
Mitochondrial Dysfunction as a Pathogenesis and Therapeutic Strategy for Metabolic-Dysfunction-Associated Steatotic Liver Disease

 , , ,
, , ,
Abstract
1. Introduction
2. Physiological Functions of Mitochondria
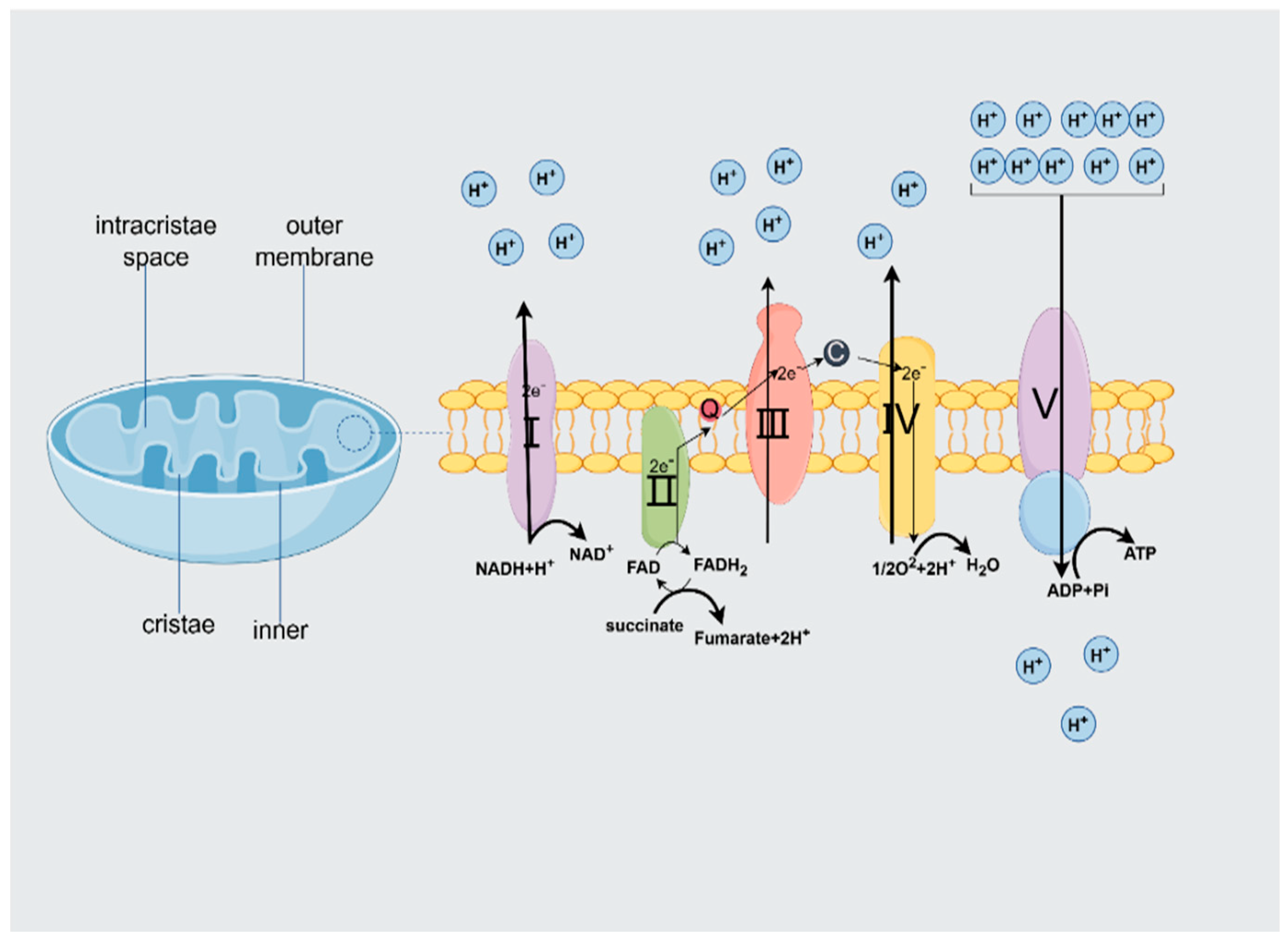
2.1. Mitochondrial Structure
2.2. ATP Production
2.2.1. Structural and Functional Characterization of the Mitochondrial Respiratory Chain
2.2.2. Factors Regulating the Efficiency of ATP Production
3. Mitochondria and MASLD
3.1. Mitochondria Function as Pivotal Metabolic Hubs Within Hepatic Tissue
3.1.1. Mitochondria and Their Function in Hepatocytes
3.1.2. Mitochondrial Dysfunction in MASLD
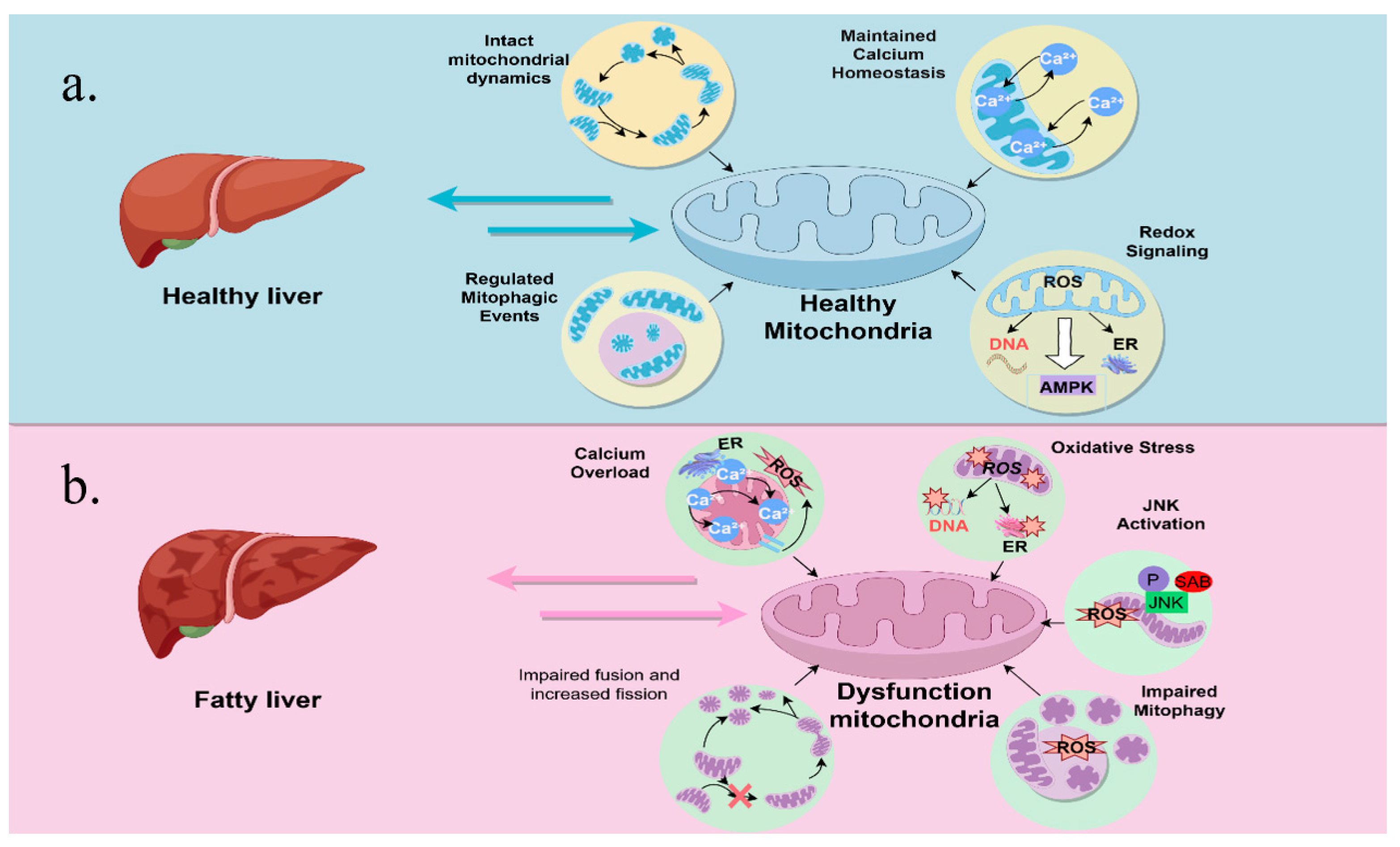
3.2. Mitochondrial Adaptation
3.3. Impaired Mitochondrial Quality Control
3.4. Mitochondrial Autophagy Defects
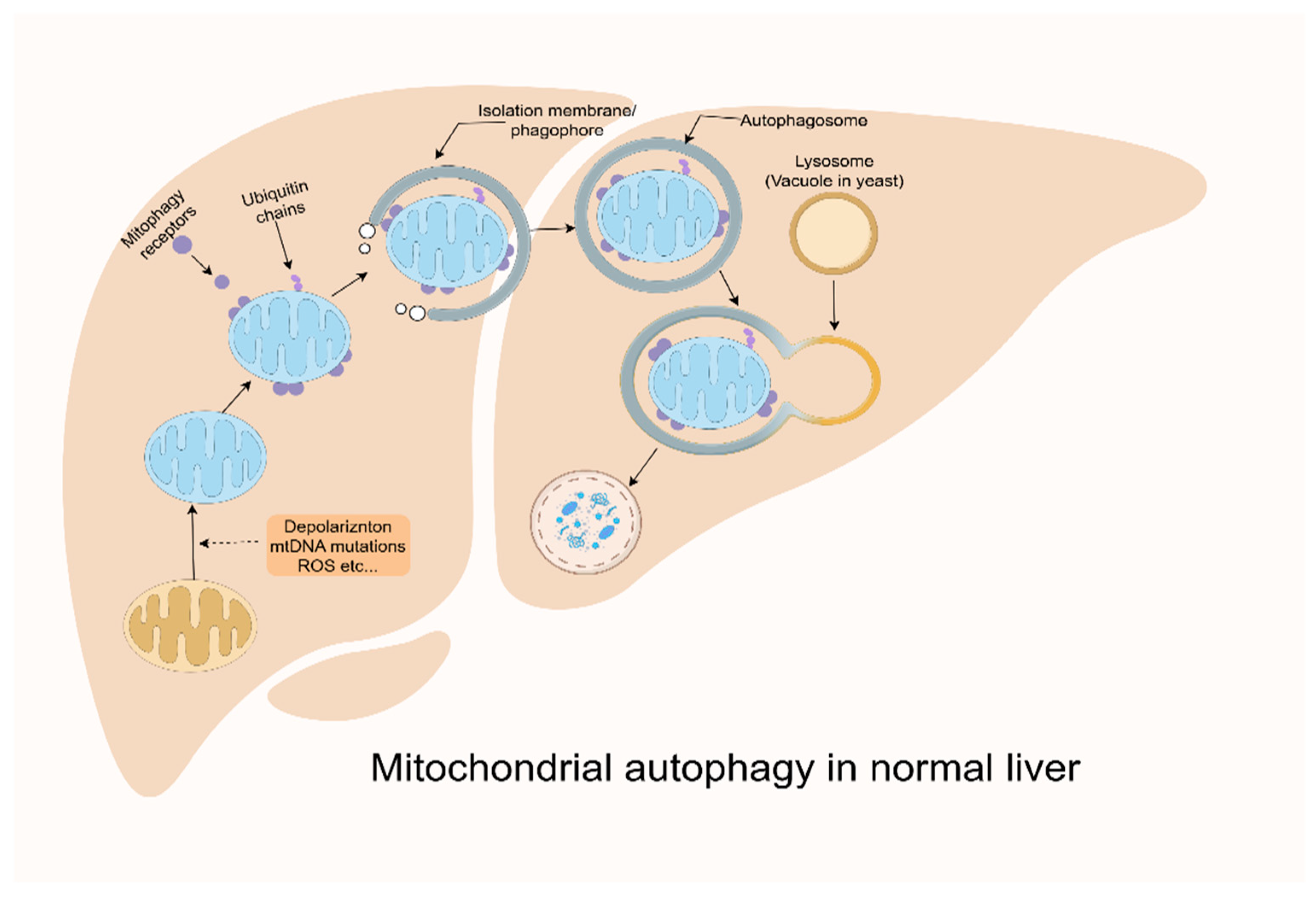
3.5. Reactive Oxygen Species and Oxidative Stress
3.6. DNA Methylation in Mitochondria
4. Mitochondrial-Based MASLD Treatment
4.1. Lifestyle Interventions
4.1.1. Dietary Modification
4.1.2. Physical Activity
4.1.3. Weight Loss
4.2. Pharmacologic and Other Therapies
4.2.1. Antidiabetics
4.2.2. Bile Acids
4.2.3. Mitochondrial-Targeting Agents
4.2.4. Mitochondrial Transplantation

{kind=link}
{kind=link}
{kind=link}
| Categories | Mechanism of Action | Clinical Effectiveness |
|---|---|---|
| Mediterranean diet [105,167,168] | Regulates lipid metabolism; reduces inflammation; improves insulin resistance. | Enhances mitochondrial biogenesis and function, resulting in improved metabolic health and reduced liver fat accumulation. |
| Low-calorie ketogenic diet [110] | Reduces liver fat synthesis; promotes fatty acid oxidation; reduces inflammatory response and oxidative stress. | Increases lipocalin; decreases levels of TNF-α, glycosylated hemoglobin (HbA1c), and lipids; increases levels of HDL and the inflammatory mediator IL-10. |
| Physical exercise [122] | Promotes fat oxidation and decomposition; improves insulin receptor function; improves insulin signaling pathway. | Mitochondrial oxidative capacity promotes fatty acid oxidation, mitochondrial biogenesis, and autophagy and reduces HDL. |
| Lose weight [137] | Enhances lipolysis and transport; increases insulin sensitivity; regulates blood glucose and lipid metabolism. | Mitochondrial function in patients with MASLD is associated with reduced hepatic inflammation and suppression of protein levels associated with hepatic neolipogenesis. |
| Antidiabetic drugs [138] | AMP-activated protein kinase; AMPK-dependent changes in cellular energy charge. | There is little beneficial effect on hepatic steatosis and inflammation and no effect on hepatic fibrosis and MASH regression. |
| Farnesol X receptor [152] | Promotes bile-acid-mediated lipid excretion; regulates glucose metabolism-related genes. | Reduces VLDL secretion and serum TG and counteracts hepatic steatosis. |
| Astragalus [162] | Mitochondria-targeted agonist. | Upregulation of Nrf2, PPAR-α, and HO-1; downregulation of mTORC1 and SREBP-1c; activation of AMPK and autophagy. |
| Mitochondrial transplantation [164,169] | Mitochondria-targeted agonist. | Direct restoration of mitochondrial function in hepatocytes. |
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| MASLD | Metabolic-dysfunction-associated steatotic liver disease |
| NAFLD | Non-alcoholic fatty liver disease |
| MAFLD | Metabolic-dysfunction-associated fatty liver disease |
| ATP | Adenosine triphosphate |
| OXPHOS | Oxidative phosphorylation |
| ADP | Adenosine diphosphate |
| TCA | Tricarboxylic acid |
| MASH | Metabolic-dysfunction-associated steatohepatitis |
| VDAC | Voltage-dependent anion-selective channels |
| O2 | Oxygen |
| CO2 | Carbon dioxide |
| NADH | Nicotinamide adenine dinucleotide |
| FADH2 | Flavin adenine dinucleotid |
| MPT | Mitochondrial permeability transition |
| ER | Endoplasmic reticulum |
| UPR | Unfolded protein response |
| JNK | C-Jun N-Terminal Kinase |
| UPRmt | Mitochondrial unfolded protein response |
| CHOP | C/EBP homologous protein |
| ATF4 | Activating transcription factor 4 |
| ATF5 | Activating transcription factor 5 |
| AMPK | AMP-activated protein kinase |
| ULK1 | Unc-51-like autophagy activating kinase 1 |
| MPTP | Mitochondrial permeability transition pore |
| DRP1 | Dynamin-related protein 1 |
| BCL2 | B-cell lymphoma 2 |
| FUNDC1 | FUN14 domain-containing protein 1 |
| GLP-1 | Glucagon-like peptide-1 |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| Mst1 | Macrophage-stimulated 1 |
| OA | Oleic acid |
| mtDNA | Mitochondrial DNA |
| TH | Thyroid hormone |
| p62 | Sequestosome 1 |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| SREBP1 | Sterol regulatory element binding protein 1 |
| FAS | Fatty acid synthase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| PNPLA3 | Patatin-like phospholipase domain-containing 3 |
| TM6SF2 | Transmembrane 6 superfamily member 2 |
| MBOAT7 | Membrane-bound O-acyltransferase structural domain 7 |
| GCKR | Glucokinase regulator |
| HSD17B13 | Hydroxysteroid 17-beta dehydrogenase-13 |
| tRNAs | Transfer RNAs |
| rRNAs | Ribosomal RNAs |
| Drp1 | Dynamin-related protein 1 |
| TLR9 | Toll-like receptor 9 |
| HSP 60 | Heat shock protein 60 |
| SNP | Single nucleotide polymorphism |
| SOD2 | Superoxide dismutase 2 |
| FIS1 | Fission, mitochondrial 1 |
| PUFA | Polyunsaturated fatty acids |
| MDA | Malondialdehyde |
| 4-HNE | 4-hydroxy-2-nonenal |
| MD | Mediterranean diet |
| LDL | Low-density lipoprotein |
| HDL | Interleukin-10 |
| KD | Ketogenic diet |
| TRF | Time-restricted feeding |
| ASTX | Astaxanthin |
| Cpt1α | Carnitine palmitoyltransferase-1 alpha |
| Acox1 | Acyl-coenzyme A oxidase 1 |
| DIO | Diet-induced obese |
| Ucp2 | Uncoupling protein 2 |
| PPAR-γ | Peroxisome proliferator-activated receptor γ |
| HIIT | High-intensity interval training |
| MIC | Moderate-intensity continuous |
| IHTG | Intrahepatic Triglyceride |
| T2DM | Type 2 diabetes mellitus |
| ACC | Acetyl coenzyme A carboxylase |
| SCD | Stearoyl coenzyme A desaturase |
| MAM | Mitochondria-associated ER membranes |
| TZD | Thiazolidinediones |
| TAG | Triacylglycerol |
| FXR | Farnesoid X receptor |
| TGR5 | Takeda G protein-coupled receptor 5 |
| OPA1 | Optic atrophy 1 |
| BA | Bile Acid |
| UDCA | Ursodeoxycholic acid |
| CDCA | Chenodeoxycholic acid |
| DCA | Deoxycholic acid |
| LCA | Lithocholic acid |
| IMM | Inner mitochondrial membrane |
| SREBP-1c | Sterol regulatory binding protein-1c |
| ChREBP | Carbohydrate response element binding protein |
| LPK | Liver pyruvate kinase |
| OCA | Obeticholic acid |
| NRF-1and-2 | Nuclear respiratory factor 1/2 |
| SDH | Succinate dehydrogenase |
| BAT | Brown adipose tissue |
| WAT | White adipose tissue |
| HO-1 | Heme oxygenase-1 |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| C3G | Cyanidin-3-glucoside |
| UCP1 | Uncoupling protein 1 |
| PM | Plasma membrane |
| PKCε | Protein kinase C epsilon |
| FFA | Free fatty acid |
| VLDL | Very-low-density lipoprotein |
| ETC | Electron transfer chain |
| TG | Triglyceride |
| NASH | Non-alcoholic fatty liver disease |
| ROS | Reactive oxygen species |
| MQC | Mitochondrial quality control |
| HDL | High-density lipoprotein |
| HFD | High-fat diet |
| BA | Bile acids |
References
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. CMLS 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Horn, P.; Wong, V.W.-S.; Ratziu, V.; Bugianesi, E.; Francque, S.; Zelber-Sagi, S.; Valenti, L.; Roden, M.; Schick, F.; et al. EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J. Hepatol. 2024, 81, 492–542. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef]
- Suomalainen, A.; Nunnari, J. Mitochondria at the crossroads of health and disease. Cell 2024, 187, 2601–2627. [Google Scholar] [CrossRef]
- Xie, L.L.; Shi, F.; Tan, Z.; Li, Y.; Bode, A.M.; Cao, Y. Mitochondrial network structure homeostasis and cell death. Cancer Sci. 2018, 109, 3686–3694. [Google Scholar] [CrossRef]
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Klopstock, T.; Kornblum, C.; Mancuso, M.; McFarland, R.; Sue, C.M.; Suomalainen, A.; Taylor, R.W.; et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. 2021, 20, 573–584. [Google Scholar] [CrossRef]
- Guan, S.; Zhao, L.; Peng, R. Mitochondrial Respiratory Chain Supercomplexes: From Structure to Function. Int. J. Mol. Sci. 2022, 23, 13880. [Google Scholar] [CrossRef]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in skin health, aging, and disease. Cell Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H.; Gerasimenko, J.V.; Gerasimenko, O.V.; Gryshchenko, O.; Peng, S. The roles of calcium and ATP in the physiology and pathology of the exocrine pancreas. Physiol. Rev. 2021, 101, 1691–1744. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Zhang, Y.; Zhou, J. Focus on Mitochondrial Respiratory Chain: Potential Therapeutic Target for Chronic Renal Failure. Int. J. Mol. Sci. 2024, 25, 949. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef]
- Radogna, F.; Gérard, D.; Dicato, M.; Diederich, M. Assessment of Mitochondrial Cell Metabolism by Respiratory Chain Electron Flow Assays. In Methods in Molecular Biology; Springer: New York, NY, USA, 2021; Volume 2276, pp. 129–141. [Google Scholar] [CrossRef]
- Patro, S.; Ratna, S.; Yamamoto, H.A.; Ebenezer, A.T.; Ferguson, D.S.; Kaur, A.; McIntyre, B.C.; Snow, R.; Solesio, M.E. ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11185. [Google Scholar] [CrossRef]
- Johnson, J.; Mercado-Ayon, E.; Mercado-Ayon, Y.; Dong, Y.N.; Halawani, S.; Ngaba, L.; Lynch, D.R. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2021, 702, 108698. [Google Scholar] [CrossRef]
- Driskill, J.H.; Pan, D. The Hippo Pathway in Liver Homeostasis and Pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 299–322. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, J.; Zhao, T.; Wang, L.; Liang, T.; Zheng, Y. Mitochondrial structure and function: A new direction for the targeted treatment of chronic liver disease with Chinese herbal medicine. J. Ethnopharmacol. 2024, 334, 118461. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Gu, X.; Zheng, Q.; Wang, J.; Zhu, H. Mitochondrial Mechanisms of Apoptosis and Necroptosis in Liver Diseases. Anal. Cell. Pathol. 2021, 2021, 8900122. [Google Scholar] [CrossRef]
- Lamanilao, G.G.; Dogan, M.; Patel, P.S.; Azim, S.; Patel, D.S.; Bhattacharya, S.K.; Eason, J.D.; Kuscu, C.; Kuscu, C.; Bajwa, A. Key hepatoprotective roles of mitochondria in liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 324, G207–G218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, X.; Hu, Q.; Wu, J.; Wang, G.; Hong, Z.; Ren, J. Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci. 2019, 236, 116464. [Google Scholar] [CrossRef]
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Morio, B.; Panthu, B.; Bassot, A.; Rieusset, J. Role of mitochondria in liver metabolic health and diseases. Cell Calcium 2021, 94, 102336. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Kawano, S.; Endo, T. Lipid homeostasis in mitochondria. Biol. Chem. 2020, 401, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Veliova, M.; Petcherski, A.; Liesa, M.; Shirihai, O.S. The biology of lipid droplet-bound mitochondria. Semin. Cell Dev. Biol. 2020, 108, 55–64. [Google Scholar] [CrossRef]
- Grattagliano, I.; Di Ciaula, A.; Baj, J.; Molina-Molina, E.; Shanmugam, H.; Garruti, G.; Wang, D.Q.; Portincasa, P. Protocols for Mitochondria as the Target of Pharmacological Therapy in the Context of Nonalcoholic Fatty Liver Disease (NAFLD). In Methods in Molecular Biology; Springer: New York, NY, USA, 2021; Volume 2310, pp. 201–246. [Google Scholar] [CrossRef]
- Xiao, J.J.; Liu, Q.; Li, Y.; Peng, F.F.; Wang, S.; Zhang, Z.; Liu, H.; Yu, H.; Tao, S.; Zhang, B.F. Regulator of calcineurin 1 deletion attenuates mitochondrial dysfunction and apoptosis in acute kidney injury through JNK/Mff signaling pathway. Cell Death Dis. 2022, 13, 774. [Google Scholar] [CrossRef]
- Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4173. [Google Scholar] [CrossRef]
- Tsilingiris, D.; Tzeravini, E.; Koliaki, C.; Dalamaga, M.; Kokkinos, A. The Role of Mitochondrial Adaptation and Metabolic Flexibility in the Pathophysiology of Obesity and Insulin Resistance: An Updated Overview. Curr. Obes. Rep. 2021, 10, 191–213. [Google Scholar] [CrossRef]
- Mao, H.; Chen, W.; Chen, L.; Li, L. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochem. Pharmacol. 2022, 199, 115011. [Google Scholar] [CrossRef]
- Shin, S.; Kim, J.; Lee, J.Y.; Kim, J.; Oh, C.M. Mitochondrial Quality Control: Its Role in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD). J. Obes. Metab. Syndr. 2023, 32, 289–302. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, S.; Wu, J.; Wang, Y. Mitochondrial metabolic dysfunction and non-alcoholic fatty liver disease: New insights from pathogenic mechanisms to clinically targeted therapy. J. Transl. Med. 2023, 21, 510. [Google Scholar] [CrossRef]
- Hung, C.M.; Lombardo, P.S.; Malik, N.; Brun, S.N.; Hellberg, K.; Van Nostrand, J.L.; Garcia, D.; Baumgart, J.; Diffenderfer, K.; Asara, J.M.; et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci. Adv. 2021, 7, eabg4544. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E. Solving mitochondrial mysteries. J. Mol. Cell. Cardiol. 2014, 78, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Qian, H.; Ding, W.X. SQSTM1/p62 and Hepatic Mallory-Denk Body Formation in Alcohol-Associated Liver Disease. Am. J. Pathol. 2023, 193, 1415–1426. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106, 17–32. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef]
- Li, A.L.; Lian, L.; Chen, X.N.; Cai, W.H.; Fan, X.B.; Fan, Y.J.; Li, T.T.; Xie, Y.Y.; Zhang, J.P. The role of mitochondria in myocardial damage caused by energy metabolism disorders: From mechanisms to therapeutics. Free Radic. Biol. Med. 2023, 208, 236–251. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells 2021, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, L.; Zheng, X.; Ge, L. Autophagosomal Membrane Origin and Formation. Autophagy Biol. Dis. Technol. Methodol. 2021, 1208, 17–42. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, Y.; Zhou, S.; Tang, P.; Xu, R.; Zhang, Y.; Wei, D.; Wen, J.; Thorne, R.F.; Zhang, X.D.; et al. TRIM27 cooperates with STK38L to inhibit ULK1-mediated autophagy and promote tumorigenesis. EMBO J. 2022, 41, e109777. [Google Scholar] [CrossRef]
- Jin, S.; Li, Y.; Xia, T.; Liu, Y.; Zhang, S.; Hu, H.; Chang, Q.; Yan, M. Mechanisms and therapeutic implications of selective autophagy in nonalcoholic fatty liver disease. J. Adv. Res. 2025, 67, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.G.; Xia, Z.; Shang, H.Y. Advances in the study of mitophagy-related receptor proteins. Acta Physiol. Sin. 2021, 73, 1025–1034. [Google Scholar]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Wang, S.; Deng, Z.; Ma, Y.; Jin, J.; Qi, F.; Li, S.; Liu, C.; Lyu, F.J.; Zheng, Q. The Role of Autophagy and Mitophagy in Bone Metabolic Disorders. Int. J. Biol. Sci. 2020, 16, 2675–2691. [Google Scholar] [CrossRef]
- Jin, K.; Shi, Y.; Zhang, H.; Zhangyuan, G.; Wang, F.; Li, S.; Chen, C.; Zhang, J.; Wang, H.; Zhang, W.; et al. A TNFα/Miz1-positive feedback loop inhibits mitophagy in hepatocytes and propagates non-alcoholic steatohepatitis. J. Hepatol. 2023, 79, 403–416. [Google Scholar] [CrossRef]
- Patel Chavez, C.; Cusi, K.; Kadiyala, S. The Emerging Role of Glucagon-like Peptide-1 Receptor Agonists for the Management of NAFLD. J. Clin. Endocrinol. Metab. 2022, 107, 29–38. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Chen, X.; Wang, Z.; Wang, X.; Zhou, Q.; Fang, W.; Zheng, C. Liraglutide prevents high glucose induced HUVECs dysfunction via inhibition of PINK1/Parkin-dependent mitophagy. Mol. Cell. Endocrinol. 2022, 545, 111560. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Dias, K.A.; Oliveira, L.A.; Pereira, S.M.S.; Abrantes, L.C.S.; Vicente, L.; Gonçalves, R.V.; Della Lucia, C.M. Anti-inflammatory and antioxidant effects of anthocyanins in Nonalcoholic fatty liver disease (NAFLD): A systematic review of in vivo studies. Crit. Rev. Food Sci. Nutr. 2025, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jian, L.; Guo, Y.; Tang, C.; Huang, Z.; Gao, J. Liver Cell Mitophagy in Metabolic Dysfunction-Associated Steatotic Liver Disease and Liver Fibrosis. Antioxidants 2024, 13, 729. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shi, Z.; Zhu, Y.; Shen, T.; Wang, H.; Shui, G.; Loor, J.J.; Fang, Z.; Chen, M.; Wang, X.; et al. Cyanidin-3-O-glucoside improves non-alcoholic fatty liver disease by promoting PINK1-mediated mitophagy in mice. Br. J. Pharmacol. 2020, 177, 3591–3607. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019, 21, 101120. [Google Scholar] [CrossRef]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e46. [Google Scholar] [CrossRef]
- Wang, H.; Luo, W.; Chen, H.; Cai, Z.; Xu, G. Mitochondrial dynamics and mitochondrial autophagy: Molecular structure, orchestrating mechanism and related disorders. Mitochondrion 2024, 75, 101847. [Google Scholar] [CrossRef] [PubMed]
- Allameh, A.; Niayesh-Mehr, R.; Aliarab, A.; Sebastiani, G.; Pantopoulos, K. Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants 2023, 12, 1653. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lu, F.; Gao, J.; Yuan, Y. Inflammation-mediated metabolic regulation in adipose tissue. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2024, 25, e13724. [Google Scholar] [CrossRef]
- Cao, P.; Wang, Y.; Zhang, C.; Sullivan, M.A.; Chen, W.; Jing, X.; Yu, H.; Li, F.; Wang, Q.; Zhou, Z.; et al. Quercetin ameliorates nonalcoholic fatty liver disease (NAFLD) via the promotion of AMPK-mediated hepatic mitophagy. J. Nutr. Biochem. 2023, 120, 109414. [Google Scholar] [CrossRef]
- Saleh Al-Maamari, J.N.; Rahmadi, M.; Panggono, S.M.; Prameswari, D.A.; Pratiwi, E.D.; Ardianto, C.; Balan, S.S.; Suprapti, B. The effects of quercetin on the expression of SREBP-1c mRNA in high-fat diet-induced NAFLD in mice. J. Basic. Clin. Physiol. Pharmacol. 2021, 32, 637–644. [Google Scholar] [CrossRef]
- Afarin, R.; Hatami, M.; Monjezi, S.; Bineshfar, F.; Ahangarpour, A. Suppression of TGF-β/Smad3 signaling pathway by Capparis spinosa and quercetin in a rat model of nonalcoholic steatohepatitis. Iran. J. Basic. Med. Sci. 2024, 27, 1096–1104. [Google Scholar] [CrossRef]
- Wei, H.; Zhao, T.; Liu, X.; Ding, Q.; Yang, J.; Bi, X.; Cheng, Z.; Ding, C.; Liu, W. Mechanism of Action of Dihydroquercetin in the Prevention and Therapy of Experimental Liver Injury. Molecules 2024, 29, 3537. [Google Scholar] [CrossRef]
- Nie, Q.; Li, M.; Huang, C.; Yuan, Y.; Liang, Q.; Ma, X.; Qiu, T.; Li, J. The clinical efficacy and safety of berberine in the treatment of non-alcoholic fatty liver disease: A meta-analysis and systematic review. J. Transl. Med. 2024, 22, 225. [Google Scholar] [CrossRef]
- Feng, X.; Sureda, A.; Jafari, S.; Memariani, Z.; Tewari, D.; Annunziata, G.; Barrea, L.; Hassan, S.T.S.; Šmejkal, K.; Malaník, M.; et al. Berberine in Cardiovascular and Metabolic Diseases: From Mechanisms to Therapeutics. Theranostics 2019, 9, 1923–1951. [Google Scholar] [CrossRef]
- Wah Kheong, C.; Nik Mustapha, N.R.; Mahadeva, S. A Randomized Trial of Silymarin for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2017, 15, 1940–1949.e1948. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Fekri, H.S.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Therapeutic and biological activities of berberine: The involvement of Nrf2 signaling pathway. J. Cell. Biochem. 2020, 121, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- García-Muñoz, A.M.; Victoria-Montesinos, D.; Ballester, P.; Cerdá, B.; Zafrilla, P. A Descriptive Review of the Antioxidant Effects and Mechanisms of Action of Berberine and Silymarin. Molecules 2024, 29, 4576. [Google Scholar] [CrossRef] [PubMed]
- Vogli, S.; Naska, A.; Marinos, G.; Kasdagli, M.I.; Orfanos, P. The Effect of Vitamin E Supplementation on Serum Aminotransferases in Non-Alcoholic Fatty Liver Disease (NAFLD): A Systematic Review and Meta-Analysis. Nutrients 2023, 15, 3733. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Cangemi, R. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 363, 1185–1186. [Google Scholar] [CrossRef] [PubMed]
- Jonas, W.; Schürmann, A. Genetic and epigenetic factors determining NAFLD risk. Mol. Metab. 2021, 50, 101111. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and Genetics in NAFLD: The Perfect Binomium. Int. J. Mol. Sci. 2020, 21, 2986. [Google Scholar] [CrossRef]
- Caputo, V.; Tarantino, G.; Santini, S.J.; Fracassi, G.; Balsano, C. The Role of Epigenetic Control of Mitochondrial (Dys)Function in MASLD Onset and Progression. Nutrients 2023, 15, 4757. [Google Scholar] [CrossRef]
- Matchett, K.P.; Paris, J.; Teichmann, S.A.; Henderson, N.C. Spatial genomics: Mapping human steatotic liver disease. Nat. reviews. Gastroenterol. Hepatol. 2024, 21, 646–660. [Google Scholar] [CrossRef]
- López-Vicario, C.; Sebastián, D.; Casulleras, M.; Duran-Güell, M.; Flores-Costa, R.; Aguilar, F.; Lozano, J.J.; Zhang, I.W.; Titos, E.; Kang, J.X.; et al. Essential lipid autacoids rewire mitochondrial energy efficiency in metabolic dysfunction-associated fatty liver disease. Hepatology 2023, 77, 1303–1318. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Dong, J.; Chen, L.; Ye, F.; Tang, J.; Liu, B.; Lin, J.; Zhou, P.H.; Lu, B.; Wu, M.; Lu, J.H.; et al. Mic19 depletion impairs endoplasmic reticulum-mitochondrial contacts and mitochondrial lipid metabolism and triggers liver disease. Nat. Commun. 2024, 15, 168. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Ngo, J.; Wang, S.P.; Williams, K.; Kramer, H.F.; Ho, G.; Rodriguez, C.; Yekkala, K.; Amuzie, C.; Bialecki, R.; et al. The mitochondrial fission protein Drp1 in liver is required to mitigate NASH and prevents the activation of the mitochondrial ISR. Mol. Metab. 2022, 64, 101566. [Google Scholar] [CrossRef] [PubMed]
- Stevanović, J.; Beleza, J.; Coxito, P.; Ascensão, A.; Magalhães, J. Physical exercise and liver “fitness”: Role of mitochondrial function and epigenetics-related mechanisms in non-alcoholic fatty liver disease. Mol. Metab. 2020, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Ju, A.; Zhang, S.; An, Q.; Xu, S.; Liu, J.; Yu, L.; Fu, Y.; Luo, Y. Albumosomes formed by cytoplasmic pre-folding albumin maintain mitochondrial homeostasis and inhibit nonalcoholic fatty liver disease. Signal Transduct. Target. Ther. 2023, 8, 229. [Google Scholar] [CrossRef]
- Weng, S.W.; Wu, J.C.; Shen, F.C.; Chang, Y.H.; Su, Y.J.; Lian, W.S.; Tai, M.H.; Su, C.H.; Chuang, J.H.; Lin, T.K.; et al. Chaperonin counteracts diet-induced non-alcoholic fatty liver disease by aiding sirtuin 3 in the control of fatty acid oxidation. Diabetologia 2023, 66, 913–930. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Bezsonov, E.E.; Baig, M.S.; Popkova, T.V.; Nedosugova, L.V.; Starodubova, A.V.; Orekhov, A.N. Mitochondrial Mutations and Genetic Factors Determining NAFLD Risk. Int. J. Mol. Sci. 2021, 22, 4459. [Google Scholar] [CrossRef]
- Patel, J.; Baptiste, B.A.; Kim, E.; Hussain, M.; Croteau, D.L.; Bohr, V.A. DNA damage and mitochondria in cancer and aging. Carcinogenesis 2020, 41, 1625–1634. [Google Scholar] [CrossRef]
- Pourteymour, S.; Drevon, C.A.; Dalen, K.T.; Norheim, F.A. Mechanisms Behind NAFLD: A System Genetics Perspective. Curr. Atheroscler. Rep. 2023, 25, 869–878. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, M.; Wang, X.; Bu, Q.; Wang, Q.; Su, W.; Li, L.; Zhou, H.; Lu, L. XBP1 deficiency promotes hepatocyte pyroptosis by impairing mitophagy to activate mtDNA-cGAS-STING signaling in macrophages during acute liver injury. Redox Biol. 2022, 52, 102305. [Google Scholar] [CrossRef]
- Zhang, N.; Tian, X.; Yan, T.; Wang, H.; Zhang, D.; Lin, C.; Liu, Q.; Jiang, S. Insights into the role of nucleotide methylation in metabolic-associated fatty liver disease. Front. Immunol. 2023, 14, 1148722. [Google Scholar] [CrossRef]
- Portincasa, P.; Khalil, M.; Mahdi, L.; Perniola, V.; Idone, V.; Graziani, A.; Baffy, G.; Di Ciaula, A. Metabolic Dysfunction-Associated Steatotic Liver Disease: From Pathogenesis to Current Therapeutic Options. Int. J. Mol. Sci. 2024, 25, 5640. [Google Scholar] [CrossRef] [PubMed]
- Thyfault, J.P.; Bergouignan, A. Exercise and metabolic health: Beyond skeletal muscle. Diabetologia 2020, 63, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Jin, X.; Li, Y. Current strategies for nonalcoholic fatty liver disease treatment (Review). Int. J. Mol. Med. 2024, 54, 88. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Anthony Sinha, R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Beygi, M.; Ahi, S.; Zolghadri, S.; Stanek, A. Management of Metabolic-Associated Fatty Liver Disease/Metabolic Dysfunction-Associated Steatotic Liver Disease: From Medication Therapy to Nutritional Interventions. Nutrients 2024, 16, 2220. [Google Scholar] [CrossRef]
- Wang, M.; Pan, W.; Xu, Y.; Zhang, J.; Wan, J.; Jiang, H. Microglia-Mediated Neuroinflammation: A Potential Target for the Treatment of Cardiovascular Diseases. J. Inflamm. Res. 2022, 15, 3083–3094. [Google Scholar] [CrossRef]
- Hassani Zadeh, S.; Mansoori, A.; Hosseinzadeh, M. Relationship between dietary patterns and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2021, 36, 1470–1478. [Google Scholar] [CrossRef]
- Cunha, G.M.; Guzman, G.; Correa De Mello, L.L.; Trein, B.; Spina, L.; Bussade, I.; Marques Prata, J.; Sajoux, I.; Countinho, W. Efficacy of a 2-Month Very Low-Calorie Ketogenic Diet (VLCKD) Compared to a Standard Low-Calorie Diet in Reducing Visceral and Liver Fat Accumulation in Patients With Obesity. Front. Endocrinol. 2020, 11, 607. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Weiskirchen, S.; Weiskirchen, R. Lipocalin 2 (LCN2) Expression in Hepatic Malfunction and Therapy. Front. Physiol. 2016, 7, 430. [Google Scholar] [CrossRef]
- Lv, H.; Liu, Y. Management of non-alcoholic fatty liver disease: Lifestyle changes. World J. Gastroenterol. 2024, 30, 2829–2833. [Google Scholar] [CrossRef]
- Cai, H.; Qin, Y.L.; Shi, Z.Y.; Chen, J.H.; Zeng, M.J.; Zhou, W.; Chen, R.Q.; Chen, Z.Y. Effects of alternate-day fasting on body weight and dyslipidaemia in patients with non-alcoholic fatty liver disease: A randomised controlled trial. BMC Gastroenterol. 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Anjum, B.; Godbole, N.M.; Rajak, S.; Shukla, P.; Tiwari, S.; Sinha, R.A.; Godbole, M.M. Time-restricted feeding reduces high-fat diet associated placental inflammation and limits adverse effects on fetal organ development. Biochem. Biophys. Res. Commun. 2019, 514, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Martín Barraza, J.I.; Bars-Cortina, D. Dietary Pattern’s Role in Hepatic Epigenetic and Dietary Recommendations for the Prevention of NAFLD. Nutrients 2024, 16, 2956. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, W.; Xiao, W. Astaxanthin: A promising therapeutic agent for organ fibrosis. Pharmacol. Res. 2023, 188, 106657. [Google Scholar] [CrossRef]
- Jia, Y.; Wu, C.; Kim, J.; Kim, B.; Lee, S.J. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J. Nutr. Biochem. 2016, 28, 9–18. [Google Scholar] [CrossRef]
- Kim, M.B.; Lee, J.; Lee, J.Y. Targeting Mitochondrial Dysfunction for the Prevention and Treatment of Metabolic Disease by Bioactive Food Components. J. Lipid Atheroscler. 2024, 13, 306–327. [Google Scholar] [CrossRef]
- Gonçalves, I.O.; Passos, E.; Diogo, C.V.; Rocha-Rodrigues, S.; Santos-Alves, E.; Oliveira, P.J.; Ascensão, A.; Magalhães, J. Exercise mitigates mitochondrial permeability transition pore and quality control mechanisms alterations in nonalcoholic steatohepatitis. Appl. Physiol. Nutr. Metab. 2016, 41, 298–306. [Google Scholar] [CrossRef]
- Vachher, M.; Bansal, S.; Kumar, B.; Yadav, S.; Burman, A. Deciphering the role of aberrant DNA methylation in NAFLD and NASH. Heliyon 2022, 8, e11119. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Neshat, S.Y.; Quiroz, V.M.; Wang, Y.; Tamayo, S.; Doloff, J.C. Liver Disease: Induction, Progression, Immunological Mechanisms, and Therapeutic Interventions. Int. J. Mol. Sci. 2021, 22, 6777. [Google Scholar] [CrossRef]
- Charatcharoenwitthaya, P.; Kuljiratitikal, K.; Aksornchanya, O.; Chaiyasoot, K.; Bandidniyamanon, W.; Charatcharoenwitthaya, N. Moderate-Intensity Aerobic vs Resistance Exercise and Dietary Modification in Patients With Nonalcoholic Fatty Liver Disease: A Randomized Clinical Trial. Clin. Transl. Gastroenterol. 2021, 12, e00316. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Sabag, A.; Hallsworth, K.; Hickman, I.J.; Macdonald, G.A.; Stine, J.G.; George, J.; Johnson, N.A. Exercise in the Management of Metabolic-Associated Fatty Liver Disease (MAFLD) in Adults: A Position Statement from Exercise and Sport Science Australia. Sports Med. 2023, 53, 2347–2371. [Google Scholar] [CrossRef] [PubMed]
- Sabag, A.; Barr, L.; Armour, M.; Armstrong, A.; Baker, C.J.; Twigg, S.M.; Chang, D.; Hackett, D.A.; Keating, S.E.; George, J.; et al. The Effect of High-intensity Interval Training vs Moderate-intensity Continuous Training on Liver Fat: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2022, 107, 862–881. [Google Scholar] [CrossRef] [PubMed]
- Rajamoorthi, A.; Arias, N.; Basta, J.; Lee, R.G.; Baldán, Á. Amelioration of diet-induced steatohepatitis in mice following combined therapy with ASO-Fsp27 and fenofibrate. J. Lipid Res. 2017, 58, 2127–2138. [Google Scholar] [CrossRef]
- Pratt, J.S.A.; Browne, A.; Browne, N.T.; Bruzoni, M.; Cohen, M.; Desai, A.; Inge, T.; Linden, B.C.; Mattar, S.G.; Michalsky, M.; et al. ASMBS pediatric metabolic and bariatric surgery guidelines, 2018. Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 2018, 14, 882–901. [Google Scholar] [CrossRef]
- Michalsky, M.; Kramer, R.E.; Fullmer, M.A.; Polfuss, M.; Porter, R.; Ward-Begnoche, W.; Getzoff, E.A.; Dreyer, M.; Stolzman, S.; Reichard, K.W. Developing criteria for pediatric/adolescent bariatric surgery programs. Pediatrics 2011, 128 (Suppl. S2), S65–S70. [Google Scholar] [CrossRef]
- Michalsky, M.P.; Inge, T.H.; Teich, S.; Eneli, I.; Miller, R.; Brandt, M.L.; Helmrath, M.; Harmon, C.M.; Zeller, M.H.; Jenkins, T.M.; et al. Adolescent bariatric surgery program characteristics: The Teen Longitudinal Assessment of Bariatric Surgery (Teen-LABS) study experience. Semin. Pediatr. Surg. 2014, 23, 5–10. [Google Scholar] [CrossRef]
- Woolford, S.J.; Clark, S.J.; Gebremariam, A.; Davis, M.M.; Freed, G.L. To cut or not to cut: Physicians’ perspectives on referring adolescents for bariatric surgery. Obes. Surg. 2010, 20, 937–942. [Google Scholar] [CrossRef]
- Sjöström, L. Review of the key results from the Swedish Obese Subjects (SOS) trial—A prospective controlled intervention study of bariatric surgery. J. Intern. Med. 2013, 273, 219–234. [Google Scholar] [CrossRef]
- Manco, M.; Mosca, A.; De Peppo, F.; Caccamo, R.; Cutrera, R.; Giordano, U.; De Stefanis, C.; Alisi, A.; Baumann, U.; Silecchia, G.; et al. The Benefit of Sleeve Gastrectomy in Obese Adolescents on Nonalcoholic Steatohepatitis and Hepatic Fibrosis. J. Pediatr. 2017, 180, 31–37.e32. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lai, H.; Chua, Y.J.; Wang, M.X.; Lee, G.H. Endoscopic Bariatric and Metabolic Therapies and Their Effects on Metabolic Syndrome and Non-alcoholic Fatty Liver Disease—A Systematic Review and Meta-Analysis. Front. Med. 2022, 9, 880749. [Google Scholar] [CrossRef]
- González, K.; Fuentes, J.; Márquez, J.L. Physical Inactivity, Sedentary Behavior and Chronic Diseases. Korean J. Fam. Med. 2017, 38, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Huang, S.Y.; Zhou, D.D.; Xiong, R.G.; Luo, M.; Saimaiti, A.; Han, M.K.; Gan, R.Y.; Zhu, H.L.; Li, H.B. Theabrownin inhibits obesity and non-alcoholic fatty liver disease in mice via serotonin-related signaling pathways and gut-liver axis. J. Adv. Res. 2023, 52, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Subramanya, S.B.; Venkataraman, B.; Meeran, M.F.N.; Goyal, S.N.; Patil, C.R.; Ojha, S. Therapeutic Potential of Plants and Plant Derived Phytochemicals against Acetaminophen-Induced Liver Injury. Int. J. Mol. Sci. 2018, 19, 3776. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Hsu, L.W.; Chen, K.D.; Chiu, K.W.; Chen, C.L.; Huang, K.T. Emerging Roles of Calcium Signaling in the Development of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 23, 256. [Google Scholar] [CrossRef]
- Bi, Y.; Liu, S.; Qin, X.; Abudureyimu, M.; Wang, L.; Zou, R.; Ajoolabady, A.; Zhang, W.; Peng, H.; Ren, J.; et al. FUNDC1 interacts with GPx4 to govern hepatic ferroptosis and fibrotic injury through a mitophagy-dependent manner. J. Adv. Res. 2024, 55, 45–60. [Google Scholar] [CrossRef]
- Linden, M.A.; Lopez, K.T.; Fletcher, J.A.; Morris, E.M.; Meers, G.M.; Siddique, S.; Laughlin, M.H.; Sowers, J.R.; Thyfault, J.P.; Ibdah, J.A.; et al. Combining metformin therapy with caloric restriction for the management of type 2 diabetes and nonalcoholic fatty liver disease in obese rats. Appl. Physiol. Nutr. Metab. 2015, 40, 1038–1047. [Google Scholar] [CrossRef]
- Wang, D.D.; Mao, Y.Z.; He, S.M.; Chen, X. Analysis of Time Course and Dose Effect From Metformin on Body Mass Index in Children and Adolescents. Front. Pharmacol. 2021, 12, 611480. [Google Scholar] [CrossRef]
- Lamos, E.M.; Kristan, M.; Siamashvili, M.; Davis, S.N. Effects of anti-diabetic treatments in type 2 diabetes and fatty liver disease. Expert Rev. Clin. Pharmacol. 2021, 14, 837–852. [Google Scholar] [CrossRef]
- EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. Diabetologia 2016, 59, 1121–1140. [CrossRef]
- Shannon, C.E.; Ragavan, M.; Palavicini, J.P.; Fourcaudot, M.; Bakewell, T.M.; Valdez, I.A.; Ayala, I.; Jin, E.S.; Madesh, M.; Han, X.; et al. Insulin resistance is mechanistically linked to hepatic mitochondrial remodeling in non-alcoholic fatty liver disease. Mol. Metab. 2021, 45, 101154. [Google Scholar] [CrossRef] [PubMed]
- Camacho, R.C.; Polidori, D.; Chen, T.; Chen, B.; Hsu, H.H.; Gao, B.; Marella, M.; Lubomirski, M.; Beavers, T.; Cabrera, J.; et al. Validation of a diet-induced Macaca fascicularis model of non-alcoholic steatohepatitis with dietary and pioglitazone interventions. Diabetes Obes. Metab. 2023, 25, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, D.; Xie, Z.; Ding, L.; Li, S.; Ma, X.; Liu, J.; Ren, J.; Xiao, C.; Yang, C.; et al. Combination of Pioglitazone and Metformin Actions on Liver Lipid Metabolism in Obese Mice. Biomolecules 2023, 13, 1199. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Cai, J.; Gonzalez, F.J. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 335–347. [Google Scholar] [CrossRef]
- Nie, Q.; Luo, X.; Wang, K.; Ding, Y.; Jia, S.; Zhao, Q.; Li, M.; Zhang, J.; Zhuo, Y.; Lin, J.; et al. Gut symbionts alleviate MASH through a secondary bile acid biosynthetic pathway. Cell 2024, 187, 2717–2734.e2733. [Google Scholar] [CrossRef]
- Masoodi, M.; Gastaldelli, A.; Hyötyläinen, T.; Arretxe, E.; Alonso, C.; Gaggini, M.; Brosnan, J.; Anstee, Q.M.; Millet, O.; Ortiz, P.; et al. Metabolomics and lipidomics in NAFLD: Biomarkers and non-invasive diagnostic tests. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 835–856. [Google Scholar] [CrossRef]
- Da Dalt, L.; Moregola, A.; Svecla, M.; Pedretti, S.; Fantini, F.; Ronzio, M.; Uboldi, P.; Dolfini, D.; Donetti, E.; Baragetti, A.; et al. The inhibition of inner mitochondrial fusion in hepatocytes reduces non-alcoholic fatty liver and improves metabolic profile during obesity by modulating bile acid conjugation. Cardiovasc. Res. 2024, 119, 2917–2929. [Google Scholar] [CrossRef]
- Chen, Y.S.; Liu, H.M.; Lee, T.Y. Ursodeoxycholic Acid Regulates Hepatic Energy Homeostasis and White Adipose Tissue Macrophages Polarization in Leptin-Deficiency Obese Mice. Cells 2019, 8, 253. [Google Scholar] [CrossRef]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular Physiology of Bile Acid Signaling in Health, Disease, and Aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Hu, H.; Lin, A.; Kong, M.; Yao, X.; Yin, M.; Xia, H.; Ma, J.; Liu, H. Intestinal microbiome and NAFLD: Molecular insights and therapeutic perspectives. J. Gastroenterol. 2020, 55, 142–158. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Hesselink, M.K.; Schrauwen, P. Therapeutic potential of resveratrol in obesity and type 2 diabetes: New avenues for health benefits? Ann. N. Y. Acad. Sci. 2013, 1290, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef]
- Kasprzak-Drozd, K.; Niziński, P.; Kasprzak, P.; Kondracka, A.; Oniszczuk, T.; Rusinek, A.; Oniszczuk, A. Does Resveratrol Improve Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD)? Int. J. Mol. Sci. 2024, 25, 3746. [Google Scholar] [CrossRef]
- Rayalam, S.; Yang, J.Y.; Ambati, S.; Della-Fera, M.A.; Baile, C.A. Resveratrol induces apoptosis and inhibits adipogenesis in 3T3-L1 adipocytes. Phytother. Res. PTR 2008, 22, 1367–1371. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Desquiret-Dumas, V.; Gueguen, N.; Leman, G.; Baron, S.; Nivet-Antoine, V.; Chupin, S.; Chevrollier, A.; Vessières, E.; Ayer, A.; Ferré, M.; et al. Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J. Biol. Chem. 2013, 288, 36662–36675. [Google Scholar] [CrossRef]
- Acharya, J.D.; Ghaskadbi, S.S. Protective effect of Pterostilbene against free radical mediated oxidative damage. BMC Complement. Altern. Med. 2013, 13, 238. [Google Scholar] [CrossRef]
- Gómez-Zorita, S.; González-Arceo, M.; Trepiana, J.; Aguirre, L.; Crujeiras, A.B.; Irles, E.; Segues, N.; Bujanda, L.; Portillo, M.P. Comparative Effects of Pterostilbene and Its Parent Compound Resveratrol on Oxidative Stress and Inflammation in Steatohepatitis Induced by High-Fat High-Fructose Feeding. Antioxidants 2020, 9, 1042. [Google Scholar] [CrossRef]
- Liu, Y.; You, Y.; Lu, J.; Chen, X.; Yang, Z. Recent Advances in Synthesis, Bioactivity, and Pharmacokinetics of Pterostilbene, an Important Analog of Resveratrol. Molecules 2020, 25, 5166. [Google Scholar] [CrossRef]
- Jones, S.A.; Ruprecht, J.J.; Crichton, P.G.; Kunji, E.R.S. Structural mechanisms of mitochondrial uncoupling protein 1 regulation in thermogenesis. Trends Biochem. Sci. 2024, 49, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, F.; Chu, Y.; Yun, Z.; Yan, Y.; Jin, J. Mitochondrial transplantation: Opportunities and challenges in the treatment of obesity, diabetes, and nonalcoholic fatty liver disease. J. Transl. Med. 2022, 20, 483. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, S.; Kim, W.K.; Han, B.S. Mitochondrial transplantation: An overview of a promising therapeutic approach. BMB Rep. 2023, 56, 488–495. [Google Scholar] [CrossRef]
- Yamada, Y.; Ito, M.; Arai, M.; Hibino, M.; Tsujioka, T.; Harashima, H. Challenges in Promoting Mitochondrial Transplantation Therapy. Int. J. Mol. Sci. 2020, 21, 6365. [Google Scholar] [CrossRef]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef]
- Dai, J.J.; Zhang, Y.F.; Zhang, Z.H. Global trends and hotspots of treatment for nonalcoholic fatty liver disease: A bibliometric and visualization analysis (2010–2023). World J. Gastroenterol. 2023, 29, 5339–5360. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Chen, W.; Jia, Z.; Xiao, Y.; Shi, A.; Ma, X. Mitochondrial Dysfunction as a Pathogenesis and Therapeutic Strategy for Metabolic-Dysfunction-Associated Steatotic Liver Disease. Int. J. Mol. Sci. 2025, 26, 4256. https://doi.org/10.3390/ijms26094256
Li X, Chen W, Jia Z, Xiao Y, Shi A, Ma X. Mitochondrial Dysfunction as a Pathogenesis and Therapeutic Strategy for Metabolic-Dysfunction-Associated Steatotic Liver Disease. International Journal of Molecular Sciences. 2025; 26(9):4256. https://doi.org/10.3390/ijms26094256
Chicago/Turabian StyleLi, Xiangqiong, Wenling Chen, Zhuangzhuang Jia, Yahui Xiao, Anhua Shi, and Xuan Ma. 2025. "Mitochondrial Dysfunction as a Pathogenesis and Therapeutic Strategy for Metabolic-Dysfunction-Associated Steatotic Liver Disease" International Journal of Molecular Sciences 26, no. 9: 4256. https://doi.org/10.3390/ijms26094256
APA StyleLi, X., Chen, W., Jia, Z., Xiao, Y., Shi, A., & Ma, X. (2025). Mitochondrial Dysfunction as a Pathogenesis and Therapeutic Strategy for Metabolic-Dysfunction-Associated Steatotic Liver Disease. International Journal of Molecular Sciences, 26(9), 4256. https://doi.org/10.3390/ijms26094256