
Paw Skin as a Translational Model for Investigating Fibrotic and Inflammatory Wound Healing Defects in Recessive Dystrophic Epidermolysis Bullosa
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. C7-Hypomorphic Mice Exhibit Growth Impairments but No Increased Mortality at Three Months of Age
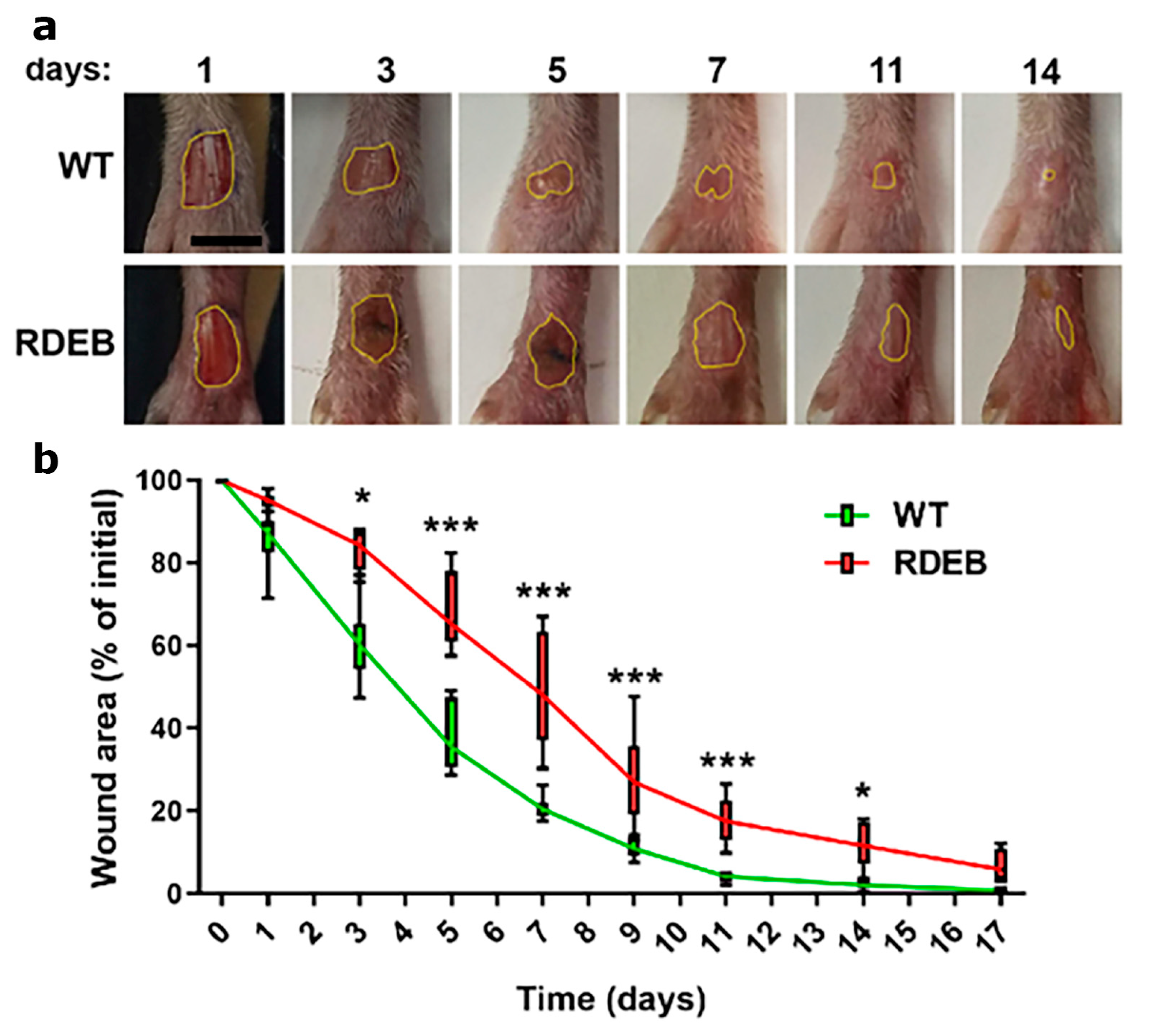
2.2. Delayed Wound Healing in C7-Hypomorphic Mouse Paw Skin
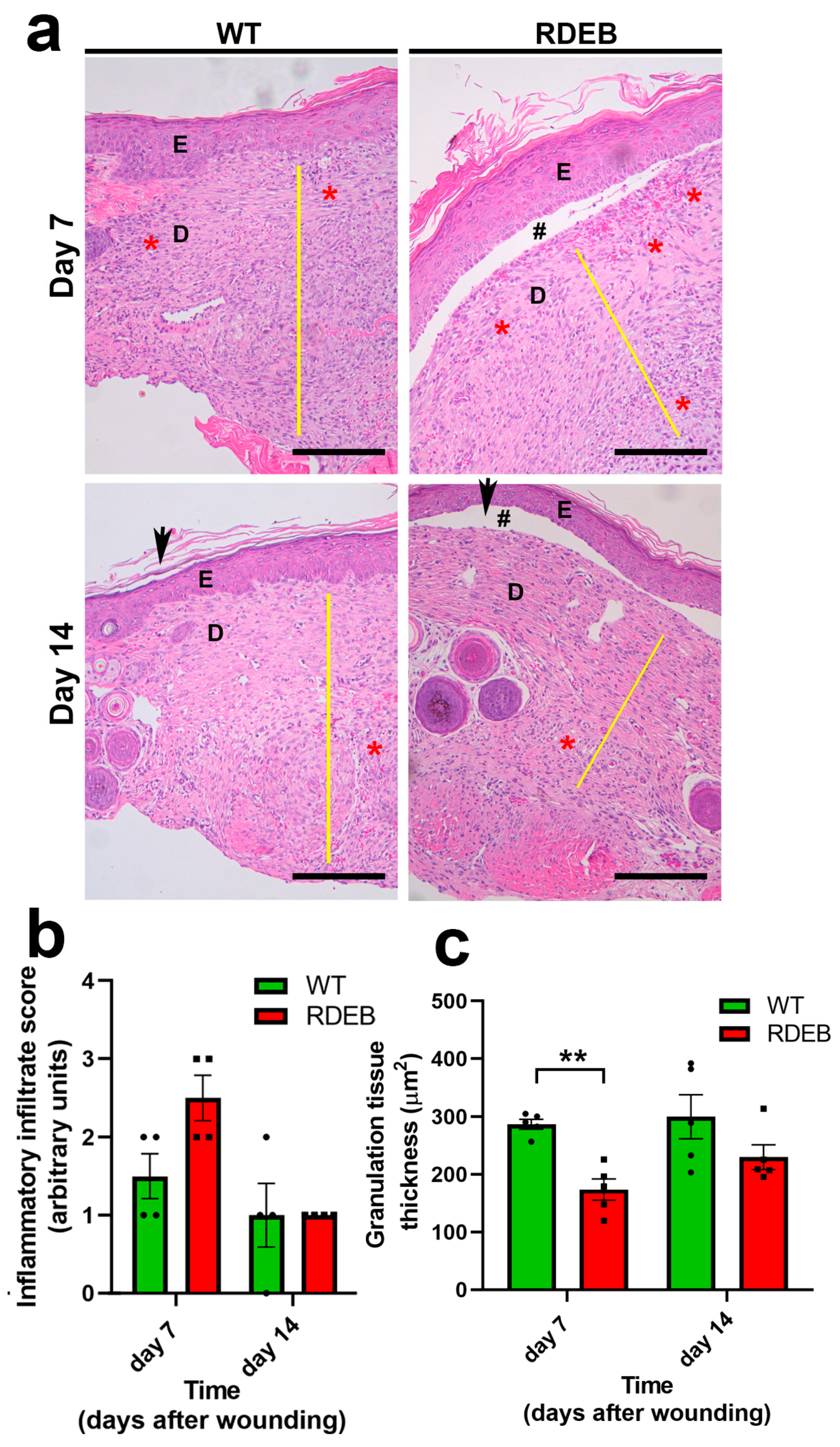
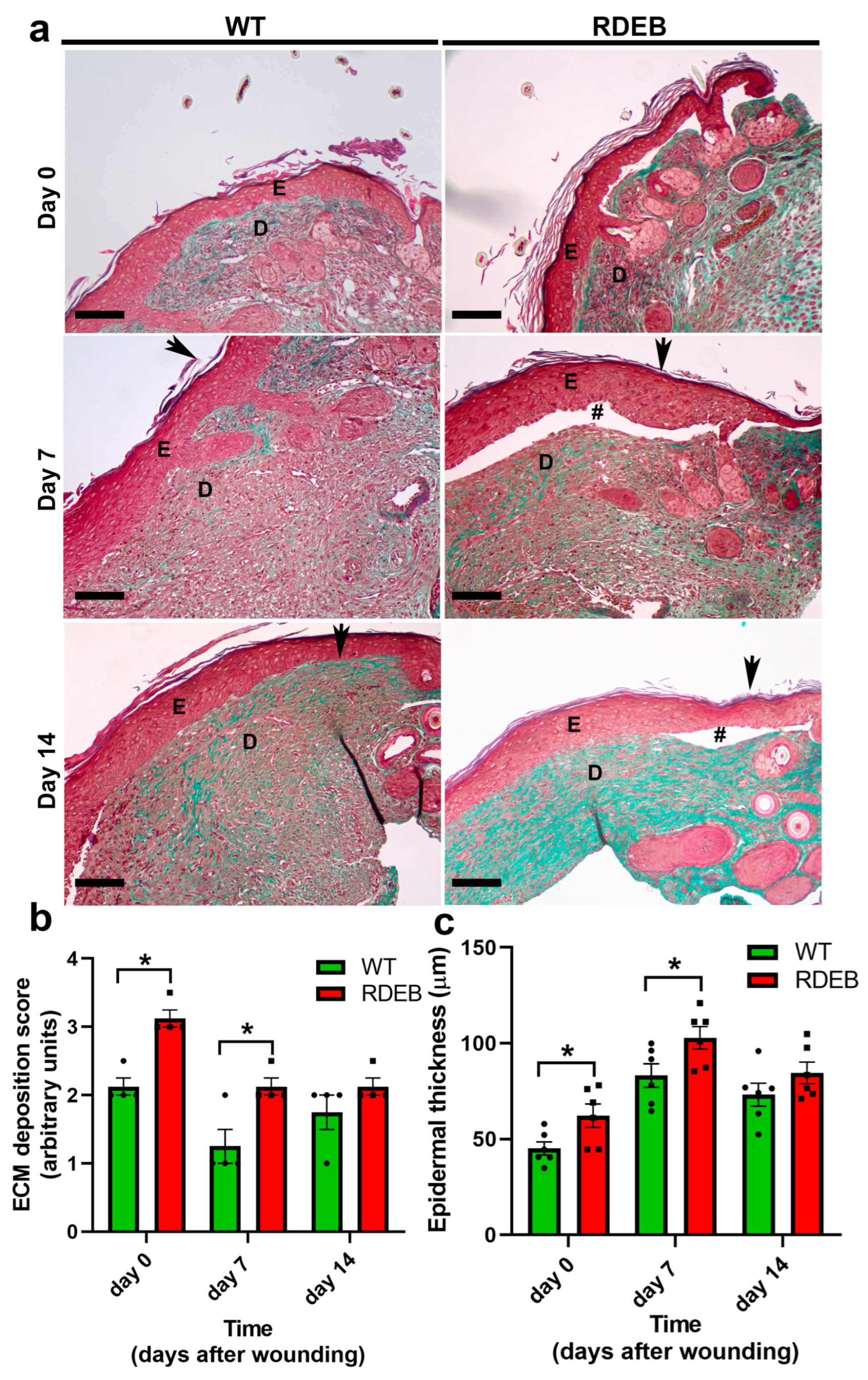
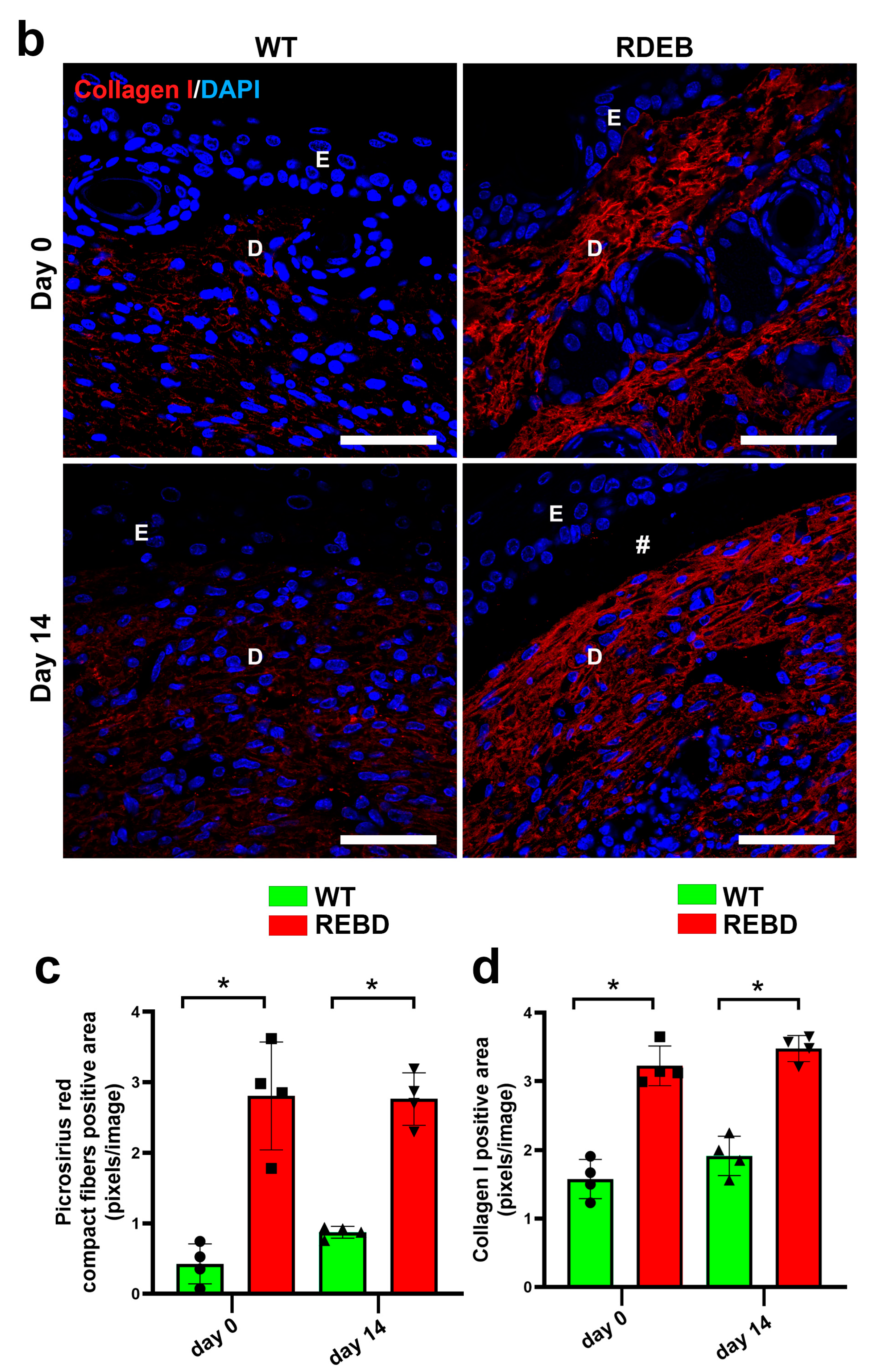
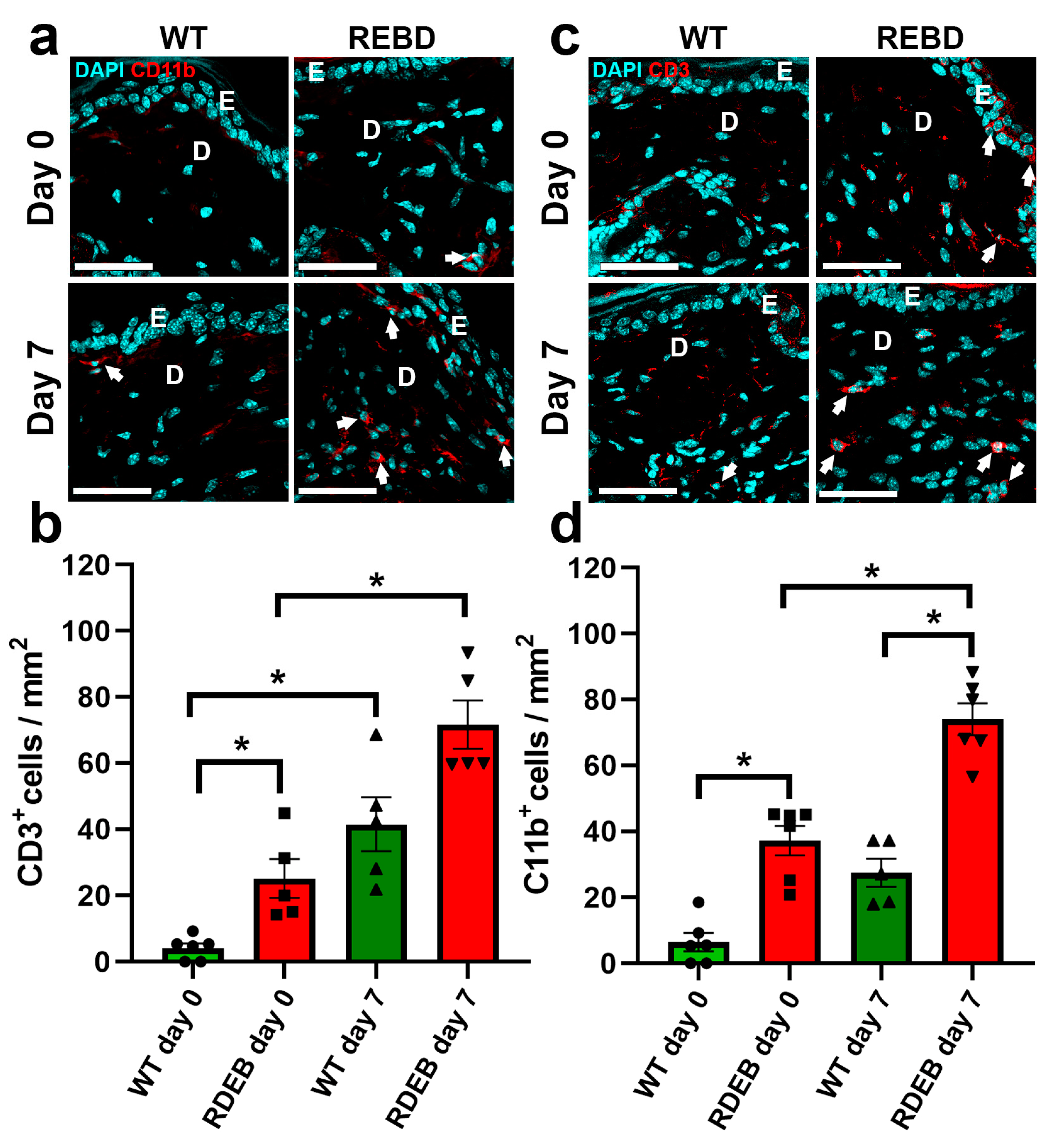
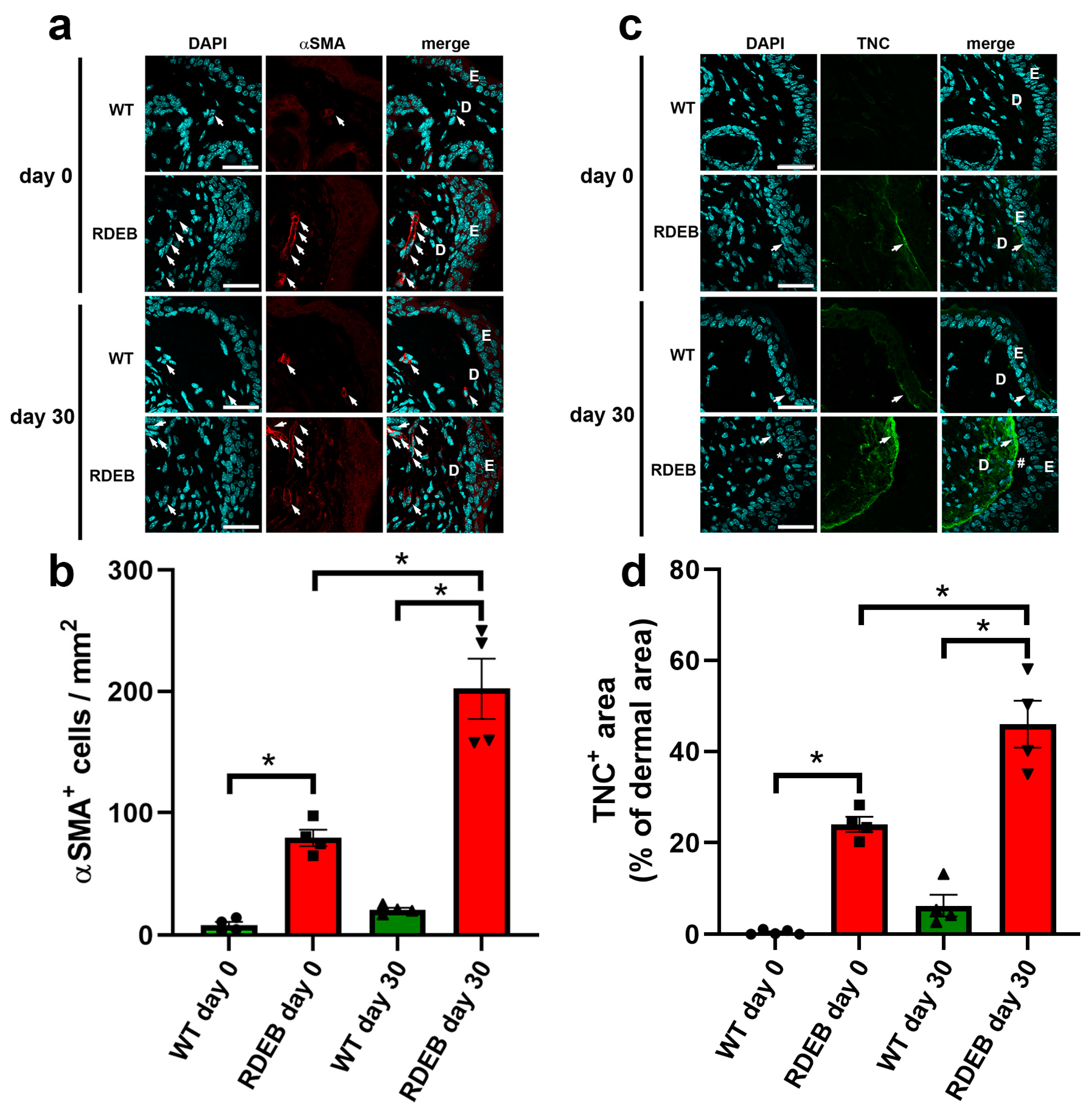
2.3. C7-Hypomorphic Mice Exhibit Increased Inflammation and Exacerbated Fibrotic Markers in Paw Skin
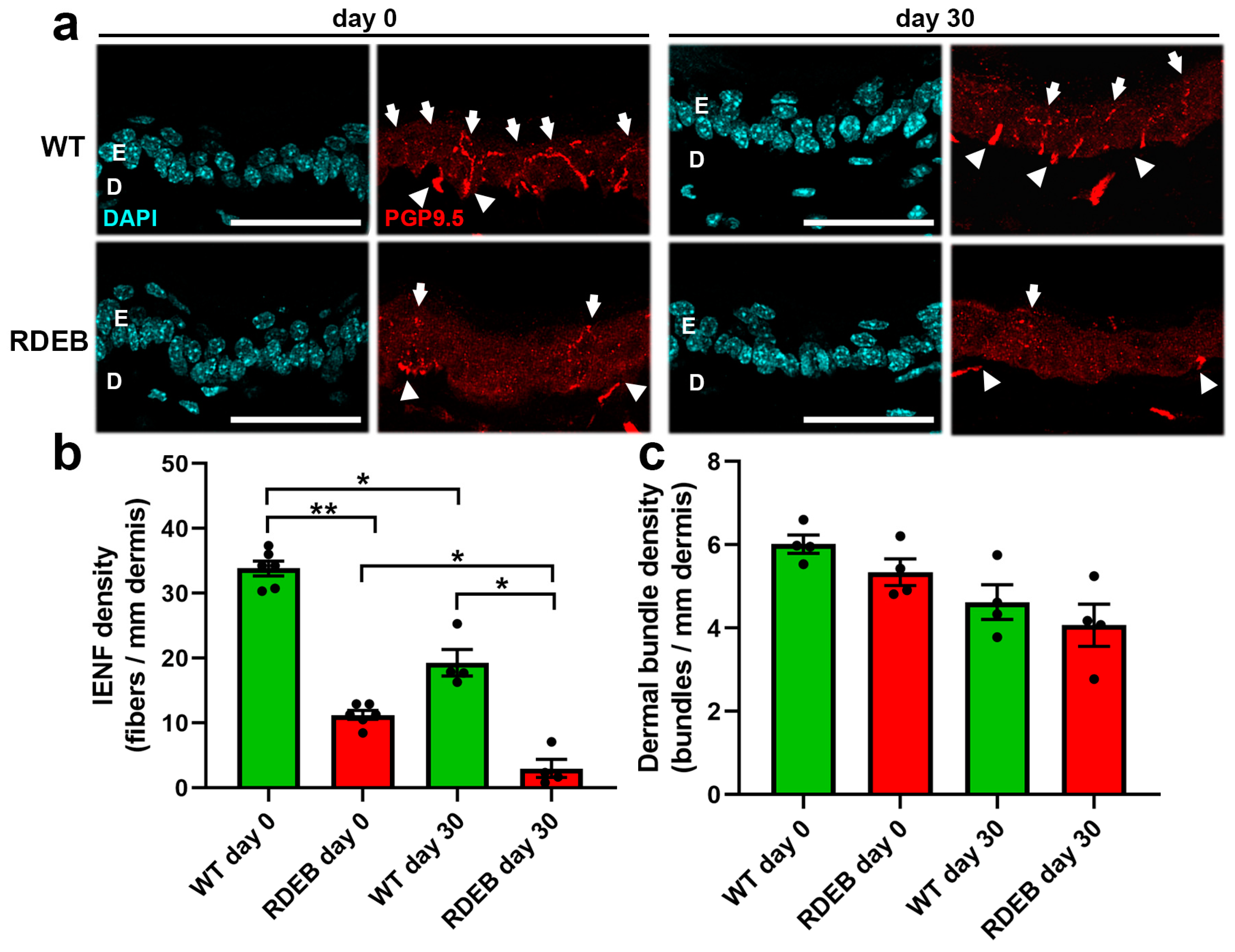
2.4. C7-Hypomorphic Mice Exhibit Reduced Intraepidermal Nerve Fiber Density in Paw Skin
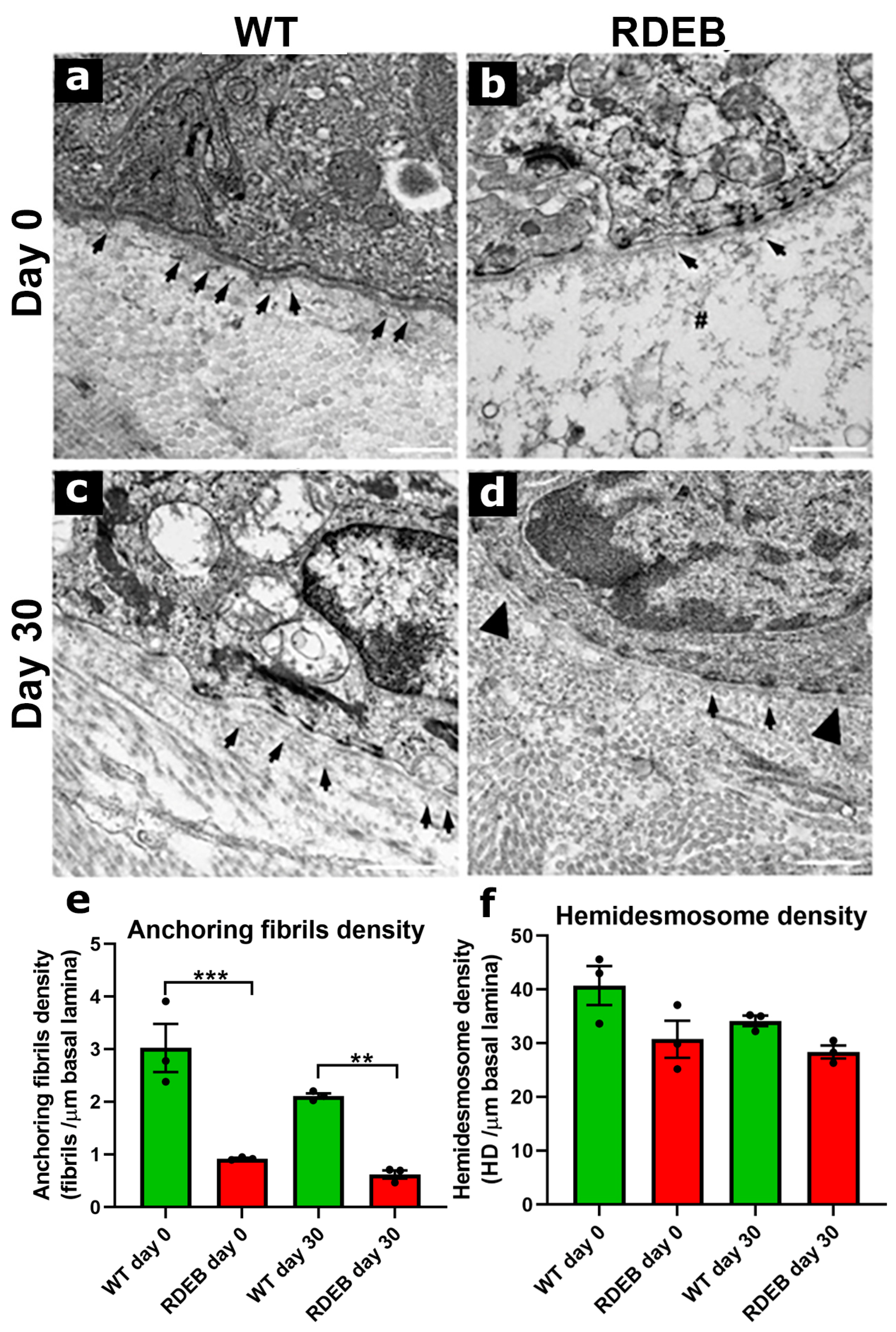
2.5. C7-Hypomorphic Mouse Paw Skin Exhibits Reduced Anchoring Fiber Density and Structural Alterations in the Dermal-Epidermal Junction After Injury
3. Discussion
3.1. Challenges in Developing a Comprehensive Therapy for RDEB
3.2. Wound Healing Under Inflammatory and Fibrotic Conditions
3.3. Limitations of the C7-Hypomorphic Mouse Model for Wound Healing Studies
3.4. Difficult-to-Heal Wounds in RDEB: Future Perspectives
3.5. Conclusions
4. Materials and Methods
4.1. Animals
4.2. Wound Induction and Measurement of Ulcer Area
4.3. Histological and Immunofluorescence Analysis of Wound Healing
4.4. Transmission Electron Microscopy Analysis of the Dermo-Epidermal Junction
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A-SMA | Alpha smooth muscle actin |
| C7 | type VII collagen |
| DEJ | dermal-epidermal junction |
| EB | epidermolysis bullosa |
| ECM | extracellular matrix |
| IENFs | intraepidermal nerve fibers |
| RDEB | recessive dystrophic EB |
| SCC | squamous cell carcinoma |
| TEM | transmission electron microscopy |
| TGFβ | transforming growth factor beta 1 |
| TNC | tenascin-C |
References
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Jevtić, M.; Löwa, A.; Nováčková, A.; Kováčik, A.; Kaessmeyer, S.; Erdmann, G.; Vávrová, K.; Hedtrich, S. Impact of intercellular crosstalk between epidermal keratinocytes and dermal fibroblasts on skin homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118722. [Google Scholar] [CrossRef] [PubMed]
- Van De Vyver, M.; Boodhoo, K.; Frazier, T.; Hamel, K.; Kopcewicz, M.; Levi, B.; Maartens, M.; Machcinska, S.; Nunez, J.; Pagani, C.; et al. Histology Scoring System for Murine Cutaneous Wounds. Stem Cells Dev. 2021, 30, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Leigh, I.M.; Eady, R.A.; Heagerty, A.H.; Purkis, P.E.; Whitehead, P.A.; Burgeson, R.E. Type VII collagen is a normal component of epidermal basement membrane, which shows altered expression in recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 1988, 90, 639–642. [Google Scholar] [CrossRef]
- Cianfarani, F.; Zambruno, G.; Castiglia, D.; Odorisio, T. Pathomechanisms of Altered Wound Healing in Recessive Dystrophic Epidermolysis Bullosa. Am. J. Pathol. 2017, 187, 1445–1453. [Google Scholar] [CrossRef]
- Laimer, M.; Prodinger, C.; Bauer, J.W. Hereditary epidermolysis bullosa. J. Dtsch. Dermatol. Ges. 2015, 13, 1125–1133. [Google Scholar] [CrossRef]
- Fine, J.-D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef]
- Cianfarani, F.; De Domenico, E.; Nyström, A.; Mastroeni, S.; Abeni, D.; Baldini, E.; Ulisse, S.; Uva, P.; Bruckner-Tuderman, L.; Zambruno, G.; et al. Decorin counteracts disease progression in mice with recessive dystrophic epidermolysis bullosa. Matrix Biol. 2019, 81, 3–16. [Google Scholar] [CrossRef]
- Nyström, A.; Velati, D.; Mittapalli, V.R.; Fritsch, A.; Kern, J.S.; Bruckner-Tuderman, L. Collagen VII plays a dual role in wound healing. J. Clin. Investig. 2013, 123, 3498–3509. [Google Scholar] [CrossRef]
- South, A.P.; Laimer, M.; Gueye, M.; Sui, J.Y.; Eichenfield, L.F.; Mellerio, J.E.; Nyström, A. Type VII Collagen Deficiency in the Oncogenesis of Cutaneous Squamous Cell Carcinoma in Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2023, 143, 2108–2119. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wang, W.; Morales-Nebreda, L.; Feng, G.; Wu, M.; Zhou, X.; Lafyatis, R.; Lee, J.; Hinchcliff, M.; Feghali-Bostwick, C.; et al. Tenascin-C drives persistence of organ fibrosis. Nat. Commun. 2016, 7, 11703. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, H.; Patel, S.B.; Pastar, I. The Role of TGFbeta Signaling in Wound Epithelialization. Adv. Wound Care 2014, 3, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Nuschke, A.; Yates, C.C. Skin tissue repair: Matrix microenvironmental influences. Matrix Biol. 2016, 49, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef]
- Nukuda, A.; Sasaki, C.; Ishihara, S.; Mizutani, T.; Nakamura, K.; Ayabe, T.; Kawabata, K.; Haga, H. Stiff substrates increase YAP-signaling-mediated matrix metalloproteinase-7 expression. Oncogenesis 2015, 4, e165. [Google Scholar] [CrossRef]
- Fritsch, A.; Loeckermann, S.; Kern, J.S.; Braun, A.; Bösl, M.R.; Bley, T.A.; Schumann, H.; von Elverfeldt, D.; Paul, D.; Erlacher, M.; et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J. Clin. Investig. 2008, 118, 1669–1679. [Google Scholar] [CrossRef]
- Nyström, A.; Thriene, K.; Mittapalli, V.; Kern, J.S.; Kiritsi, D.; Dengjel, J.; Bruckner-Tuderman, L. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol. Med. 2015, 7, 1211–1228. [Google Scholar] [CrossRef]
- Ingen-Housz-Oro, S.; Blanchet-Bardon, C.; Vrillat, M.; Dubertret, L. Vitamin and trace metal levels in recessive dystrophic epidermolysis bullosa. J. Eur. Acad. Dermatol. Venereol. 2004, 18, 649–653. [Google Scholar] [CrossRef]
- Reimer, A.; Hess, M.; Schwieger-Briel, A.; Kiritsi, D.; Schauer, F.; Schumann, H.; Bruckner-Tuderman, L.; Has, C. Natural history of growth and anaemia in children with epidermolysis bullosa: A retrospective cohort study. Br. J. Dermatol. 2020, 182, 1437–1448. [Google Scholar] [CrossRef]
- Zomer, H.D.; Trentin, A.G. Skin wound healing in humans and mice: Challenges in translational research. J. Dermatol. Sci. 2018, 90, 3–12. [Google Scholar] [CrossRef]
- Griffiths, C.E.; Barker, J.N.W.N. Pathogenesis and clinical features of psoriasis. Lancet 2007, 370, 263–271. [Google Scholar] [CrossRef]
- Matar, D.Y.; Ng, B.; Darwish, O.; Wu, M.; Orgill, D.P.; Panayi, A.C. Skin Inflammation with a Focus on Wound Healing. Adv. Wound Care 2023, 12, 269–287. [Google Scholar] [CrossRef] [PubMed]
- Raziyeva, K.; Kim, Y.; Zharkinbekov, Z.; Kassymbek, K.; Jimi, S.; Saparov, A. Immunology of Acute and Chronic Wound Healing. Biomolecules 2021, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.Z.; Stevenson, A.W.; Prêle, C.M.; Fear, M.W.; Wood, F.M. The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing. Biomedicines 2020, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bruckner-Tuderman, L. Injury- and inflammation-driven skin fibrosis: The paradigm of epidermolysis bullosa. Matrix Biol. 2018, 68–69, 547–560. [Google Scholar] [CrossRef]
- Udalova, I.A.; Ruhmann, M.; Thomson, S.J.; Midwood, K.S. Expression and immune function of tenascin-C. Crit. Rev. Immunol. 2011, 31, 115–145. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Midwood, K.S.; Varga, J. Tenascin-C in fibrosis in multiple organs: Translational implications. Semin. Cell Dev. Biol. 2022, 128, 130–136. [Google Scholar] [CrossRef]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Suchindran, C. Assessment of mobility, activities and pain in different subtypes of epidermolysis bullosa. Clin. Exp. Dermatol. 2004, 29, 122–127. [Google Scholar] [CrossRef]
- Jeffs, E.; Pillay, E.I.; Ledwaba-Chapman, L.; Bisquera, A.; Robertson, S.J.; McGrath, J.A.; Wang, Y.; Martinez, A.E.; Mellerio, J.E. Pain in recessive dystrophic epidermolysis bullosa (RDEB): Findings of the Prospective Epidermolysis Bullosa Longitudinal Evaluation Study (PEBLES). Orphanet J. Rare Dis. 2024, 19, 375. [Google Scholar] [CrossRef]
- von Bischhoffshausen, S.; Ivulic, D.; Alvarez, P.; Schuffeneger, V.C.; Idiaquez, J.; Fuentes, C.; Morande, P.; Fuentes, I.; Palisson, F.; Bennett, D.L.H.; et al. Recessive dystrophic epidermolysis bullosa results in painful small fibre neuropathy. Brain 2017, 140, 1238–1251. [Google Scholar] [CrossRef]
- Ebenezer, G.J.; Laast, V.A.; Dearman, B.; Hauer, P.; Tarwater, P.M.; Adams, R.J.; Zink, M.C.; McArthur, J.C.; Mankowski, J.L. Altered cutaneous nerve regeneration in a simian immunodeficiency virus/macaque intracutaneous axotomy model. J. Comp. Neurol. 2009, 514, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Díaz, P.; Muñoz, D.; Espinoza, F.; Nystrom, A.; Fuentes, I.; Ezquer, M.; Bennett, D.L.; Calvo, M. Characterisation of the pathophysiology of neuropathy and sensory dysfuncion in a mouse model of recessive dystrophic epidermolysis bullosa. Pain 2022, 163, 2052–2060. [Google Scholar] [CrossRef]
- Amano, S. Possible involvement of basement membrane damage in skin photoaging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.; Rittie, L. Restoration of the basement membrane after wounding: A hallmark of young human skin altered with aging. J. Cell Commun. Signal. 2018, 12, 401–411. [Google Scholar] [CrossRef]
- Behm, B.; Babilas, P.; Landthaler, M.; Schreml, S. Cytokines, chemokines and growth factors in wound healing. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 812–820. [Google Scholar] [CrossRef]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.V.; Soulika, A.M. The Dynamics of the Skin’s Immune System. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef]
- Odorisio, T.; Di Salvio, M.; Orecchia, A.; Di Zenzo, G.; Piccinni, E.; Cianfarani, F.; Travaglione, A.; Uva, P.; Bellei, B.; Conti, A.; et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-beta signalling in modifying disease severity. Hum. Mol. Genet. 2014, 23, 3907–3922. [Google Scholar] [CrossRef]
- Berberich, B.; Thriene, K.; Gretzmeier, C.; Kühl, T.; Bayer, H.; Athanasiou, I.; Rafei-Shamsabadi, D.A.; Bruckner-Tuderman, L.; Nyström, A.; Kiritsi, D.; et al. Proteomic Profiling of Fibroblasts Isolated from Chronic Wounds Identifies Disease-Relevant Signaling Pathways. J. Investig. Dermatol. 2020, 140, 2280–2290. [Google Scholar] [CrossRef]
- Bernasconi, R.; Thriene, K.; Romero-Fernández, E.; Gretzmeier, C.; Kühl, T.; Maler, M.; Nauroy, P.; Kleiser, S.; Rühl-Muth, A.C.; Stumpe, M.; et al. Pro-inflammatory immunity supports fibrosis advancement in epidermolysis bullosa: Intervention with Ang-(1–7). EMBO Mol. Med. 2021, 13, e14392. [Google Scholar] [CrossRef]
- Küttner, V.; Mack, C.; Gretzmeier, C.; Bruckner-Tuderman, L.; Dengjel, J. Loss of collagen VII is associated with reduced transglutaminase 2 abundance and activity. J. Investig. Dermatol. 2014, 134, 2381–2389. [Google Scholar] [CrossRef]
- Guo, J.; Lin, Q.; Shao, Y.; Rong, L.; Zhang, D. miR-29b promotes skin wound healing and reduces excessive scar formation by inhibition of the TGF-beta1/Smad/CTGF signaling pathway. Can. J. Physiol. Pharmacol. 2017, 95, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Kotzki, S.; Savina, Y.; Bouvet, R.; Gil, H.; Blaise, S.; Cracowski, J.-L.; Roustit, M. Iontophoresis of treprostinil promotes wound healing in a murine model of scleroderma-related ulcers. Rheumatology 2022, 61, 2704–2708. [Google Scholar] [CrossRef]
- Maeda, T.; Yamamoto, T.; Imamura, T.; Tsuboi, R. Impaired wound healing in bleomycin-induced murine scleroderma: A new model of wound retardation. Arch. Dermatol. Res. 2016, 308, 87–94. [Google Scholar] [CrossRef]
- Yang, L.; Chan, T.; Demare, J.; Iwashina, T.; Ghahary, A.; Scott, P.G.; Tredget, E.E. Healing of burn wounds in transgenic mice overexpressing transforming growth factor-beta 1 in the epidermis. Am. J. Pathol. 2001, 159, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.L.; Caley, M.P.; Moore, K.; Szentpetery, Z.; Marsh, S.T.; Murrell, D.F.; Kim, M.H.; Avari, M.; McGrath, J.A.; Cerio, R.; et al. Suppression of TGFbeta and Angiogenesis by Type VII Collagen in Cutaneous SCC. J. Natl. Cancer Inst. 2016, 108, djv293. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bruckner-Tuderman, L.; Kiritsi, D. Dystrophic Epidermolysis Bullosa: Secondary Disease Mechanisms and Disease Modifiers. Front. Genet. 2021, 12, 737272. [Google Scholar] [CrossRef]
- Wong, V.W.; Sorkin, M.; Glotzbach, J.P.; Longaker, M.T.; Gurtner, G.C. Surgical approaches to create murine models of human wound healing. J. Biomed. Biotechnol. 2011, 2011, 969618. [Google Scholar] [CrossRef]
- Dunn, L.; Prosser, H.C.; Tan, J.T.; Vanags, L.Z.; Ng, M.K.; Bursill, C.A. Murine model of wound healing. J. Vis. Exp. 2013, 75, e50265. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, X.; Ge, J.; Tredget, E.E.; Wu, Y. The mouse excisional wound splinting model, including applications for stem cell transplantation. Nat. Protoc. 2013, 8, 302–309. [Google Scholar] [CrossRef]
- Grada, A.; Mervis, J.; Falanga, V. Research Techniques Made Simple: Animal Models of Wound Healing. J. Investig. Dermatol. 2018, 138, 2095–2105. [Google Scholar] [CrossRef]
- Yang, G.; Cintina, I.; Pariser, A.; Oehrlein, E.; Sullivan, J.; Kennedy, A. The national economic burden of rare disease in the United States in 2019. Orphanet J. Rare Dis. 2022, 17, 163. [Google Scholar] [CrossRef] [PubMed]
- Chateau, A.V.; Blackbeard, D.; Aldous, C. The impact of epidermolysis bullosa on the family and healthcare practitioners: A scoping review. Int. J. Dermatol. 2023, 62, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Dissemond, J.; Romanelli, M. Inflammatory skin diseases and wounds. Br. J. Dermatol. 2022, 187, 167–177. [Google Scholar] [CrossRef]
- Babakoohi, S.; Sipe, A.; Zamanifekri, M.; Hunter, W.D. Small fiber neuropathy in epidermolysis bullosa simplex. JAAD Case Rep. 2024, 48, 23–25. [Google Scholar] [CrossRef]
- Bardhan, A.; Bruckner-Tuderman, L.; Chapple, I.L.; Fine, J.D.; Harper, N.; Has, C.; Magin, T.M.; Marinkovich, M.P.; Marshall, J.F.; McGrath, J.A.; et al. Epidermolysis bullosa. Nat. Rev. Dis. Primers 2020, 6, 78. [Google Scholar] [CrossRef]
- Thien, C.I.; Bessa, V.R.; Miotto, I.Z.; Samorano, L.P.; Rivitti-Machado, M.C.; de Oliveira, Z.N.P. Hereditary epidermolysis bullosa: Clinical-epidemiological profile of 278 patients at a tertiary hospital in Sao Paulo, Brazil. An. Bras. Dermatol. 2024, 99, 380–390. [Google Scholar] [CrossRef]
- Lau, T.; Sahota, D.; Lau, C.; Chan, C.; Lam, F.; Ho, Y.; Fung, K.; Lau, C.; Leung, P. An in vivo investigation on the wound-healing effect of two medicinal herbs using an animal model with foot ulcer. Eur. Surg. Res. 2008, 41, 15–23. [Google Scholar] [CrossRef]
- Olivares, B.; Martínez, F.A.; Ezquer, M.; Morales, B.J.; Fuentes, I.; Calvo, M.; Campodónico, P.R. Betaine-urea deep eutectic solvent improves imipenem antibiotic activity. J. Mol. Liq. 2022, 350, 118551. [Google Scholar] [CrossRef]
- Shi, R.; Jin, Y.; Cao, C.; Han, S.; Shao, X.; Meng, L.; Cheng, J.; Zhang, M.; Zheng, J.; Xu, J.; et al. Localization of human adipose-derived stem cells and their effect in repair of diabetic foot ulcers in rats. Stem Cell Res. Ther. 2016, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, C.; Contador, D.; Campero, M.; Ezquer, M.; Ezquer, F. Characterization of diabetic neuropathy progression in a mouse model of type 2 diabetes mellitus. Biol. Open 2018, 7, bio036830. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, C.; Contador, D.; Díaz, D.; Cárcamo, C.; Santapau, D.; Lobos-Gonzalez, L.; Acosta, C.; Campero, M.; Carpio, D.; Gabriele, C.; et al. Human adipose-derived mesenchymal stem cell-conditioned medium ameliorates polyneuropathy and foot ulceration in diabetic BKS db/db mice. Stem Cell Res. Ther. 2020, 11, 168. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Gregorio, C.; Ramos-Gonzalez, G.; Morales-Catalán, B.; Ezquer, F.; Ezquer, M. Paw Skin as a Translational Model for Investigating Fibrotic and Inflammatory Wound Healing Defects in Recessive Dystrophic Epidermolysis Bullosa. Int. J. Mol. Sci. 2025, 26, 4281. https://doi.org/10.3390/ijms26094281
De Gregorio C, Ramos-Gonzalez G, Morales-Catalán B, Ezquer F, Ezquer M. Paw Skin as a Translational Model for Investigating Fibrotic and Inflammatory Wound Healing Defects in Recessive Dystrophic Epidermolysis Bullosa. International Journal of Molecular Sciences. 2025; 26(9):4281. https://doi.org/10.3390/ijms26094281
Chicago/Turabian StyleDe Gregorio, Cristian, Giselle Ramos-Gonzalez, Bernardo Morales-Catalán, Fernando Ezquer, and Marcelo Ezquer. 2025. "Paw Skin as a Translational Model for Investigating Fibrotic and Inflammatory Wound Healing Defects in Recessive Dystrophic Epidermolysis Bullosa" International Journal of Molecular Sciences 26, no. 9: 4281. https://doi.org/10.3390/ijms26094281
APA StyleDe Gregorio, C., Ramos-Gonzalez, G., Morales-Catalán, B., Ezquer, F., & Ezquer, M. (2025). Paw Skin as a Translational Model for Investigating Fibrotic and Inflammatory Wound Healing Defects in Recessive Dystrophic Epidermolysis Bullosa. International Journal of Molecular Sciences, 26(9), 4281. https://doi.org/10.3390/ijms26094281