENO2, F5, FOSB, FOSL1, G6PC1, GATA3, GDF15, GPR158, GRIA1, GSTA5, GZMA, H19, HAS1, HMOX1, HNF4A, IFI16, IL1B, IL6, ITGAX, Kap, KCNQ3, KNG1, KRT14, KRT5, KRT8, L1CAM, LDHA, LIF, MAOA, MID1, MSTN, MUC5B, Mx1, MYH7, MYL11, MYL3, MYOCD, NOS1, OSM, PAK3, PGAM2, PNMT, PPARGC1A, PRSS3, RND1, RSAD2, SCGB1A1, SELE, SERPINA1, SLC4A1, SOD2, SPOCK1, SQLE, STAR, D4, TNFAIP6, TNFRSF14, TNFSF13B |
| Prostaglandin E2 | Chemical–endogenous mammalian | Inhibited | −3.527 | 0.000226 | ADAMTS4, ALOX15, CCL5, CD40, CSF3, CST7, CXCL3, Cxcl9, CXCR3, FOSB, FOSL1, GBP6, H2-M2, HDC, HMOX1, IL1B, IL6, MYOZ1, OSM, SCN9A, Tcstv4, TNFAIP6, TREM1 |
| AGT | Growth factor | Inhibited | −3.291 | 1.01 × 10−15 | ACTA1, ADAM23, ADAMTS4, ALOX12, ALOX15, AMY2B, ANXA1, ATP6V0A4, CAV3, CCKAR, CCL5, Ccl7, Ccl9, CEL, CELA2A, CLPS, COL10A1, CPA1, CPA2, CTRB2, CXCL2, CXCL3, CYP4A11, DBH, DCUN1D3, DIO2, DMBT1, FOSL1, GABRB3, GALNT13, GDF15, GSTA5, HMOX1, IFI16, IL1B, IL6, ITGAX, KCNJ2, L1CAM, LIF, MAP2, MATN2, MMP8, MYH7, NCALD, NOS1, NPY, PM20D1, PNLIP, PNLIPRP1, PNLIPRP2, PPARGC1A, PRSS2, PTGIS, REG1A, RNASE1, SCG2, SELE, SERPINB2, SIGLEC1, SLC12A3, SLC13A1, SLC6A2, SNCA, SOD2, SRGAP3, SYT4, TH, TNFRSF18, TTN, XIRP1, ZG16 |
| IL1B | Cytokine | Inhibited | −3.266 | 1.8 × 10−12 | ADAMTS4, ALOX12, ALOX15, ANXA1, BPIFB1, CALB1, CCKAR, CCL5, Ccl7, Ccl9, CD40, CD40LG, COL10A1, CSF2, CSF3, CXCL2, CXCL3, CXCL6, Cxcl9, CYP2B6, FOSB, FOSL1, G0S2, GATA3, GBP6, GDF15, GRIA1, H19, HAS1, HLA-E, HMOX1, HNF4A, HOMER1, IFI16, IL1B, IL6, INSRR, ISG20, ISL1, ITGB8, KRT5, LDHA, LIF, MMP8, MUC5B, NOS1, NPY, NTRK1, OSM, PCSK1, PPARGC1A, Ppbp, PSMB9, PTGIS, RAB3C, RNASE1, RSAD2, SAA1, SCGB1A1, SCN9A, SELE, SERPINB2, SMAD9, SNAP25, SNCA, SOD2, STMN2, TFPI2, TNFAIP6, TNFSF13B, TOB1, TREM1, UBE2L6 |
| NFAT5 | Transcription regulator | Inhibited | −3.18 | 9.68 × 10−5 | ACTN2, CSF2, CSF3, CXCL3, Cxcl9, IFI16, IL1B, IL6, LIF, Mx1, Oas1c (includes others), PLIN2, PTGIS, RSAD2 |
| REST | Transcription regulator | Inhibited | −3.177 | 8.68 × 10−12 | CHGB, FUT9, GRIA2, KCNQ2, KCNQ3, KIAA1549L, L1CAM, NCAM2, NEFH, Nefm, PCLO, PCSK1, PPARGC1A, SCG2, SCG3, SLC13A1, SLC7A14, SNAP25, SRRM4, STMN2, STMN3, SYT1, SYT4, TNNT1 |
| E. coli B4 lipopolysaccharide | Chemical toxicant | Inhibited | −3.174 | 1.06 × 10−6 | ANXA1, CCL5, CD40, CD40LG, Cd52, CSF2, CSF3, CXCL2, CXCL3, CXCL6, F5, FOSB, HMOX1, IFI16, IL1B, IL6, Mx1, RSAD2, SELE, SNCA, SOD2, Stfa2/Stfa2l1, TH, TNFSF8 |
| Cholesterol | Chemical–endogenous mammalian | Inhibited | −3.124 | 0.000116 | ACACA, Ccl7, CXCL2, CXCL3, DDC, GP1BA, GSTA5, IL1B, IL6, ITGAX, KCNA5, MAP2, MMP8, PLIN2, PPARGC1A, SAA1, SQLE, STARD4 |
| RELA | Transcription regulator | Inhibited | −3.002 | 2.39 × 10−5 | ALPK2, ANKRD2, BEX1, CCL5, CD40, CSF2, CXCL2, CXCL3, CXCL6, CYP2B6, DIO2, FOSB, GDF15, HMOX1, HNF4A, IL1B, IL6, ISG20, KRT8, Madcam1, Mx1, NEFH, PSMB9, REG3G, SAA1, SCN9A, SELE, SOD2, TFPI2, TREM1 |
| SP2509 | Chemical reagent | Activated | 3.939 | 0.0116 | CCL5, CNTN1, CNTNAP2, CRMP1, Cxcl9, IGF2BP2, ISL1, KIF5C, L1CAM, MAP2, NEFL, NRXN1, PAK3, SPOCK1, STMN2, SYT1 |
| SB203580 | Chemical drug | Activated | 3.604 | 8.52 × 10−8 | ADAMTS4, ANXA1, BPGM, CAV3, CCL5, Ccl7, CD40, CSF2, CST7, CXCL3, Cxcl3, EGF, ENO1, FOSB, G6PC1, HAS1, HMOX1, HPCA, IL1B, IL6, ISG20, MMP8, MSTN, MUC5B, MYL11, MYOCD, NEFH, PPARGC1A, RND1, SELE, SMYD1, SOD2, TFPI2, TNFAIP6, TNFSF8, TREM1 |
| KDM5A | Enzyme | Activated | 3.441 | 3.71 × 10−9 | ACTN2, Actn3, CACNA1S, HMOX1, HOMER1, MYBPC1, MYH2, MYH4, MYH7, MYOM2, PGAM2, REEP1, RYR1, SOD2, TCAP, TNNC2, TNNI2, TNNT1, TRIM72 |
| SP600125 | Chemical drug | Activated | 3.315 | 0.0023 | ADAMTS4, CCL5, CSF2, CSF3, CXCL2, CXCL3, CXCL6, FOSB, HAS1, HMOX1, HNF4A, IL1B, IL6, MUC5B, MYH7, Oas1c (includes others), SELE, SOD2, TNFRSF18 |
| FBXO32 | Enzyme | Activated | 3.24 | 5.81 × 10−8 | CSF2, CXCL2, CXCL3, Cxcl3, CXCL6, IL1B, IL6, MYOCD, PDIA2, PTGIS, REG3G, SERPINB2, SOD2, TFPI2 |
| N-acetyl-L-cysteine | Chemical drug | Activated | 3.185 | 0.00919 | ACTA1, CCL5, CXCL3, HDC, HMOX1, IL1B, IL6, MIOX, MYOCD, PPARGC1A, SELE, SNCA, SOD2, TRIM63 |
| IMMUNOGLOBULIN (complex) | Complex | Activated | 2.842 | 1.6 × 10−10 | ACACA, ACTA1, ALAS2, APOL2, BC147527, BPGM, CCL5, Ccl7, Ccl9, CD40, CD40LG, Cd52, CHST2, CSF2, CST7, CXCL2, CXCL3, CXCL6, Cxcl9, CXCR3, ENO1, ENO2, FAIM2, FOSB, G6PC1, GATA3, GZMA, HEMGN, HLA-E, HMOX1, IFI16, IL1B, IL6, ISG20, ITGAX, ITGB8, JCHAIN, MFSD2A, MPO, Mx1, MZB1, NEB, NOS1, OSM, PGAM2, PPARGC1A, PRDX5, PYGM, RHAG, RSAD2, SCG2, SELE, SLC25A37, SOD2, SQLE, TIGIT, TNFRSF18, TNFSF13B, TNFSF8, TTN, UGP2 |
| ARID1A | Transcription regulator | Activated | 2.813 | 0.00079 | ANXA1, CAV3, CKM, CXCL2, GATA3, IL1B, KRT14, KRT5, KRT8, SERPINA1 |
| ALPHA CATENIN (family) | Group | Activated | 2.81 | 0.0185 | ADAMTS4, CXCL2, CXCL6, IL1B, IL6, SAA1, SELE, TNFAIP6 |
| LTBR | Transmembrane receptor | Activated | 2.789 | 4.85 × 10−5 | CCL21, CSF2, GPM6B, IL6, Klra4 (includes others), Madcam1, SERPINA1, TNFSF13B |
| Diphenyleneiodonium | Chemical reagent | Activated | 2.759 | 0.000457 | CXCL2, CXCL3, GDF15, HMOX1, IL1B, IL6, SELE, SOD2 |
| PTF1A | Transcription regulator | Activated | 2.728 | 7.66 × 10−8 | AMY2B, CACNA2D3, CEL, CPA1, CPA2, CTRB2, GRIK2, ISL1, KLHL14, NPY, PRSS3, TFAP2B |
| Sb202190 | Chemical drug | Activated | 2.671 | 0.00141 | CAV3, Ccl9, CKM, CSF2, HAS1, HMOX1, IL1B, IL6, PPARGC1A, SCN9A, SELE |
| TP53 | Transcription regulator | Activated | 2.654 | 0.0468 | ADGRB3, ALDOC, ALOX15, ANXA1, APOBEC2, CCL5, CEL, CKM, CKMT2, CSF2, CSMD3, CXCL2, CXCL3, DHRS9, DNASE1, EGF, ENO2, ESRRB, F5, FOSL1, G0S2, G6PC1, GDF15, GPM6B, H19, HDC, HMOX1, IFI16, IGF2BP2, IL1B, IL6, ITGB1BP2, KCNJ2, KRT14, KRT8, LDHA, LIF, LRAT, MB, MRPS2, Mx1, MYL2, MYL3, MYOCD, NFE2, NOS1, NR2F1, NRAP, PAK3, PGAM2, PPARGC1A, PSMB9, SCN3B, SELE, SERPINB2, SLC5A8, SOD2, SQLE, SRGAP3, STARD4, TCAP, TFPI2, TMOD4, TNFRSF18, TOB1, TTN |
| NR5A2 | Ligand-dependent nuclear receptor | Activated | 2.607 | 6.61 × 10−5 | Ccl7, CEL, CELA3B, CPA1, CPA2, CTRL, Cxcl9, HNF4A, IL1B, IL6, PNLIP, SAA1, SYCN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, C.-C.; Chen, C.-Y.; Chuu, C.-P.; Huang, C.-Y.; Lu, C.-J.; Lu, H.-Y. Transcriptome Insights into Protective Mechanisms of Ferroptosis Inhibition in Aortic Dissection. Int. J. Mol. Sci. 2025, 26, 4338. https://doi.org/10.3390/ijms26094338
Shih C-C, Chen C-Y, Chuu C-P, Huang C-Y, Lu C-J, Lu H-Y. Transcriptome Insights into Protective Mechanisms of Ferroptosis Inhibition in Aortic Dissection. International Journal of Molecular Sciences. 2025; 26(9):4338. https://doi.org/10.3390/ijms26094338
Chicago/Turabian StyleShih, Chun-Che, Chi-Yu Chen, Chih-Pin Chuu, Chun-Yang Huang, Chia-Jung Lu, and Hsin-Ying Lu. 2025. "Transcriptome Insights into Protective Mechanisms of Ferroptosis Inhibition in Aortic Dissection" International Journal of Molecular Sciences 26, no. 9: 4338. https://doi.org/10.3390/ijms26094338
APA StyleShih, C.-C., Chen, C.-Y., Chuu, C.-P., Huang, C.-Y., Lu, C.-J., & Lu, H.-Y. (2025). Transcriptome Insights into Protective Mechanisms of Ferroptosis Inhibition in Aortic Dissection. International Journal of Molecular Sciences, 26(9), 4338. https://doi.org/10.3390/ijms26094338