
Rapid Dereplication of Trunk Bark Constituents of Croton sylvaticus and Molecular Docking of Terpenoids from Three Congolese Croton Species
 , , , ,
, , , , 
Abstract
1. Introduction
2. Results and Discussion
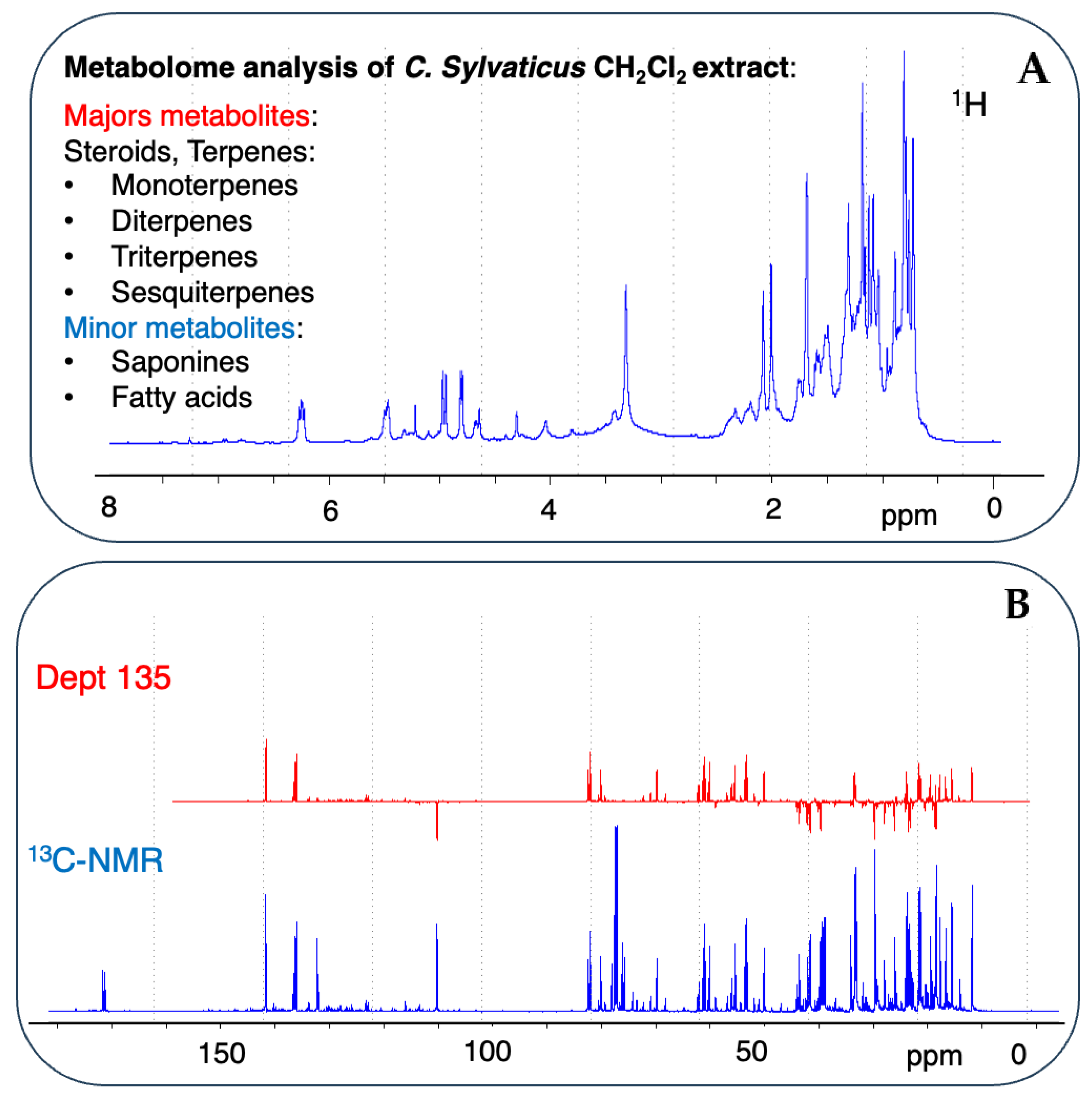
2.1. Chemical Screening
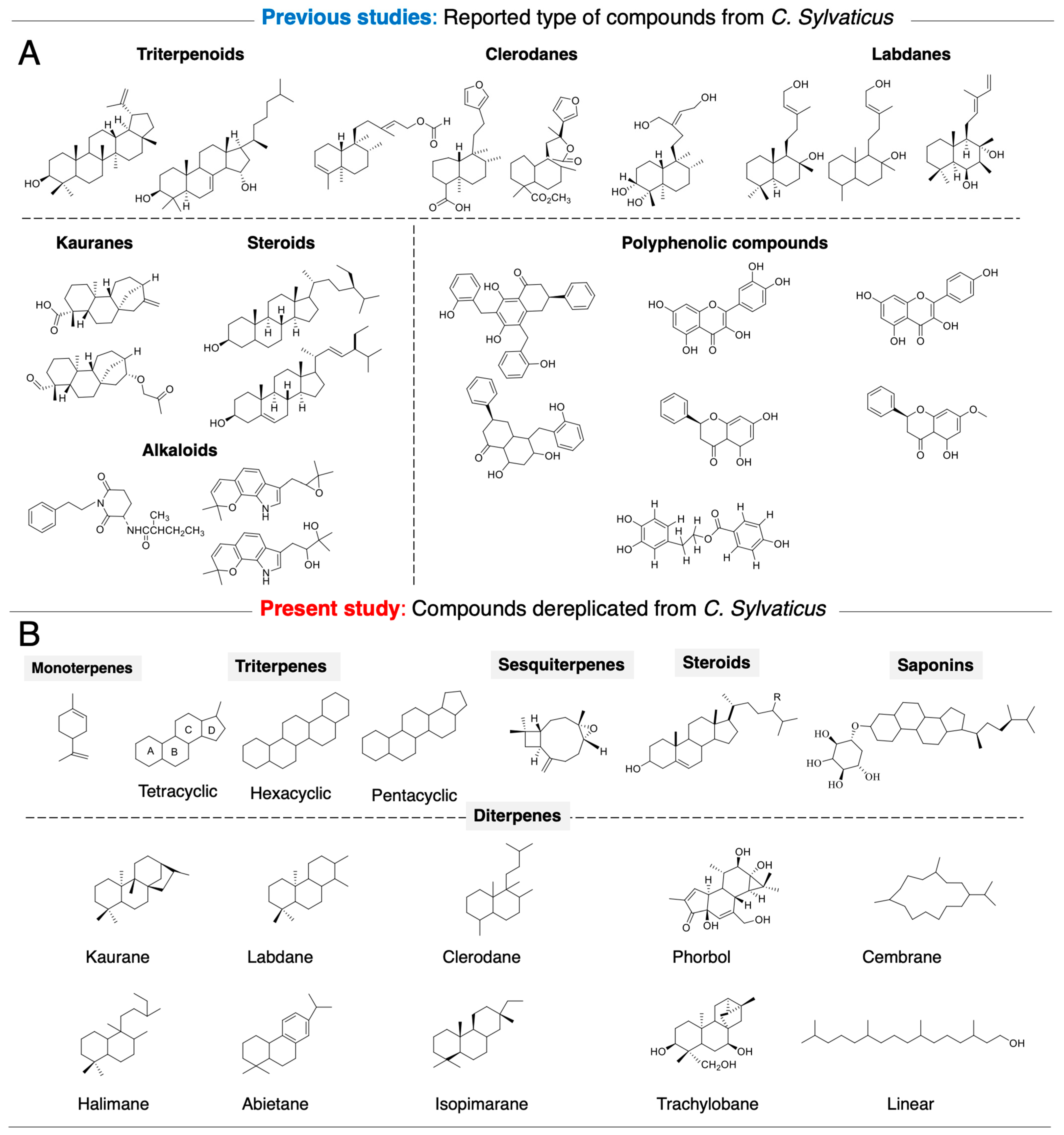
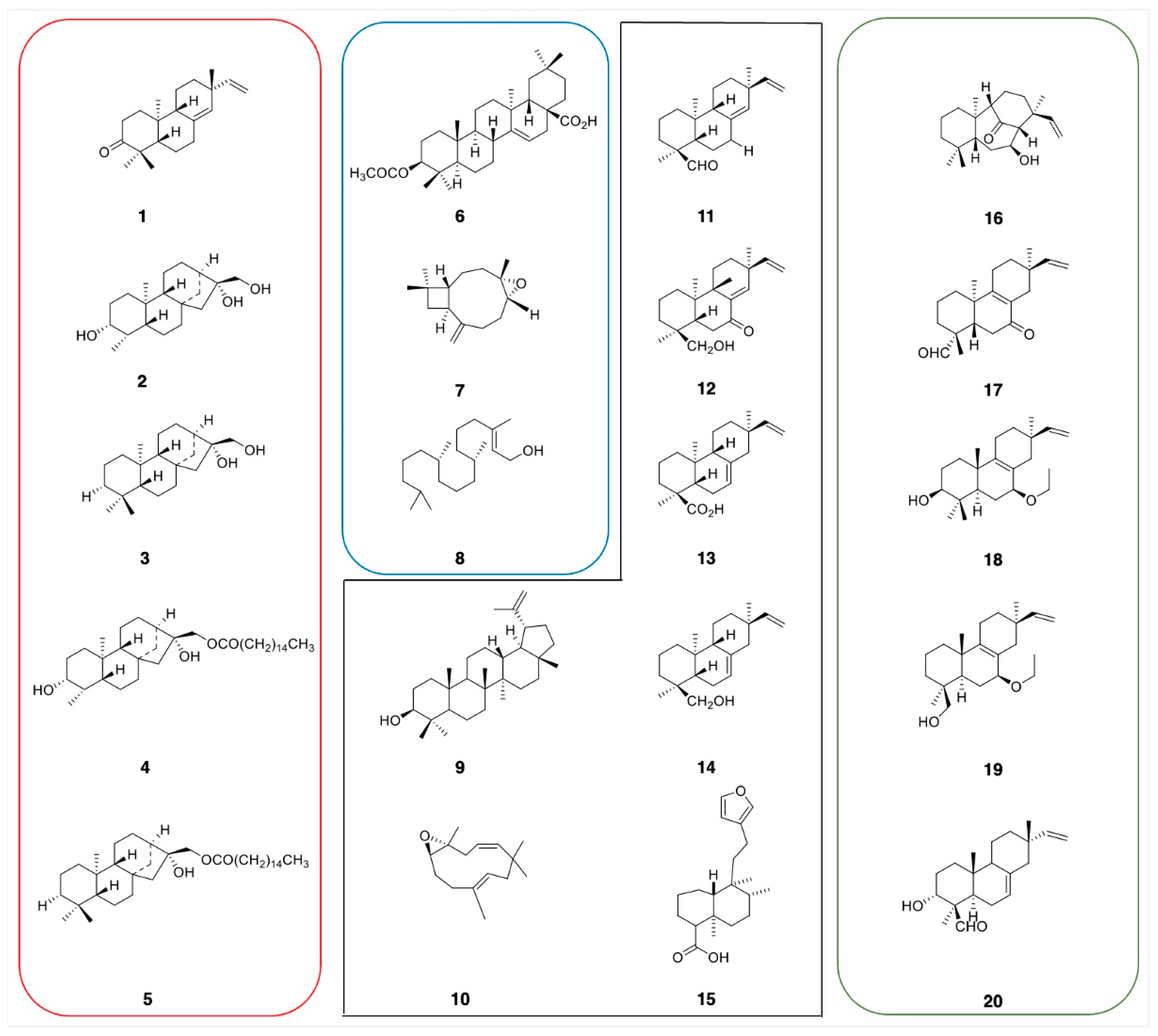
2.2. Rapid Dereplication of EDCM from C. sylvaticus
2.3. Phytochemical Analysis
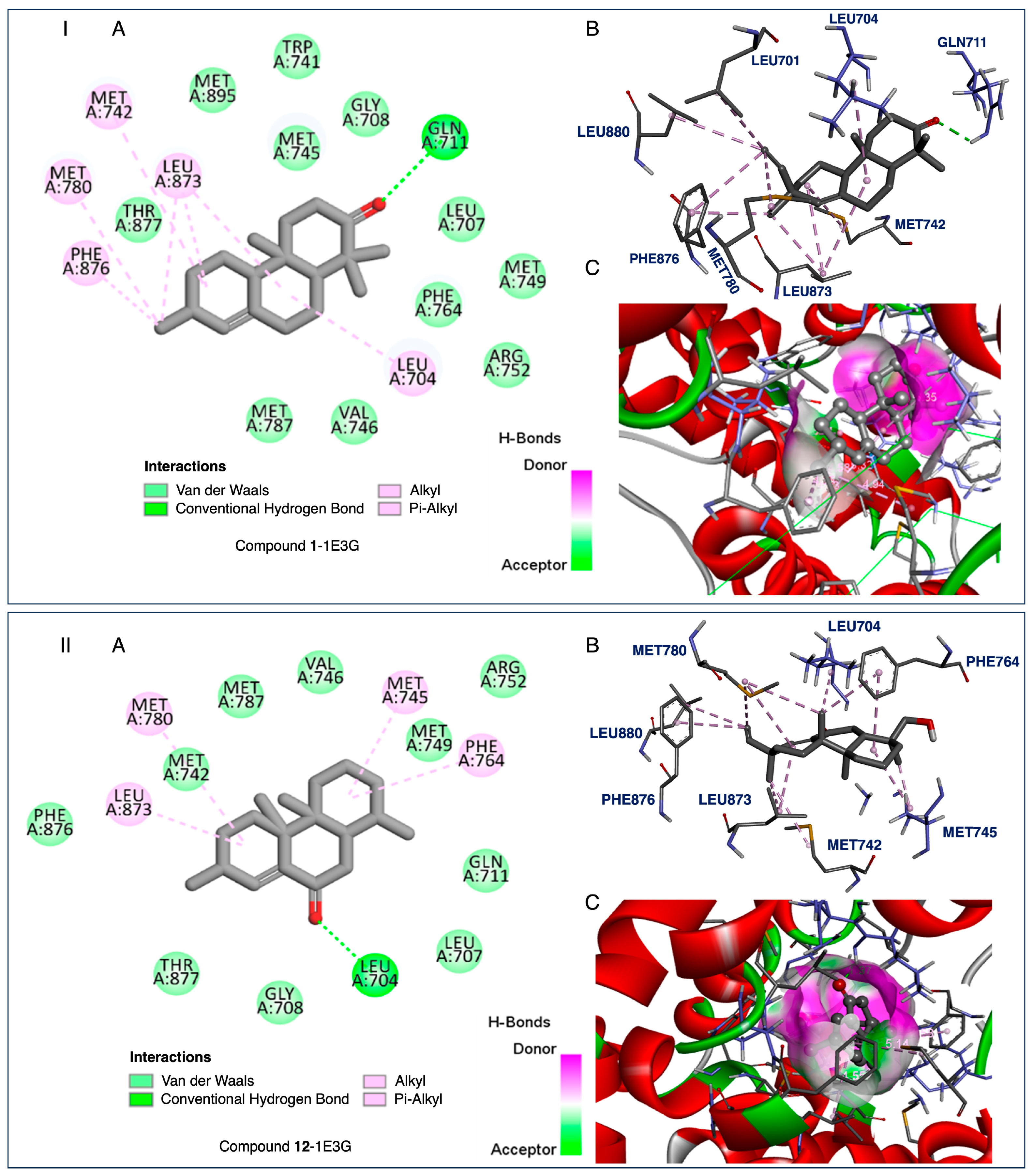
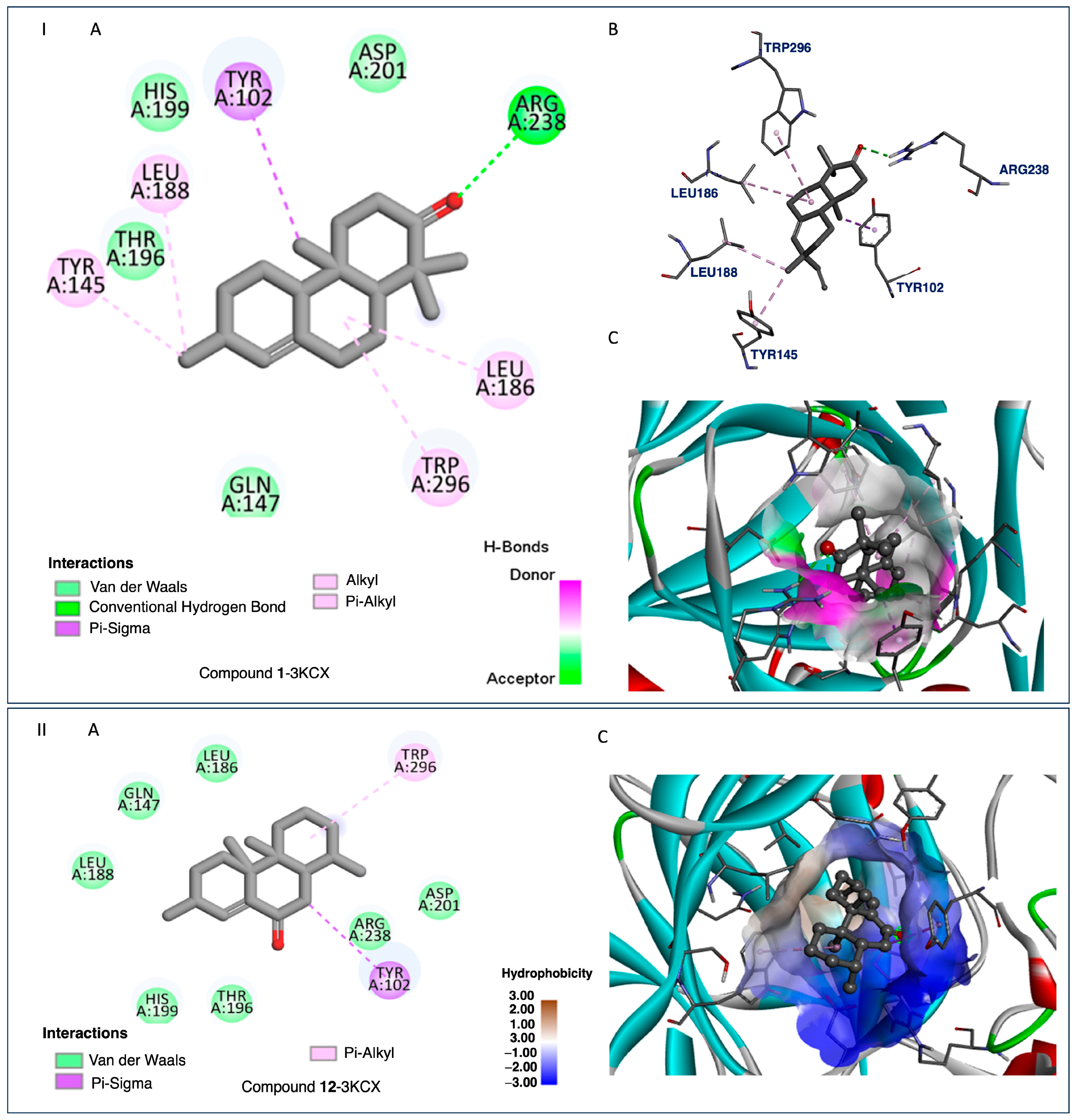
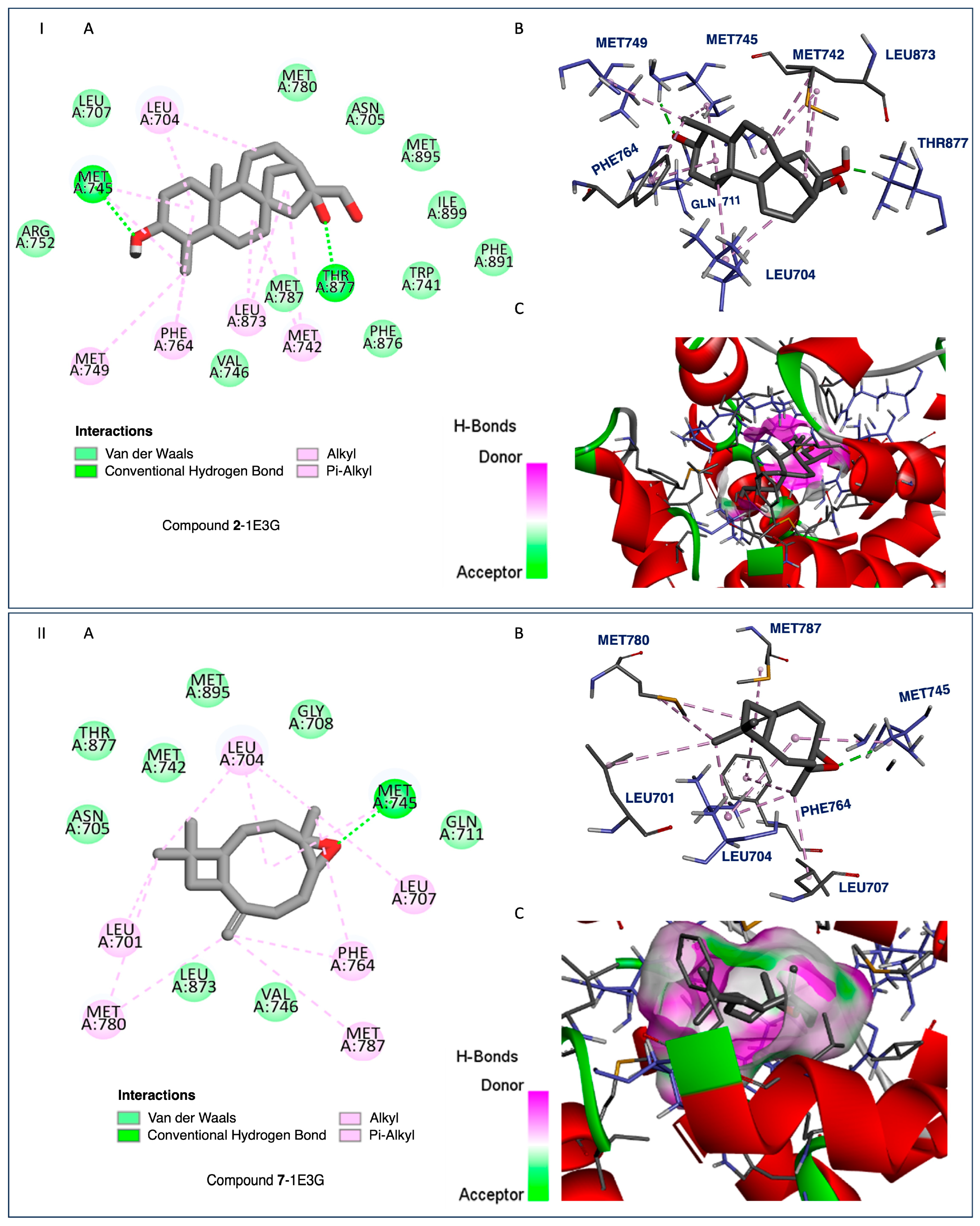
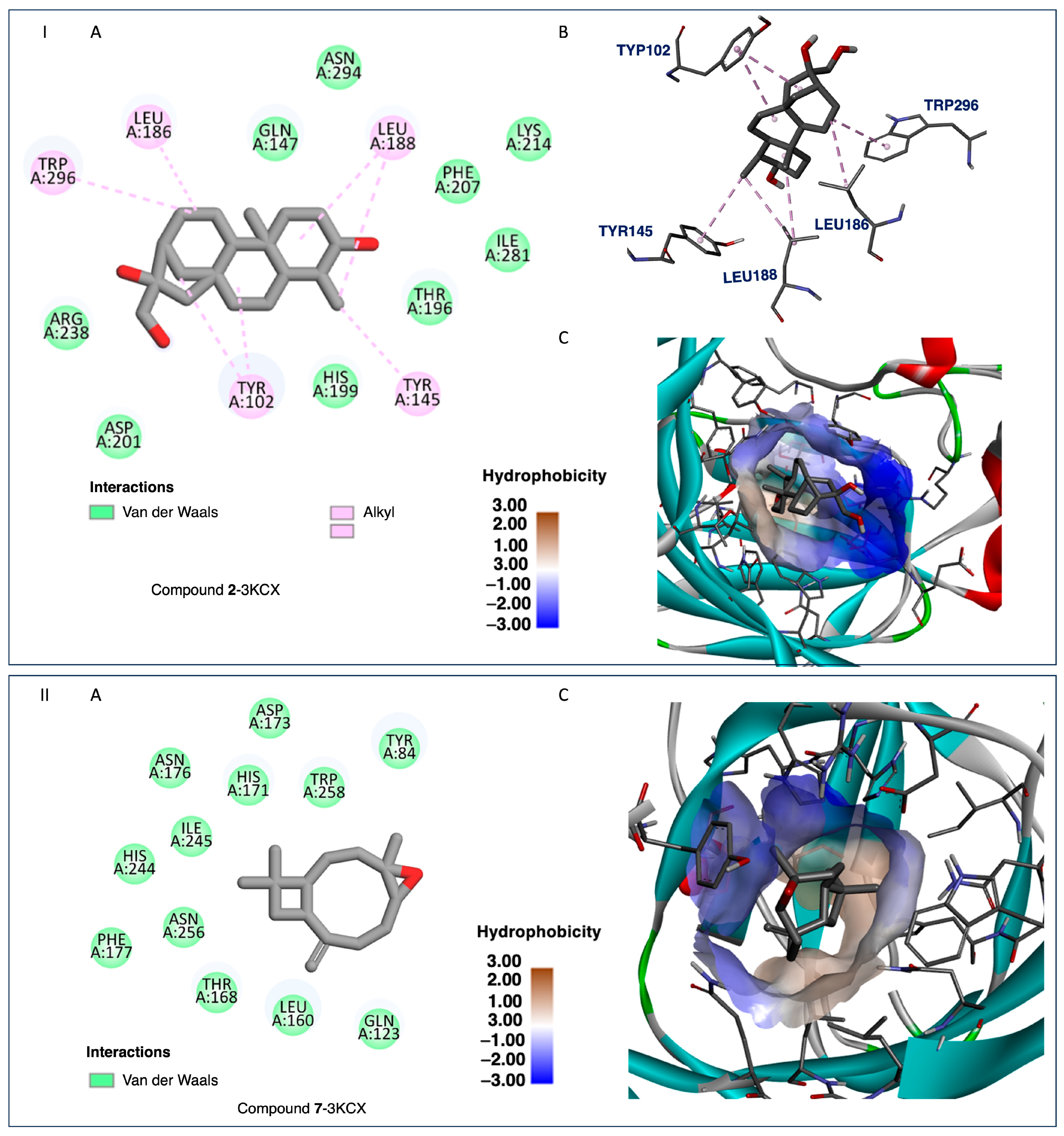
2.4. Docking and SAR Studies of Selected Terpenoids (1–15)
2.5. Limitations, Strengths, and Future Prospects
3. Material and Methods
3.1. Material and Chemicals
3.2. Methods
3.2.1. Extraction
3.2.2. Phytochemical Screening Test
3.2.3. Metabolome Profiling
3.2.4. Purification and Structure Elucidation of Major Constituents from C. sylvaticus
3.2.5. Molecular Docking
3.2.6. Docking Strategies
3.2.7. ADMET Profiling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paul, E.B.; Andrew, L.H.; Kenneth, J.W.; Benjamin, V.E.; Ricarda, R. Molecular phylogenetics of the giant genus Croton and tribe Crotoneae (Euphorbiaceae sensu stricto) using ITS and trnL-trnF DNA sequence data. Am. J. Bot. 2005, 92, 1520–1534. [Google Scholar] [CrossRef]
- Maroyi, A. Croton megalocarpus Hutch. in Tropical Africa: Phytochemistry, Pharmacology and Medicinal Potential. Res. J. Med. Plants 2017, 11, 124–133. [Google Scholar] [CrossRef]
- Salatino, A.; Salatino, M.L.F.; Negri, G. Traditional uses, Chemistry and Pharmacology of Croton species (Euphorbiaceae). J. Braz. Chem. Soc. 2007, 18, 11–33. [Google Scholar] [CrossRef]
- Robyns, W. Flore du Congo et du Rwanda-Urundi: Spermatophytes, Tableau analytique des familles; Bruxelles: Brussels, Belgium, 1958; 70p. [Google Scholar]
- Liu, C.P.; Xu, J.B.; Zhao, J.X.; Xu, C.H.; Dong, L.; Ding, J.; Yue, J.M. Diterpenoids from Croton laui and their cytotoxic and antimicrobial activities. J. Nat. Prod. 2014, 77, 1013–1020. [Google Scholar] [CrossRef]
- Wang, G.C.; Li, J.G.; Li, G.Q.; Xu, J.J.; Wu, X.; Ye, W.C.; Li, Y.L. Clerodane diterpenoids from Croton crassifolius. J. Nat. Prod. 2012, 75, 2188–2192. [Google Scholar] [CrossRef] [PubMed]
- Furlan, C.M.; Santos, K.P.; Sedano-Partida, M.D.; Barbosa da Motta, L.; Santos, D.Y.A.C.; Salatinoet, M.L.F.; Negri, G.; Berry, P.E.; van Ee, B.W.; Salatino, A. Flavonoids and antioxidant potential of nine Argentinian species of Croton (Euphorbiaceae). Braz. J. Bot. 2015, 38, 693–702. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Yang, K.X.; Yang, X.W.; Khan, A.; Liu, L.; Wang, B.; Zhao, Y.L.; Liu, Y.P.; Li, Y.; Luo, X.D. New Cytotoxic Tigliane Diterpenoids from Croton caudatus. Planta Med. 2016, 82, 729–733. [Google Scholar] [CrossRef]
- Sani, M.I.; Langat, M.K.; Mas-Claret, E.; Mbala, M.B.; Mvingu, K.B.; Dulcie, A.M. Ent-abietane and ent-pimarane diterpenoids from Croton mubango (Euphorbiaceae). Phytochemistry 2020, 170, 112–217. [Google Scholar] [CrossRef]
- Sani, M.I.; Mas-Claret, E.; Langat, M.K.; Thomas, H.; Bethany, S.; Mbala, M.B.; Mvingu, K.B.; Dulcie, A.M. Cytotoxic diterpenoids from the leaves and stem bark of Croton haumanianus (Euphorbiaceae). Phytochemistry 2020, 178, 112455. [Google Scholar] [CrossRef]
- Ndunda, B.; Langat, M.K.; Jacob, O.; Midiwo and Leonidah, K.O. Diterpenoid Derivatives of Kenyan Croton sylvaticus. Nat. Prod. Commun. 2015, 10, 557–558. [Google Scholar] [CrossRef]
- Selowa, S.C.; Shai, L.J.; Masoko, P.; Mokgotho, M.P.; Magano, S.R. Antibacterial activity of extracts of three Croton species collected in Mpumalanga region in South Africa. Afr. J. Tradit. Complement. Altern. Med. 2010, 7, 98–103. [Google Scholar] [CrossRef]
- Mokoka, T.A.; McGaw, L.J.; Eloff, J.N. Antifungal efficacy of ten selected South African plant species against Cryptococcus neoformans. Pharm. Biol. 2010, 48, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Frum, Y.; Viljoen, A.M. In vitro 5-lipoxygenase and anti-oxidant activities of South African medicinal plants commonly used topically for skin diseases. Ski. Pharm. Physiol. 2006, 19, 329–335. [Google Scholar] [CrossRef]
- Mutalib, A.A.; Ashwell, R.N.; Johannes, V.S. Acetylcholinesterase inhibitors from Croton sylvaticus ethyl acetate leaf extract and their mutagenic effects. Nat. Prod. Commun. 2013, 8, 795–798. [Google Scholar] [CrossRef]
- Kihampa, C.; Joseph, C.C.; Nkunya, M.H.H.; Magesa, S.M.; Hassanali, A.; Heydenreich, M. Larvicidal and IGR activity of extract of Tanzanian plants against malaria vector mosquitoes. J. Vect. Borne Dis. 2009, 46, 145–152. [Google Scholar]
- Wu, Z.; Xia, F.; Lin, R. Global burden of cancer and associated risk factors in 204 countries and territories, 1980-2021: A systematic analysis for the GBD 2021. J. Hematol. Oncol. 2024, 17, 119–131. [Google Scholar] [CrossRef]
- Swati, S.; Veda, P.P.; Kusum, Y.; Anurag, Y.; Dwivedi, U.N. Natural Products as Anti-Cancerous Therapeutic Molecules Targeted towards Topoisomerases. Curr. Protein Pept. Sci. 2020, 21, 1103–1142. [Google Scholar] [CrossRef]
- Syeda, T.A.; Ulas, A.; Kálmán, I.; Adriana, M.; Syed, R.A.S.; Syed, Z.H.; Damla, A.-A.; Hayri, D.; Zehra, H.-M.; Fatih, R.I.; et al. Natural Products/Bioactive Compounds as a Source of Anticancer Drugs. Cancers 2022, 14, 6203. [Google Scholar] [CrossRef]
- Chunarkar-Patil, P.; Mohammed, K.; Richa, M.; Subhasree, R.; Aftab, A.; Devvret, V.; Sagar, B.; Rajni, D.; Himanshu, N.S.; Sanjay, K. Anticancer Drug Discovery Based on Natural Products: From Computational Approaches to Clinical Studies. Biomedicines 2024, 12, 201. [Google Scholar] [CrossRef]
- Tshibangu, D.S.; Divakar, S.; Ramanathan, M.; Syamala, G.G.; Ngbolua, K.-T.; Mudogo, V.; Tshilanda, D.D.; Gbolo, B.Z.; Mpiana, P.T. In vitro Screening of the Leaf Extracts from Gardenia ternifolia (Forest Gardenia) for their Anticancer Activity. J. Complement. Altern. Med. Res. 2016, 1, 1–7. [Google Scholar] [CrossRef]
- Li, Q.; Xu, C.S.; Yu, H.L.; Zhang, Y.; Wang, J.; Shen, Y.B.; Wu, H.W.; Gu, M.M.; Liao, Z.X. A Tricyclo[4.3.1]decane Diterpenoid Skeleton from Croton laui: Isolation and 1H NMR-Based Metabolomic Profiling. Org Lett. 2025, 27, 4300–4304. [Google Scholar] [CrossRef]
- Shi, Y.; Gilkes, D.M. HIF-1 and HIF-2 in cancer: Structure, regulation, and therapeutic prospects. Cell. Mol. Life Sci. 2025, 82, 44. [Google Scholar] [CrossRef]
- Moon, H.; Han, S.; Park, H.; Choe, J. Crystal structures of human FIH-1 in complex with quinol family inhibitors. Mol. Cells 2010, 29, 471–474, Erratum in Mol. Cells 2010, 29, 625. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Han, L.; Sun, H.; Zhou, L.; Wei, Z. Androgen receptor expression and clinical significance in breast cancer. World J. Surg. Oncol. 2025, 23, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Bastos, D.A.; Soares, A.; Schutz, F.A.B.; Cronemberger, E.; de Almeida Luz, M.; Martins, S.P.D.S.; Muniz, D.Q.B.; Cárcano, F.M.; Smaletz, O.; Peixoto, F.A.; et al. Androgen Receptor Pathway Inhibitor Therapy for Advanced Prostate Cancer: Secondary Analysis of a Randomized Clinical Trial. JAMA Netw. Open 2025, 8, e2454253. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Elshan, N.G.R.D.; Rettig, M.B.; Jung, M.E. Molecules targeting the androgen receptor (AR) signaling axis beyond the AR-Ligand binding domain. Med. Res. Rev. 2019, 39, 910–960. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Matias, P.M.; Donner, P.; Coelho, R.; Thomaz, M.; Peixoto, C.; Macedo, S.; Otto, N.; Joschko, S.; Scholz, P.; Wegg, A.; et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J. Biol. Chem. 2000, 275, 26164–26171. [Google Scholar] [CrossRef] [PubMed]
- Andolfi, C.; Bartolini, C.; Morales, E.; Gündoğdu, B.; Puhr, M.; Guzman, J.; Wach, S.; Taubert, H.; Aigner, A.; Eder, I.E.; et al. MED12 and CDK8/19 Modulate Androgen Receptor Activity and Enzalutamide Response in Prostate Cancer. Endocrinology 2024, 165, bqae114. [Google Scholar] [CrossRef]
- Nuzillard, J.M. Taxonomy focused natural product databases for carbon-13 NMR based dereplication. Analytica 2021, 2, 50–56. [Google Scholar] [CrossRef]
- Mwangi, J.W.; Thoithi, G.N.; Addae-Mensah, I.; Achenbach, H.; Lwande, W.; Hassanali, H. Aromatic plants of Kenya III: Volatile and some non-volatile constituents of Croton sylvaticus. East Cent. Afr. J. Pharm. Sci. 1998, 1, 41–43. [Google Scholar]
- Kapingu, M.C.; Mbwambo, Z.H.; Moshi, M.J.; Magadula, J.J. Brine shrimp lethality of a glutarimide alkaloid from Croton sylvaticus Hochst. East Cent. Afr. J. Pharm. Sci. 2006, 8, 3–5. [Google Scholar] [CrossRef]
- Khan, R.; Khan, H.; Abdullah, Y.; Ping, Q.D. Feasibility of Repurposing Clioquinol for Cancer Therapy. Recent Pat. Anti-Cancer Drug Discov. 2020, 15, 14–31. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, L. Progress in research on paclitaxel and tumor immunotherapy. Cell Mol. Biol. Lett. 2019, 24, 40. [Google Scholar] [CrossRef] [PubMed]
- Teresa, M.B.B. Extraction and Qualitative Phytochemical Screening of Medicinal Plants: A Brief Summary. Int. J. Pharm. 2018, 8, 137–143. [Google Scholar]
- Junaid, R.S.; Patil, M.K. Qualitative tests for preliminary phytochemical Screening: An overview. Int. J. Chem. Stud. 2020, 8, 603–608. [Google Scholar] [CrossRef]
- Bruguière, A.; Derbré, S.; Dietsch, J.; Leguy, J.; Rahier, V.; Pottier, Q.; Bréard, D.; Suor-Cherer, S.; Viault, G.; Le Ray, A.M.; et al. MixONat, a Software for the Dereplication of Mixtures Based on 13C NMR Spectroscopy. Anal. Chem. 2020, 92, 8793–8801. [Google Scholar] [CrossRef]
- Anshika, N.S.; Meghna, M.B.; Neeti, S. Structure Based docking studies towards exploring potential anti-androgen activity of selected phytochemicals against prostate cancer. Sci. Rep. 2017, 7, 1955. [Google Scholar] [CrossRef]
- VidyaB, M.; Madhu, S.S.; Batoul, F.; Mastan, M.; Afroz, A.; Ganj, P.N. Molecular docking of angiogenesis target protein HIF-1α and genistein in breast cancer. Gene 2019, 701, 169–172. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Selvaraj, A.; Antony, S.; Hakdong, S. Anti-methanogenic efect of rhubarb (Rheum spp.)-An in silico docking studies on methyl-coenzyme reductase (MCR). Saudi J. Biol. Sci. 2019, 26, 1458–1462. [Google Scholar] [CrossRef]
- Nissink, J.W.M.; Murray, C.; Hartshorn, M.; Verdonk, M.L.; Cole, J.C.; Taylor, R. A new test set for validating predictions of protein–ligand interaction. Proteins 2002, 49, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Matondo, A.; Kilembe, J.T.; Mwanangombo, D.T.; Nsimba, B.M.; Mawete, D.T.; Gbolo, B.Z.; Bongo, G.N.; Ngbolua, K.N. Facing COVID-19 via anti-infammator mechanism of action: Molecular docking and pharmacokinetic studies of six-anti-infammatory compounds derived from Passifora edulis. J. Complement. Altern. Med Res. 2020, 12, 35–51. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ∆Gbinding (kcal/mol) | (ΔG Difference kcal/mol) | Molecular Weight | logP | Rotatable Bonds | H-Bond Acceptors | H-Bond Donors | Surface Area (Å2) | Water Solubility | |
|---|---|---|---|---|---|---|---|---|---|---|
| HAR (1E3G) Prostate Cancer | HIF-1α (3KCX) Breast Cancer | |||||||||
| 1 | −7.7 | −8.4 | 0.7 | 286.4 | 5.3 | 1 | 1 | 0 | 129.4 | −6.1 |
| 2 | −7.3 | −8.3 | 1 | 308.4 | 2.7 | 1 | 3 | 3 | 133.6 | −3.7 |
| 3 | −5.9 | −8.0 | 2.1 | 306.4 | 4.1 | 1 | 2 | 2 | 135.2 | −4.7 |
| 4 | −1.5 | −7.4 | 5.9 | 546.8 | 8.7 | 16 | 4 | 2 | 239.9 | −5.8 |
| 5 | −1.6 | −7.4 | 5.8 | 544.9 | 10.1 | 16 | 3 | 1 | 241.5 | −5.5 |
| 6 | −6.6 | −8.1 | 1.5 | 484.7 | 7.4 | 2 | 3 | 1 | 212.2 | −4.3 |
| 7 | −7.2 | −7.2 | 0 | 220.3 | 3.9 | 0 | 1 | 0 | 99.2 | −4.3 |
| 8 | −6.2 | −6.6 | 0.4 | 296.5 | 6.3 | 13 | 1 | 1 | 133.7 | −7.5 |
| 9 | −6.1 | −9.1 | 3 | 426.7 | 8.0 | 1 | 1 | 1 | 192.3 | −6 |
| 10 | −6.7 | −7.1 | 0.4 | 220.3 | 4.2 | 0 | 1 | 0 | 99.5 | −4.1 |
| 11 | −6.8 | −7.8 | 1 | 286.4 | 5.3 | 2 | 1 | 0 | 129.4 | −6.1 |
| 12 | −7.4 | −8.4 | 1 | 316.4 | 4.6 | 2 | 2 | 1 | 140.5 | −5.1 |
| 13 | −5.8 | −8.0 | 2.2 | 302.4 | 5.2 | 2 | 1 | 1 | 134.2 | −4.3 |
| 14 | −5.9 | −8.2 | 2.3 | 288.4 | 5.1 | 2 | 1 | 1 | 130.0 | −5.7 |
| 15 | −6.6 | −8.7 | 2.1 | 318.4 | 5.1 | 4 | 2 | 1 | 139.0 | −4.1 |
| REF 1 | −11.7 | −8.8 | 2.9 | 284.4 | 3.7 | 0 | 2 | 1 | 126.2 | −4.1 |
| REF 2 | −6.5 | −6.4 | 0.1 | 305.5 | 3.2 | 0 | 2 | 1 | 93.7 | −3.4 |
| REF 3 | −6.1 | −9.8 | 3.7 | 853.9 | 3.7 | 10 | 14 | 4 | 357.9 | −3.2 |
| 16 | −7.2 | −8.3 | 1.1 | 304.5 | 4.4 | 1 | 2 | 1 | 134.9 | −4.3 |
| 17 | −7.6 | −8.0 | 0.4 | 300.4 | 4.6 | 2 | 2 | 0 | 133.6 | −5.5 |
| 18 | −5.3 | −8.2 | 2.9 | 332.5 | 5.3 | 3 | 2 | 1 | 147.9 | −4.8 |
| 19 | −5.6 | −8.2 | 2.6 | 332.5 | 5.3 | 4 | 2 | 1 | 147.9 | −5.3 |
| 20 | −5.8 | −8.2 | 2.4 | 302.5 | 4.3 | 2 | 2 | 1 | 134.2 | −5.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mvingu, B.K.; Nsiama, T.; Kanga, O.N.; Taba, K.M.; Kilembe, J.T.; Mputu, J.-N.K.; Garifo, S.; Henoumont, C.; Dibwe, D.F.; Mbala, B.M.; et al. Rapid Dereplication of Trunk Bark Constituents of Croton sylvaticus and Molecular Docking of Terpenoids from Three Congolese Croton Species. Int. J. Mol. Sci. 2025, 26, 4305. https://doi.org/10.3390/ijms26094305
Mvingu BK, Nsiama T, Kanga ON, Taba KM, Kilembe JT, Mputu J-NK, Garifo S, Henoumont C, Dibwe DF, Mbala BM, et al. Rapid Dereplication of Trunk Bark Constituents of Croton sylvaticus and Molecular Docking of Terpenoids from Three Congolese Croton Species. International Journal of Molecular Sciences. 2025; 26(9):4305. https://doi.org/10.3390/ijms26094305
Chicago/Turabian StyleMvingu, Bienvenu Kamalandua, Tienabe Nsiama, Obed Nsemi Kanga, Kalulu Muzele Taba, Jason Thambwe Kilembe, Jean-Noël Kanyinda Mputu, Sarah Garifo, Céline Henoumont, Dya Fita Dibwe, Blaise Mavinga Mbala, and et al. 2025. "Rapid Dereplication of Trunk Bark Constituents of Croton sylvaticus and Molecular Docking of Terpenoids from Three Congolese Croton Species" International Journal of Molecular Sciences 26, no. 9: 4305. https://doi.org/10.3390/ijms26094305
APA StyleMvingu, B. K., Nsiama, T., Kanga, O. N., Taba, K. M., Kilembe, J. T., Mputu, J.-N. K., Garifo, S., Henoumont, C., Dibwe, D. F., Mbala, B. M., & Laurent, S. (2025). Rapid Dereplication of Trunk Bark Constituents of Croton sylvaticus and Molecular Docking of Terpenoids from Three Congolese Croton Species. International Journal of Molecular Sciences, 26(9), 4305. https://doi.org/10.3390/ijms26094305