
Determination of Dronedarone and Debutyldronedarone in Human Plasma by HPLC-UV

Abstract
1. Introduction
2. Results
2.1. Method Development
2.1.1. Chromatographic Separation
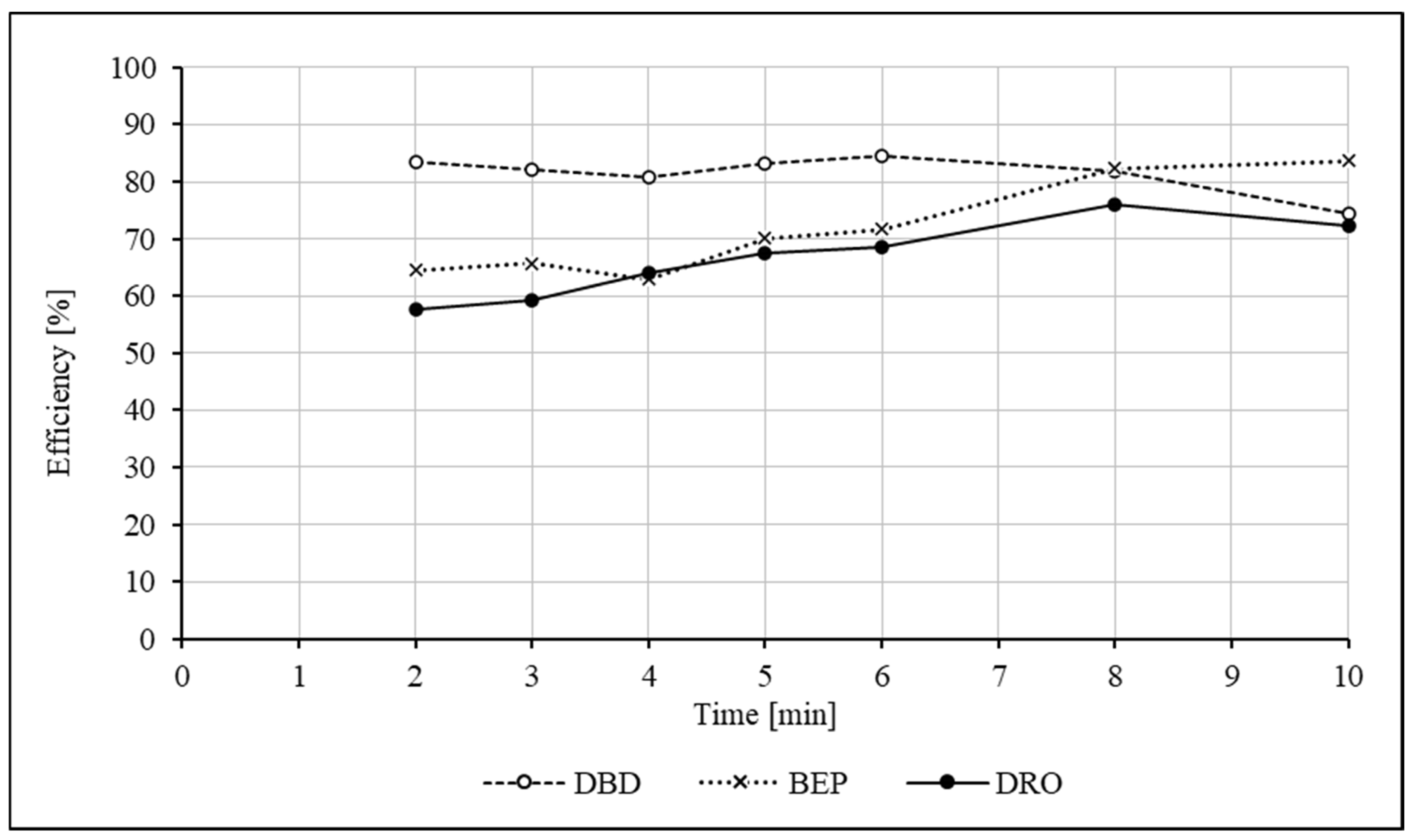
2.1.2. Extraction
2.1.3. Analytical Wavelength
2.2. Method Validation
2.2.1. Specificity
2.2.2. Calibration and Linearity
2.2.3. Precision and Accuracy
2.2.4. Limit of Quantification, Range, Dilution Integrity and Carry-Over
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Instrumentation
4.3. Chromatographic Conditions
4.4. Stock and Working Solutions, Calibration and Quality Controls
4.5. Sample Preparation
4.6. Method Validation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HPLC | high-performance liquid chromatography |
| UV | ultraviolet |
| LC-MS/MS | liquid chromatography–tandem mass spectrometry |
| LC-CN | cyanopropyl derivatized silica phase |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| C18 | octadecyl column |
| CYP | cytochrome P450 |
| PK | pharmacokinetic |
References
- Patel, C.; Yan, G.-X.; Kowey, P.R. Dronedarone. Circulation 2009, 120, 636–644. [Google Scholar] [CrossRef]
- Hohnloser, S.H.; Crijns, H.J.; van Eickels, M.; Gaudin, C.; Page, R.L.; Torp-Pedersen, C.; Connolly, S.J.; ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N. Engl. J. Med. 2009, 360, 668–678. [Google Scholar] [CrossRef]
- Vamos, M.; Hohnloser, S.H. Amiodarone and dronedarone: An update. Trends Cardiovasc. Med. 2016, 26, 597–602. [Google Scholar] [CrossRef]
- Blomström-Lundqvist, C.; Naccarelli, G.V.; McKindley, D.S.; Bigot, G.; Wieloch, M.; Hohnloser, S.H. Effect of dronedarone vs. placebo on atrial fibrillation progression: A post hoc analysis from ATHENA trial. Europace 2023, 25, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Multaq: EPAR—Product Information. Available online: https://www.ema.europa.eu (accessed on 10 March 2025).
- Duray, G.Z.; Torp-Pedersen, C.; Connolly, S.J.; Hohnloser, S.H. Effects of dronedarone on clinical outcomes in patients with lone atrial fibrillation: Pooled post hoc analysis from the ATHENA/EURIDIS/ADONIS studies. J. Cardiovasc. Electrophysiol. 2011, 22, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Le Heuzey, J.Y.; De Ferrari, G.M.; Radzik, D.; Santini, M.; Zhu, J.; Davy, J.M. A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: The DIONYSOS study. J. Cardiovasc. Electrophysiol. 2010, 21, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Køber, L.; Torp-Pedersen, C.; McMurray, J.J.; Gøtzsche, O.; Lévy, S.; Crijns, H.; Amlie, J.; Carlsen, J. Dronedarone Study Group, Increased mortality after dronedarone therapy for severe heart failure. N. Engl. J. Med. 2008, 358, 2678–2687. [Google Scholar] [CrossRef] [PubMed]
- Thind, M.; McKindley, D.S.; Reiffel, J.A.; Naccarelli, G.V.; Stewart, J.; Kowey, P.R. Predictors of dronedarone plasma drug concentrations and effect on atrial fibrillation/atrial flutter recurrence: Analyses from the EURIDIS and ADONIS studies. Clin. Cardiol. 2022, 45, 119–128. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency Recommends Restricting Use of Multaq. Available online: https://www.ema.europa.eu (accessed on 10 March 2025).
- Naccarelli, G.V.; McKindley, D.S.; Rashkin, J.; Ollier, C.; Reiffel, J.A. Bioavailability of dronedarone tablets administered with or without food in healthy participants. Am. Heart J. Plus 2024, 45, 100423. [Google Scholar] [CrossRef] [PubMed]
- Bolderman, R.W.; Hermans, J.R.R.; Maessen, J.G. Determination of the class III antiarrhythmic drugs dronedarone and amiodarone, and their principal metabolites in plasma and myocardium by high-performance liquid chromatography and UV-detection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Bin Jardan, Y.A.; Gabr, R.Q.; Brocks, D.R. Pharmacokinetics of dronedarone in rat using a newly developed high-performance liquid chromatographic assay method. Biomed. Chromatogr. 2014, 28, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Chadha, R.; Bali, A.; Bansal, G. Bioanalytical method validation for dronedarone and duloxetine in blood serum. J. AOAC Int. 2017, 100, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Yang, S.; Zhong, D.; Dai, X.; Chen, X. Simultaneous determination of dronedarone and its active metabolite debutyldronedarone in human plasma by liquid chromatography-tandem mass spectrometry: Application to a pharmacokinetic study. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 3071–3075. [Google Scholar] [CrossRef] [PubMed]
- Parekh, J.M.; Sanyal, M.; Yadav, M.; Shrivastav, P.S. Systematic evaluation of plasma phospholipids for reliable and precise determination of dronedarone and desbutyldronedarone by LC-MS/MS. Bioanalysis 2014, 6, 2635–2650. [Google Scholar] [CrossRef] [PubMed]
- Baek, I.H. Dose proportionality and pharmacokinetics of dronedarone following intravenous and oral administration to rat. Xenobiotica 2019, 49, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, M.; Xie, S.; Ye, X.; Xu, X. Simultaneous determination of amiodarone, dronedarone, and their principal metabolites in SD rat plasma by UPLC-MS/MS and its application in pharmacokinetics. Arab. J. Chem. 2021, 14, 103300. [Google Scholar] [CrossRef]
- ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis. 2022. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (accessed on 10 March 2025).
- Bioanalytical Method Validation. Guidance for Industry. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 10 March 2025).
- Kunicki, P.K.; Sitkiewicz, D. High-performance liquid chromatographic analysis of some antiarrhythmic drugs in human serum using cyanopropyl derivatized silica phase. J. Liq. Chromatogr. Relat. Technol. 1996, 19, 1169–1181. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| INTRA-ASSAY | INTER-ASSAY | ||||||
|---|---|---|---|---|---|---|---|
| Analyte | Level/Concentration Added [ng/mL] | Concentration Determined (mean ± SD) [ng/mL] | Precision (RSD) [%] | Inaccuracy [%] | Concentration Determined (mean ± SD) [ng/mL] | Precision (RSD) [%] | Inaccuracy [%] |
| DRO | LLOQ—10 | 8.8 ± 1.0 | 11.0 | −12.5 | 10.5 ± 1.4 | 13.7 | +4.5 |
| QC-L—30 | 31.3 ± 3.3 | 10.6 | +4.4 | 31.1 ± 1.5 | 4.9 | +3.8 | |
| QC-M—250 | 246.4 ± 5.8 | 2.4 | −1.4 | 245.4 ± 5.2 | 2.1 | −1.9 | |
| QC-H—800 | 824.3 ± 54.6 * | 6.6 * | +3.0 * | 812.6 ± 47.1 | 5.8 | +1.6 | |
| ULOQ—1000 | 1054.4 ± 25.2 | 2.4 | +5.4 | 1051.2 ± 22.8 | 2.2 | +5.1 | |
| DBD | LLOQ—10 | 8.8 ± 1.5 | 17.3 | −12.2 | 9.3 ± 1.3 | 13.8 | −6.9 |
| QC-L—30 | 30.7 ± 3.2 | 10.3 | +2.3 | 30.2 ± 3.3 | 11.0 | +0.8 | |
| QC-M—250 | 254.0 ± 13.5 | 5.3 | +1.6 | 254.2 ± 14.3 | 5.6 | +1.7 | |
| QC-H—800 | 865.7 ± 32.8 * | 3.8 * | +8.2 * | 881.7 ± 24.6 | 2.8 | +10.2 | |
| ULOQ—1000 | 1042.8 ± 47.8 | 4.6 | +4.3 | 1039.9 ± 43.3 | 4.2 | +4.0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunicki, P.K.; Stocki, A. Determination of Dronedarone and Debutyldronedarone in Human Plasma by HPLC-UV. Int. J. Mol. Sci. 2025, 26, 4304. https://doi.org/10.3390/ijms26094304
Kunicki PK, Stocki A. Determination of Dronedarone and Debutyldronedarone in Human Plasma by HPLC-UV. International Journal of Molecular Sciences. 2025; 26(9):4304. https://doi.org/10.3390/ijms26094304
Chicago/Turabian StyleKunicki, Paweł K., and Adam Stocki. 2025. "Determination of Dronedarone and Debutyldronedarone in Human Plasma by HPLC-UV" International Journal of Molecular Sciences 26, no. 9: 4304. https://doi.org/10.3390/ijms26094304
APA StyleKunicki, P. K., & Stocki, A. (2025). Determination of Dronedarone and Debutyldronedarone in Human Plasma by HPLC-UV. International Journal of Molecular Sciences, 26(9), 4304. https://doi.org/10.3390/ijms26094304