
Dynamics of SARS-CoV-2 Mutations in Wastewater Provide Insights into the Circulation of Virus Variants in the Population

 , and
, and
Abstract
1. Introduction
2. Results
2.1. SARS-CoV-2 S Gene Fragments Are Efficiently Amplified in Wastewater Samples
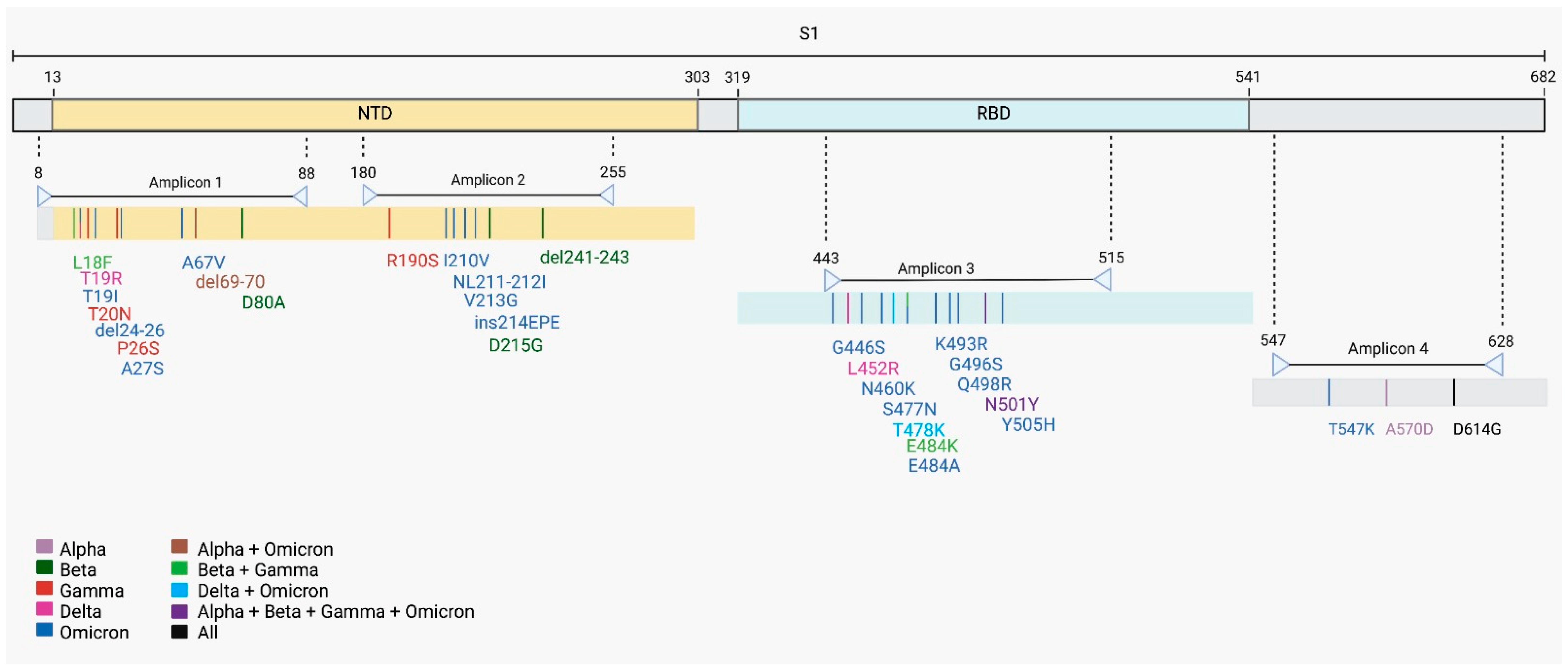
2.2. Identification of 29 Mutations in SARS-CoV-2 S Gene
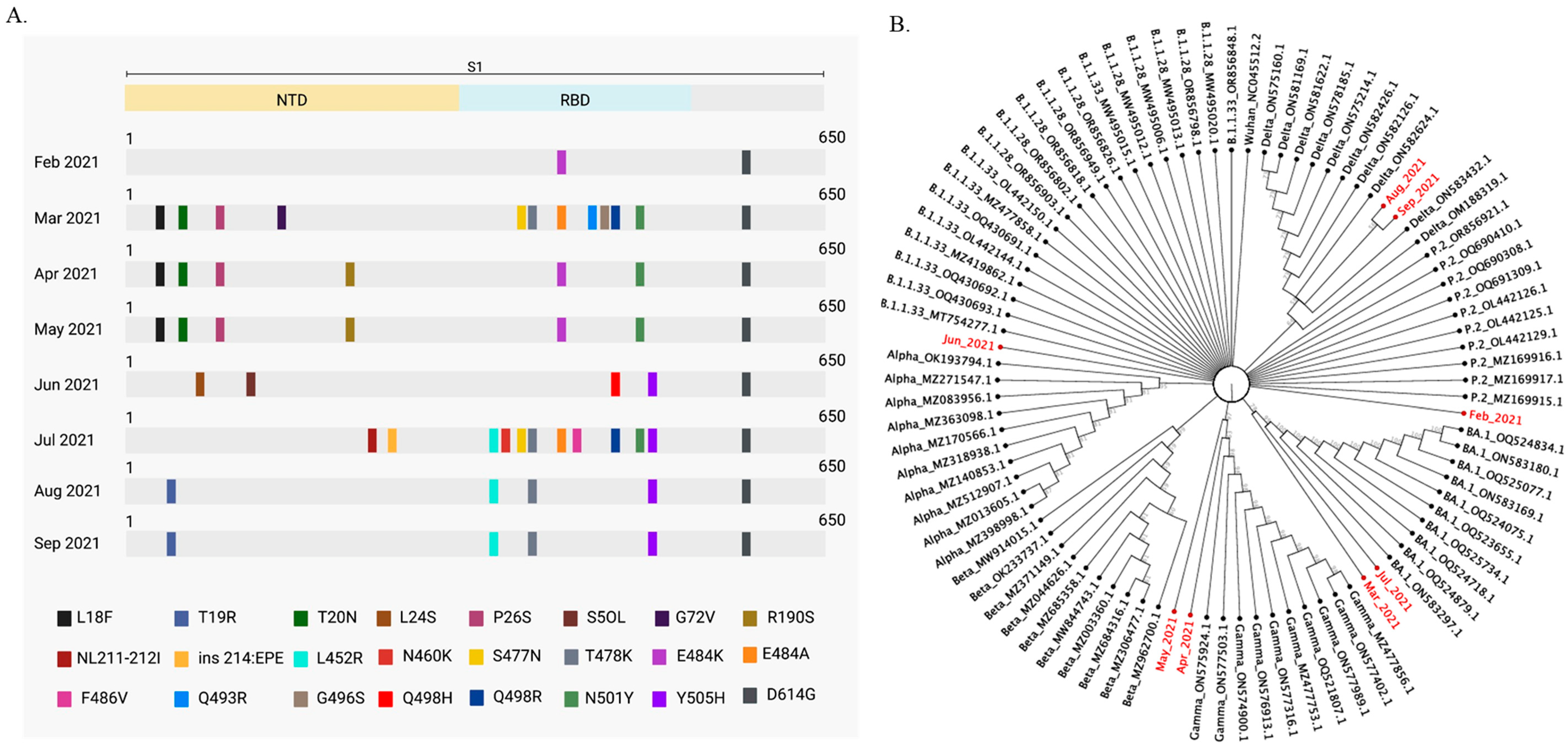
2.3. SARS-CoV-2 Variants Classification in Wastewater
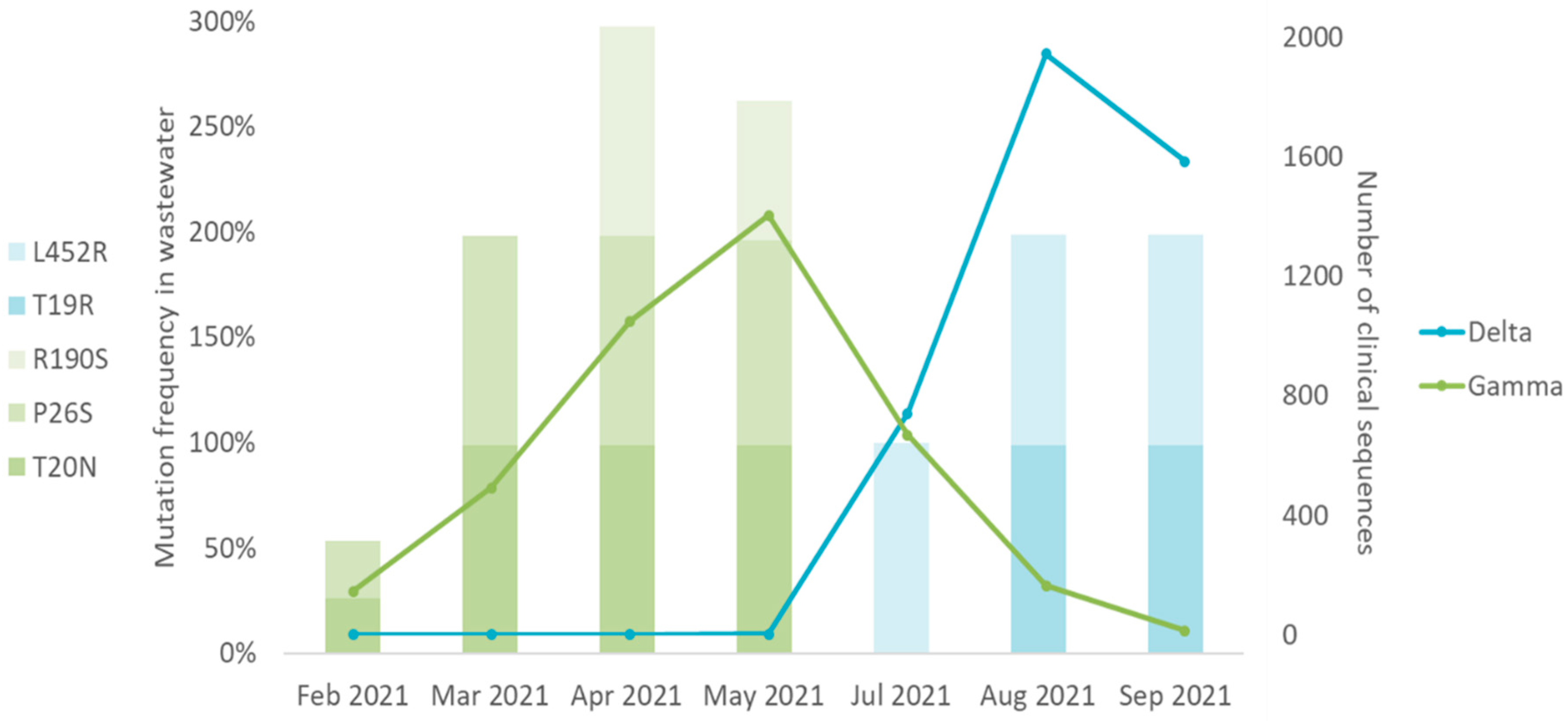
2.4. SARS-CoV-2 Mutations Found in Wastewater Samples Match SARS-CoV-2 Variants in Clinical Samples
3. Discussion
4. Materials and Methods
4.1. Samples Information
4.2. Virus Concentration
4.3. RNA Extraction and SARS-CoV-2 RT-qPCR
4.4. SARS-CoV-2 S Gene Targeted Amplification
4.5. Next-Generation Sequencing (NGS)
4.6. Data Analysis
4.7. Phylogenetic Tree
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO COVID-19 Dashboard. Available online: https://data.who.int/dashboards/covid19/cases?n=c (accessed on 6 March 2025).
- Mistry, P.; Barmania, F.; Mellet, J.; Peta, K.; Strydom, A.; Viljoen, I.M.; James, W.; Gordon, S.; Pepper, M.S. SARS-CoV-2 Variants, Vaccines, and Host Immunity. Front. Immunol. 2022, 12, 809244. [Google Scholar] [CrossRef]
- World Health Organization. Historical Working Definitions and Primary Actions for SARS-CoV-2 Variants. (n.d.-g). Available online: https://www.who.int/publications/m/item/historical-working-definitions-and-primary-actions-for-sars-cov-2-variants (accessed on 6 February 2024).
- World Health Organization. Updated Working Definitions and Primary Actions for SARS-CoV-2 Variants. (n.d.-d). Available online: https://www.who.int/publications/m/item/updated-working-definitions-and-primary-actions-for--sars-cov-2-variants (accessed on 6 February 2024).
- Voloch, C.M.; da Silva Francisco, R.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.D.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C.; et al. Genomic Characterization of a Novel SARS-CoV-2 Lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95, e00119-21. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E.; et al. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations; (2020, December 18). Virological. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563/1 (accessed on 29 April 2024).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Nomoto, H.; Kutsuna, S.; Ujiie, M.; Suzuki, T.; Sato, R.; Fujimoto, T.; Kuroda, M.; Wakita, T.; Ohmagari, N. Novel SARS-CoV-2 Variant in Travelers from Brazil to Japan—Volume 27, Number 4—April 2021—Emerging Infectious Diseases Journal—CDC. Emerg. Infect. Dis. 2021, 27, 1243–1245. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, G. Sequence Analysis of the Emerging SARS-CoV-2 Variant Omicron in South Africa. J. Méd. Virol. 2022, 94, 1728–1733. [Google Scholar] [CrossRef]
- Parums, D.V. Editorial: A Rapid Global Increase in COVID-19 Is Due to the Emergence of the EG.5 (Eris) Subvariant of Omicron SARS-CoV-2. Méd. Sci. Monit. Int. Méd. J. Exp. Clin. Res. 2023, 29, e942244-1–e942244-4. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Arora, P.; Sidarovich, A.; Graichen, L.; Hörnich, B.; Hahn, A.; Hoffmann, M.; Pöhlmann, S. Functional Analysis of Polymorphisms at the S1/S2 Site of SARS-CoV-2 Spike Protein. PLoS ONE 2022, 17, e0265453. [Google Scholar] [CrossRef]
- Liao, Y.-C.; Chen, F.-J.; Chuang, M.-C.; Wu, H.-C.; Ji, W.-C.; Yu, G.-Y.; Huang, T.-S. High-Integrity Sequencing of Spike Gene for SARS-CoV-2 Variant Determination. Int. J. Mol. Sci. 2022, 23, 3257. [Google Scholar] [CrossRef] [PubMed]
- Salles, T.S.; Cavalcanti, A.C.; da Costa, F.B.; Dias, V.Z.; de Souza, L.M.; de Meneses, M.D.F.; da Silva, J.A.S.; Amaral, C.D.; Felix, J.R.; Pereira, D.A.; et al. Genomic Surveillance of SARS-CoV-2 Spike Gene by Sanger Sequencing. PLoS ONE 2022, 17, e0262170. [Google Scholar] [CrossRef]
- Agrawal, S.; Orschler, L.; Schubert, S.; Zachmann, K.; Heijnen, L.; Tavazzi, S.; Gawlik, B.M.; de Graaf, M.; Medema, G.; Lackner, S. Prevalence and Circulation Patterns of SARS-CoV-2 Variants in European Sewage Mirror Clinical Data of 54 European Cities. Water Res. 2022, 214, 118162. [Google Scholar] [CrossRef]
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA 2020, 323, 1843–1844. [Google Scholar] [CrossRef]
- Ahmed, W.; Angel, N.; Edson, J.; Bibby, K.; Bivins, A.; O’Brien, J.W.; Choi, P.M.; Kitajima, M.; Simpson, S.L.; Li, J.; et al. First Confirmed Detection of SARS-CoV-2 in Untreated Wastewater in Australia: A Proof of Concept for the Wastewater Surveillance of COVID-19 in the Community. Sci. Total Environ. 2020, 728, 138764. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, A.; Kantor, R.S.; Olm, M.R.; Whitney, O.N.; Al-Shayeb, B.; Lou, Y.C.; Flamholz, A.; Kennedy, L.C.; Greenwald, H.; Hinkle, A.; et al. Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. mBio 2021, 12, e02703-20. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Bertsch, P.M.; Bivins, A.; Bibby, K.; Farkas, K.; Gathercole, A.; Haramoto, E.; Gyawali, P.; Korajkic, A.; McMinn, B.R.; et al. Comparison of Virus Concentration Methods for the RT-QPCR-Based Recovery of Murine Hepatitis Virus, a Surrogate for SARS-CoV-2 from Untreated Wastewater. Sci. Total Environ. 2020, 739, 139960. [Google Scholar] [CrossRef]
- Sims, N.; Kasprzyk-Hordern, B. Future Perspectives of Wastewater-Based Epidemiology: Monitoring Infectious Disease Spread and Resistance to the Community Level. Environ. Int. 2020, 139, 105689. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Tedla, N.; Bull, R.A. Broadly-Neutralizing Antibodies Against Emerging SARS-CoV-2 Variants. Front. Immunol. 2021, 12, 752003. [Google Scholar] [CrossRef]
- Mercier, P.L.; Hulo, C.; Masson, P.; Castro, E. de SARS-CoV-2 Variants~ViralZone. Available online: https://viralzone.expasy.org/9556 (accessed on 24 April 2025).
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource—Improved Response to Emergent Viral Outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Fiocruz. Dashboard-Pt. (n.d.). Genomahcov. Available online: https://www.genomahcov.fiocruz.br/dashboard-pt/ (accessed on 13 December 2023).
- Rosa, G.L.; Mancini, P.; Ferraro, G.B.; Veneri, C.; Iaconelli, M.; Lucentini, L.; Bonadonna, L.; Brusaferro, S.; Brandtner, D.; Fasanella, A.; et al. Rapid Screening for SARS-CoV-2 Variants of Concern in Clinical and Environmental Samples Using Nested RT-PCR Assays Targeting Key Mutations of the Spike Protein. Water Res. 2021, 197, 117104. [Google Scholar] [CrossRef] [PubMed]
- Carcereny, A.; Garcia-Pedemonte, D.; Martínez-Velázquez, A.; Quer, J.; Garcia-Cehic, D.; Gregori, J.; Antón, A.; Andrés, C.; Pumarola, T.; Chacón-Villanueva, C.; et al. Dynamics of SARS-CoV-2 Alpha (B.1.1.7) Variant Spread: The Wastewater Surveillance Approach. Environ. Res. 2022, 208, 112720. [Google Scholar] [CrossRef] [PubMed]
- Knierim, E.; Lucke, B.; Schwarz, J.M.; Schuelke, M.; Seelow, D. Systematic Comparison of Three Methods for Fragmentation of Long-Range PCR Products for Next Generation Sequencing. PLoS ONE 2011, 6, e28240. [Google Scholar] [CrossRef]
- Costello, M.; Pugh, T.J.; Fennell, T.J.; Stewart, C.; Lichtenstein, L.; Meldrim, J.C.; Fostel, J.L.; Friedrich, D.C.; Perrin, D.; Dionne, D.; et al. Discovery and Characterization of Artifactual Mutations in Deep Coverage Targeted Capture Sequencing Data Due to Oxidative DNA Damage during Sample Preparation. Nucleic Acids Res. 2013, 41, e67. [Google Scholar] [CrossRef]
- Wilton, T.; Bujaki, E.; Klapsa, D.; Majumdar, M.; Zambon, M.; Fritzsche, M.; Mate, R.; Martin, J. Rapid Increase of SARS-CoV-2 Variant B.1.1.7 Detected in Sewage Samples from England between October 2020 and January 2021. mSystems 2021, 6, 10–128. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Levy, J.I.; Hoff, P.D.; Humphrey, G.; Birmingham, A.; Jepsen, K.; Farmer, S.; Tubb, H.M.; Valles, T.; Tribelhorn, C.E.; et al. Wastewater Sequencing Reveals Early Cryptic SARS-CoV-2 Variant Transmission. Nature 2022, 609, 101–108. [Google Scholar] [CrossRef]
- Barnes, K.G.; Levy, J.I.; Gauld, J.; Rigby, J.; Kanjerwa, O.; Uzzell, C.B.; Chilupsya, C.; Anscombe, C.; Tomkins-Tinch, C.; Mbeti, O.; et al. Utilizing River and Wastewater as a SARS-CoV-2 Surveillance Tool in Settings with Limited Formal Sewage Systems. Nat. Commun. 2023, 14, 7883. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, R.; Yamaguchi, K.; Katayama, K.; Ando, H.; Setsukinai, K.; Kobayashi, H.; Okabe, S.; Imoto, S.; Kitajima, M. Identification of SARS-CoV-2 Variants in Wastewater Using Targeted Amplicon Sequencing during a Low COVID-19 Prevalence Period in Japan. Sci. Total Environ. 2023, 887, 163706. [Google Scholar] [CrossRef]
- Shempela, D.M.; Muleya, W.; Mudenda, S.; Daka, V.; Sikalima, J.; Kamayani, M.; Sandala, D.; Chipango, C.; Muzala, K.; Musonda, K.; et al. Wastewater Surveillance of SARS-CoV-2 in Zambia: An Early Warning Tool. Int. J. Mol. Sci. 2024, 25, 8839. [Google Scholar] [CrossRef]
- Mautner, L.; Hoyos, M.; Dangel, A.; Berger, C.; Ehrhardt, A.; Baiker, A. Replication Kinetics and Infectivity of SARS-CoV-2 Variants of Concern in Common Cell Culture Models. Virol. J. 2022, 19, 76. [Google Scholar] [CrossRef]
- Pan American Health Organization (PAHO). Brazil Will Receive the First Vaccines Against COVID-19 Through the COVAX Mechanism. (n.d.). Available online: https://www.paho.org/en/news/21-3-2021-brazil-will-receive-first-vaccines-against-covid-19-through-covax-mechanism (accessed on 29 April 2024).
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.-M.; et al. Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef]
- Zeng, B.; Gao, L.; Zhou, Q.; Yu, K.; Sun, F. Effectiveness of COVID-19 Vaccines against SARS-CoV-2 Variants of Concern: A Systematic Review and Meta-Analysis. BMC Med. 2022, 20, 200. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Slavov, S.N.; Fonseca, V.; Wilkinson, E.; Tegally, H.; Patané, J.S.L.; Viala, V.L.; San, E.J.; Rodrigues, E.S.; Santos, E.V.; et al. Genomic Epidemiology of the SARS-CoV-2 Epidemic in Brazil. Nat. Microbiol. 2022, 7, 1490–1500. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. 16 March 2023. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 12 March 2024).
- Pastorino, B.; Touret, F.; Gilles, M.; Luciani, L.; de Lamballerie, X.; Charrel, R.N. Evaluation of Chemical Protocols for Inactivating SARS-CoV-2 Infectious Samples. Viruses 2020, 12, 624. [Google Scholar] [CrossRef]
- Batéjat, C.; Grassin, Q.; Manuguerra, J.-C.; Leclercq, I. Heat Inactivation of the Severe Acute Respiratory Syndrome Coronavirus. J. Biosaf. Biosecurity 2021, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ct |
|---|---|
| February 2021 | 32.3 |
| March 2021 | 33.4 |
| April 2021 | 32.4 |
| May 2021 | 31.8 |
| June 2021 | 28.4 |
| July 2021 | 34.1 |
| August 2021 | 31.3 |
| September 2021 | 31.2 |
| NTD | RBD | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L18F | T19R | T20N | L24S | P26S | S50L | I68V | G72V | T76A | R190S | NL211-212I | ins214EPE | G446S | L452R | N460K | S477N | T478K | E484A | E484K | F486V | Q493R | S494A | G496S | Q498H | Q498R | N501Y | Y505H | T547K | D614G | |
| February 2021 | 26 | 26 | 27 | 26 | 100 | 48 | 38 | 100 | |||||||||||||||||||||
| March 2021 | 99 | 99 | 99 | 99 | 31 | 100 | 99 | 100 | 99 | 100 | 100 | 100 | 53 | 41 | 100 | ||||||||||||||
| April 2021 | 99 | 99 | 99 | 99 | 28 | 98 | 100 | 49 | 31 | 100 | |||||||||||||||||||
| May 2021 | 99 | 99 | 97 | 66 | 24 | 100 | 100 | 51 | 52 | 100 | |||||||||||||||||||
| June 2021 | 57 | 98 | 31 | 17 | 96 | 55 | 34 | 100 | |||||||||||||||||||||
| July 2021 | 98 | 97 | 17 | 100 | 100 | 100 | 100 | 100 | 99 | 100 | 100 | 71 | 47 | 100 | |||||||||||||||
| August 2021 | 99 | 11 | 26 | 100 | 99 | 58 | 35 | 100 | |||||||||||||||||||||
| September 2021 | 99 | 28 | 100 | 99 | 56 | 42 | 100 | ||||||||||||||||||||||
| Primer Name | Nucleotide Sequence (5′-3′) | Orientation | Reaction | Amplicon Size (bp) |
|---|---|---|---|---|
| S-1F | CTTGTTTTATTGCCACTAGTCTCTAG | Sense | First PCR | 273 |
| S-1R | CARTGGAAGCAAAATAAACACCATC | Anti-sense | ||
| S-1.1F | GCCACTAGTCTCTAGTCAGTGTG | Sense | Nested-PCR | 241 |
| S-1.1R | CATCATTAAATGGTAGGACAGGGT | Anti-sense | ||
| S-2F | CTTATGGACCTTGAAGGAAAACAGG | Sense | First PCR | 259 |
| S-2R | GTCCAACCTGAAGAAGAATCACCA | Anti-sense | ||
| S-2.2F | AGGAAAACAGGGTAATTTCAAAAATCT | Sense | Nested-PCR | 226 |
| S-2.2R | CACCAGGAGTCAAATAACTTCTATGT | Anti-sense | ||
| S-3F | GGAATTCTAACAAKCTTGATTCTAAGGT | Sense | First PCR | 245 |
| S-3R | GAAGTTCAAAAGAAAGTACTACTACTCT | Anti-sense | ||
| S-3.3F | CTTGATTCTAAGGTTRGTGGTAATT | Sense | Nested-PCR | 216 |
| S-3.3R | GTACTACTACTCTGTATGGTTGGTRAC | Anti-sense | ||
| S-4F | CAATTTCAACTTCAATGGTTTAAMAGG | Sense | First PCR | 275 |
| S-4R | GGAGTAAGTTGATCTGCATGAATAGC | Anti-sense | ||
| S-4.4F | GGTTTAAMAGGCACAGGTGTTC | Sense | Nested-PCR | 244 |
| S-4.4R | GCATGAATAGCAACAGGGACTTC | Anti-sense |
| SARS-CoV-2 Lineage | Sequence ID |
|---|---|
| Wuhan | NC_045512.2 |
| B.1.1.33 | OL442150.1; MZ477858.1; OQ430691.1; OL442144.1; MZ419862.1; OQ430692.1; OQ430693.1; MT754277.1; OR856848.1 |
| B.1.1.28 | MW495020.1; OR856798.1; MW495013.1; MW495006.1; MW495012.1; MW495015.1; OR856826.1; OR856949.1; OR856818.1; OR856802.1; OR856903.1 |
| P.2 | MZ169915.1; MZ169917.1; MZ169916.1; OL442129.1; OL442125.1; OL442126.1; OQ691309.1; OQ690308.1; OQ690410.1; OR856921.1 |
| Alpha | OK193794.1; MZ271547.1; MZ083956.1; MZ363098.1; MZ170566.1; MZ318938.1; MZ140853.1; MZ512907.1; MZ013605.1; MZ398998.1 |
| Beta | MZ685358.1; MW844743.1; MZ003360.1; MZ684316.1; MZ306477.1; MW914015.1; OK233737.1; MZ371149.1; MZ044626.1; MZ962700.1 |
| Gamma | ON575924.1; ON577503.1; ON574900.1; ON576913.1; ON577316.1; MZ477753.1; OQ521807.1; ON577989.1; ON577402.1; MZ477856.1 |
| Delta | ON582624.1; ON583432.1; OM188319.1; ON582126.1; ON582426.1; ON575214.1; ON578185.1; ON581622.1; ON581169.1; ON575160.1 |
| Omicron (BA.1) | ON583297.1; OQ524879.1; OQ524718.1; OQ525734.1; OQ523655.1; OQ524075.1; ON583169.1; OQ525077.1; ON583180.1; OQ524834.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, S.M.; Simas, M.C.d.C.; da Costa, L.J.; Silva, R. Dynamics of SARS-CoV-2 Mutations in Wastewater Provide Insights into the Circulation of Virus Variants in the Population. Int. J. Mol. Sci. 2025, 26, 4324. https://doi.org/10.3390/ijms26094324
Costa SM, Simas MCdC, da Costa LJ, Silva R. Dynamics of SARS-CoV-2 Mutations in Wastewater Provide Insights into the Circulation of Virus Variants in the Population. International Journal of Molecular Sciences. 2025; 26(9):4324. https://doi.org/10.3390/ijms26094324
Chicago/Turabian StyleCosta, Sara Mesquita, Maria Clara da Costa Simas, Luciana Jesus da Costa, and Rosane Silva. 2025. "Dynamics of SARS-CoV-2 Mutations in Wastewater Provide Insights into the Circulation of Virus Variants in the Population" International Journal of Molecular Sciences 26, no. 9: 4324. https://doi.org/10.3390/ijms26094324
APA StyleCosta, S. M., Simas, M. C. d. C., da Costa, L. J., & Silva, R. (2025). Dynamics of SARS-CoV-2 Mutations in Wastewater Provide Insights into the Circulation of Virus Variants in the Population. International Journal of Molecular Sciences, 26(9), 4324. https://doi.org/10.3390/ijms26094324