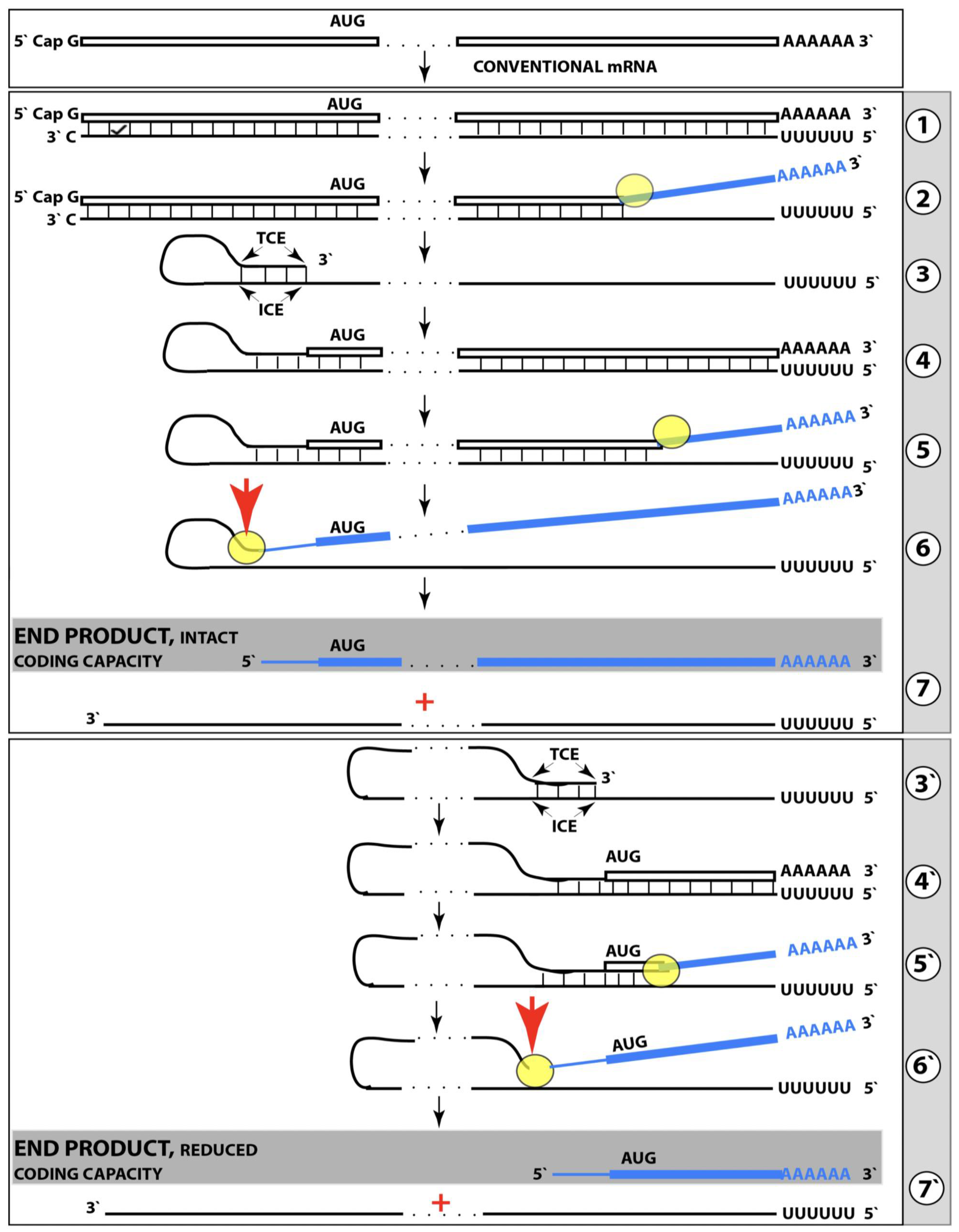
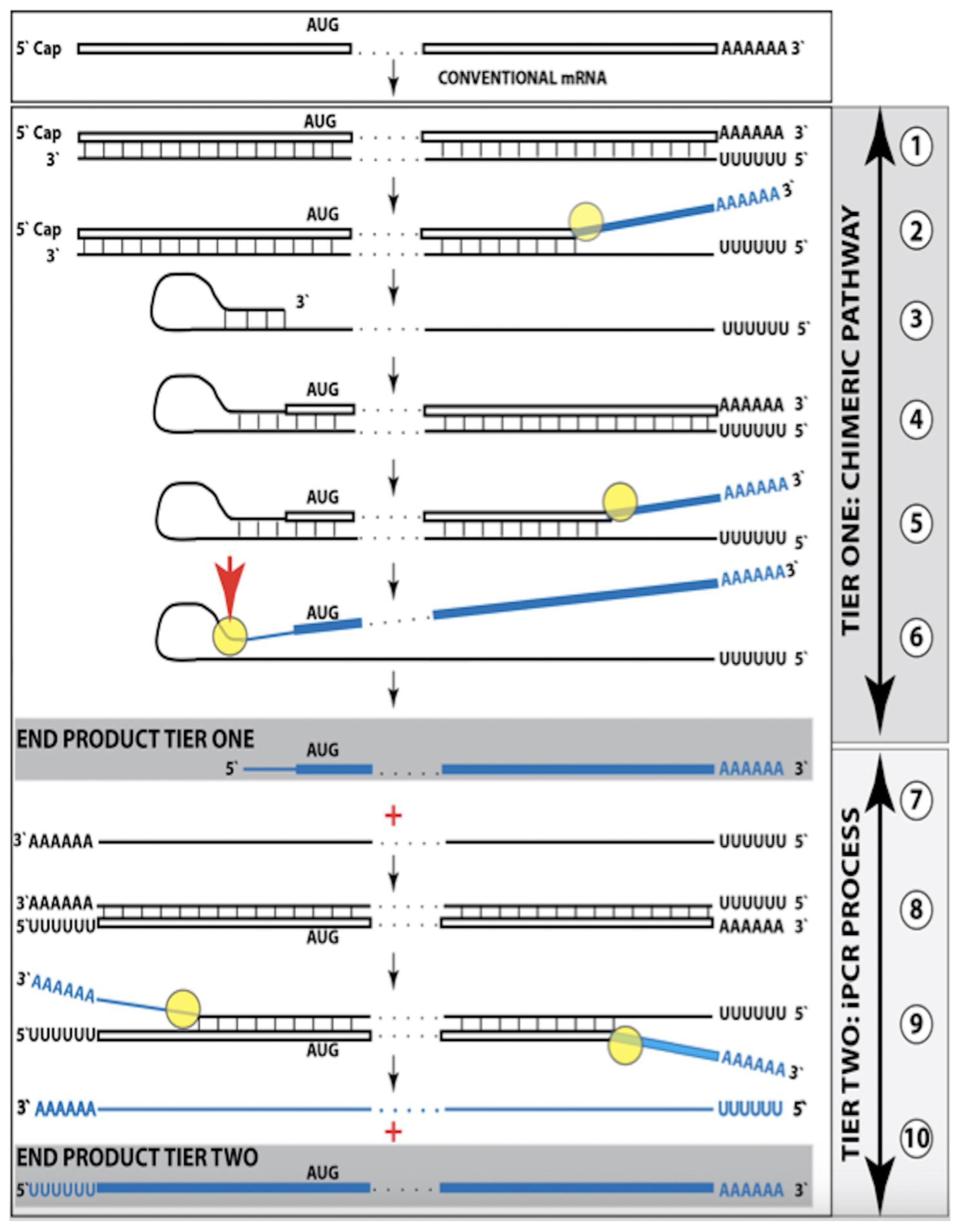
Figure 1.
Mammalian RNA-dependent mRNA amplification: Principal stages. Single lines: Antisense RNA.
Boxed lines: Sense RNA.
Blue boxed lines: Single-stranded RNA separated from its complementary RNA strand by helicase.
Yellow circles: Helicase complex containing helicase strand-separating activity, nucleotide modifying activity and RNA cleaving activity.
Red arrows: positions of the cleavage of the intermediate generating RNA end products of the amplification process.
RdRp: RNA-dependent RNA polymerase.
AUG: Translation initiation codon.
TCE: 3′ Terminal Complementary Element of the antisense RNA strands.
ICE: Internal Complementary Element of the antisense RNA strands.
Top panel: Conventionally transcribed mRNA molecule referred to as progenitor mRNA.
Middle panel: Stages of the chimeric pathway of the RNA-dependent mRNA amplification; the ICE is located within a portion of antisense RNA corresponding to the 5′ UTR of the progenitor mRNA molecule. (
1): The progenitor mRNA is transcribed by RdRp into antisense RNA. (
2): Complementary RNA strands are separated by the helicase activity. Helicase complex mounts the 3′ poly(A) segment and moves along the sense RNA strand modifying on average ever fifth nucleotide. (
3): Folding of the antisense RNA into self-priming configuration is guided by the interaction of its TCE and ICE elements. (
4): RdRp extends the 3′ terminus of self-primed antisense RNA. This produces a hairpin-like molecule containing both sense and antisense RNA components and referred to as the chimeric RNA intermediate. (
5): Complementary strands of the chimeric RNA intermediate are separated by the helicase activity. Nucleotide modifications introduced during the separation prevent the re-annealing of the sense and antisense RNA strands. (
6): When the helicase reaches the single-stranded portion of he chimeric intermediate (either the 5′ end of the TCE or a TCE/ICE mismatch) it cleaves the RNA molecule. (
7): End products of RNA-dependent mRNA amplification. Antisense RNA is truncated at its 3′ end and sense portion of the chimeric RNA is truncated at the 5′ end and acquires the cleaved-off antisense RNA fragment; its translation would result in the complete original polypeptide.
Bottom panel: (
3′–
7′) correspond to (
3–
7) of the middle panel. The ICE element is situated within a segment of antisense RNA corresponding to the coding region of the progenitor mRNA. The chimeric RNA end product is 5′-truncated within its coding region. Potential outcome of its translation are described in the main text. Please note that this figure was shown in [
10].
Figure 1.
Mammalian RNA-dependent mRNA amplification: Principal stages. Single lines: Antisense RNA.
Boxed lines: Sense RNA.
Blue boxed lines: Single-stranded RNA separated from its complementary RNA strand by helicase.
Yellow circles: Helicase complex containing helicase strand-separating activity, nucleotide modifying activity and RNA cleaving activity.
Red arrows: positions of the cleavage of the intermediate generating RNA end products of the amplification process.
RdRp: RNA-dependent RNA polymerase.
AUG: Translation initiation codon.
TCE: 3′ Terminal Complementary Element of the antisense RNA strands.
ICE: Internal Complementary Element of the antisense RNA strands.
Top panel: Conventionally transcribed mRNA molecule referred to as progenitor mRNA.
Middle panel: Stages of the chimeric pathway of the RNA-dependent mRNA amplification; the ICE is located within a portion of antisense RNA corresponding to the 5′ UTR of the progenitor mRNA molecule. (
1): The progenitor mRNA is transcribed by RdRp into antisense RNA. (
2): Complementary RNA strands are separated by the helicase activity. Helicase complex mounts the 3′ poly(A) segment and moves along the sense RNA strand modifying on average ever fifth nucleotide. (
3): Folding of the antisense RNA into self-priming configuration is guided by the interaction of its TCE and ICE elements. (
4): RdRp extends the 3′ terminus of self-primed antisense RNA. This produces a hairpin-like molecule containing both sense and antisense RNA components and referred to as the chimeric RNA intermediate. (
5): Complementary strands of the chimeric RNA intermediate are separated by the helicase activity. Nucleotide modifications introduced during the separation prevent the re-annealing of the sense and antisense RNA strands. (
6): When the helicase reaches the single-stranded portion of he chimeric intermediate (either the 5′ end of the TCE or a TCE/ICE mismatch) it cleaves the RNA molecule. (
7): End products of RNA-dependent mRNA amplification. Antisense RNA is truncated at its 3′ end and sense portion of the chimeric RNA is truncated at the 5′ end and acquires the cleaved-off antisense RNA fragment; its translation would result in the complete original polypeptide.
Bottom panel: (
3′–
7′) correspond to (
3–
7) of the middle panel. The ICE element is situated within a segment of antisense RNA corresponding to the coding region of the progenitor mRNA. The chimeric RNA end product is 5′-truncated within its coding region. Potential outcome of its translation are described in the main text. Please note that this figure was shown in [
10].
![Ijms 26 04252 g001]()
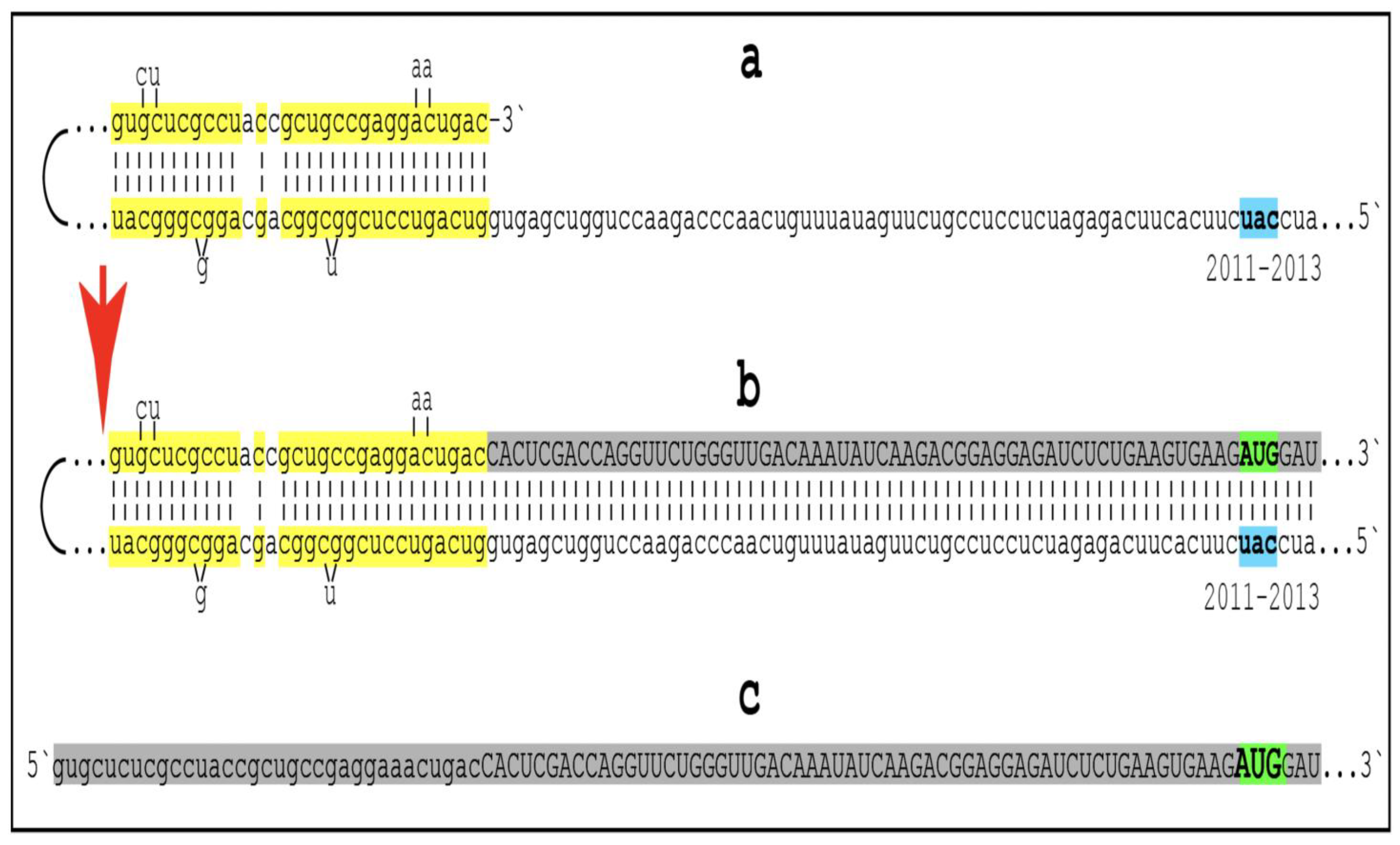
Figure 2.
Human AβPP mRNA Is a Legitimate Template for the Asymmetric RNA-dependent Amplification. Small letters: nucleotide sequences of the relevant segments of the human antisense AβPP RNA molecule.
Large letters: nucleotide sequence of a portion of human AβPP mRNA produced by the extension of the 3′ end of self-primed antisense RNA.
Highlighted in yellow: the TCE and ICE components of the human antisense AβPP RNA.
2011–2013: Positions counted in numbers of nucleotides from the 3′ terminus of the human AβPP antisense RNA. Nucleotides at these positions form the “uac” (
shown in blue) corresponding to the “AUG” (
shown in green) encoding Met671 of human AβPP mRNA. Segments (
a) to (
c) correspond to stages 3′ to 7′ of
Figure 1. (
a): Folding of human antisense AβPP RNA into self-primed structure is guided by the interaction of its TCE and ICE components. (
b): The extension of the 3′ end of self-primed human antisense AβPP RNA produces 3′-terminal portion of AβPP RNA mRNA (
shown in gray). Following the separation of strands by the helicase complex the hairpin-like intermediate is cleaved. (
c) The cleavage (denoted by red arrow) produces the chimeric RNA end product (
shown in gray). Its sense orientation portion is truncated deep within the coding region of AβPP mRNA; its translation would start from the AUG encoding Met 671 of AβPP and produce the C99 fragment independently of AβPP. Please note that this figure was shown in [
10].
Figure 2.
Human AβPP mRNA Is a Legitimate Template for the Asymmetric RNA-dependent Amplification. Small letters: nucleotide sequences of the relevant segments of the human antisense AβPP RNA molecule.
Large letters: nucleotide sequence of a portion of human AβPP mRNA produced by the extension of the 3′ end of self-primed antisense RNA.
Highlighted in yellow: the TCE and ICE components of the human antisense AβPP RNA.
2011–2013: Positions counted in numbers of nucleotides from the 3′ terminus of the human AβPP antisense RNA. Nucleotides at these positions form the “uac” (
shown in blue) corresponding to the “AUG” (
shown in green) encoding Met671 of human AβPP mRNA. Segments (
a) to (
c) correspond to stages 3′ to 7′ of
Figure 1. (
a): Folding of human antisense AβPP RNA into self-primed structure is guided by the interaction of its TCE and ICE components. (
b): The extension of the 3′ end of self-primed human antisense AβPP RNA produces 3′-terminal portion of AβPP RNA mRNA (
shown in gray). Following the separation of strands by the helicase complex the hairpin-like intermediate is cleaved. (
c) The cleavage (denoted by red arrow) produces the chimeric RNA end product (
shown in gray). Its sense orientation portion is truncated deep within the coding region of AβPP mRNA; its translation would start from the AUG encoding Met 671 of AβPP and produce the C99 fragment independently of AβPP. Please note that this figure was shown in [
10].
![Ijms 26 04252 g002]()
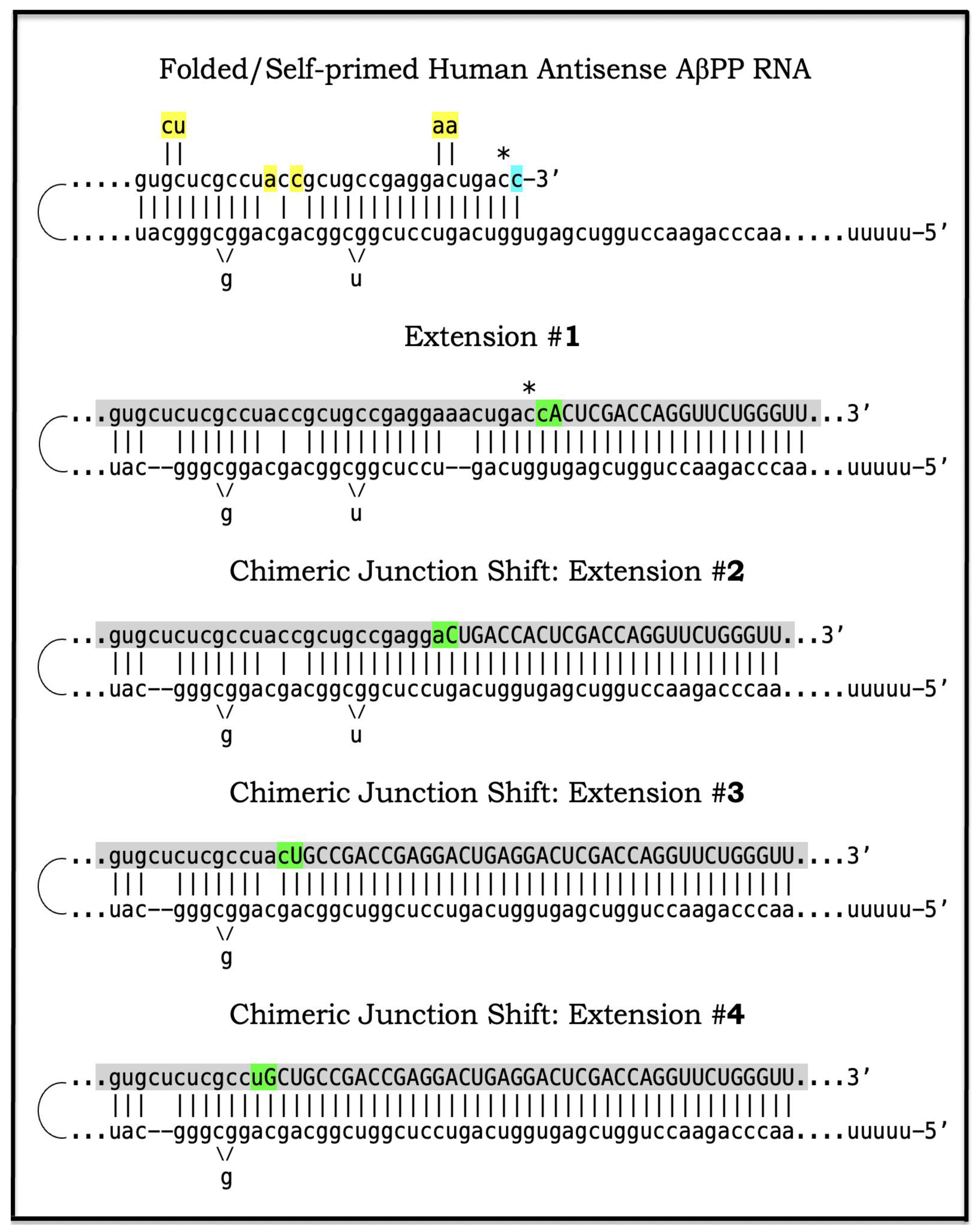
Figure 3.
Anticipated nucleotide sequences of chimeric junction regions of chimeric human AβPP mRNA amplification intermediates. Small letters: Nucleotide sequences of the relevant portions of self-primed antisense AβPP RNA. Large letters: Nucleotide sequences of sense RNA generated by the extension of self-primed human AβPP RNA. Yellow boxes: Mismatches within the TCE/ICE double-stranded complex. Green boxes: Chimeric junctions consisting of the 3′-terminal nucleotide of antisense RNA and 5′-terminal nucleotide of the sense RNA strand. Asterisk: The nucleotide of the antisense RNA molecule corresponding to the transcription start site positioned 149 nucleotides upstream from the translation-initiating AUG codon. Shown in blue: “c” transcribed from the 5′-terminal cap “G” of AβPP mRNA; it is accommodated within the self primed antisense RNA. Shown in gray: anticipated nucleotide sequences of the regions containing chimeric junctions. Extension #1 of the self-primed RNA generates chimeric RNA intermediate containing the full-size TCE element of antisense RNA. Extension #2: Cleavage of the chimeric RNA intermediate occurs at the first TCE/ICE mismatch. Self-primed antisense RNA configuration remains stable and is extended; the chimeric junction shifts upstream from the original one. Extension #3: Cleavage of the chimeric RNA intermediate occurs at the second TCE/ICE mismatch. Self-primed antisense RNA configuration remains stable and is extended again; the chimeric junction shifts upstream. Extension #4: Cleavage of the chimeric RNA intermediate occurs at the third TCE/ICE mismatch. Self-primed antisense RNA conformation remains stable and is extended once more; the chimeric junction shifts as shown.
Figure 3.
Anticipated nucleotide sequences of chimeric junction regions of chimeric human AβPP mRNA amplification intermediates. Small letters: Nucleotide sequences of the relevant portions of self-primed antisense AβPP RNA. Large letters: Nucleotide sequences of sense RNA generated by the extension of self-primed human AβPP RNA. Yellow boxes: Mismatches within the TCE/ICE double-stranded complex. Green boxes: Chimeric junctions consisting of the 3′-terminal nucleotide of antisense RNA and 5′-terminal nucleotide of the sense RNA strand. Asterisk: The nucleotide of the antisense RNA molecule corresponding to the transcription start site positioned 149 nucleotides upstream from the translation-initiating AUG codon. Shown in blue: “c” transcribed from the 5′-terminal cap “G” of AβPP mRNA; it is accommodated within the self primed antisense RNA. Shown in gray: anticipated nucleotide sequences of the regions containing chimeric junctions. Extension #1 of the self-primed RNA generates chimeric RNA intermediate containing the full-size TCE element of antisense RNA. Extension #2: Cleavage of the chimeric RNA intermediate occurs at the first TCE/ICE mismatch. Self-primed antisense RNA configuration remains stable and is extended; the chimeric junction shifts upstream from the original one. Extension #3: Cleavage of the chimeric RNA intermediate occurs at the second TCE/ICE mismatch. Self-primed antisense RNA configuration remains stable and is extended again; the chimeric junction shifts upstream. Extension #4: Cleavage of the chimeric RNA intermediate occurs at the third TCE/ICE mismatch. Self-primed antisense RNA conformation remains stable and is extended once more; the chimeric junction shifts as shown.
![Ijms 26 04252 g003]()
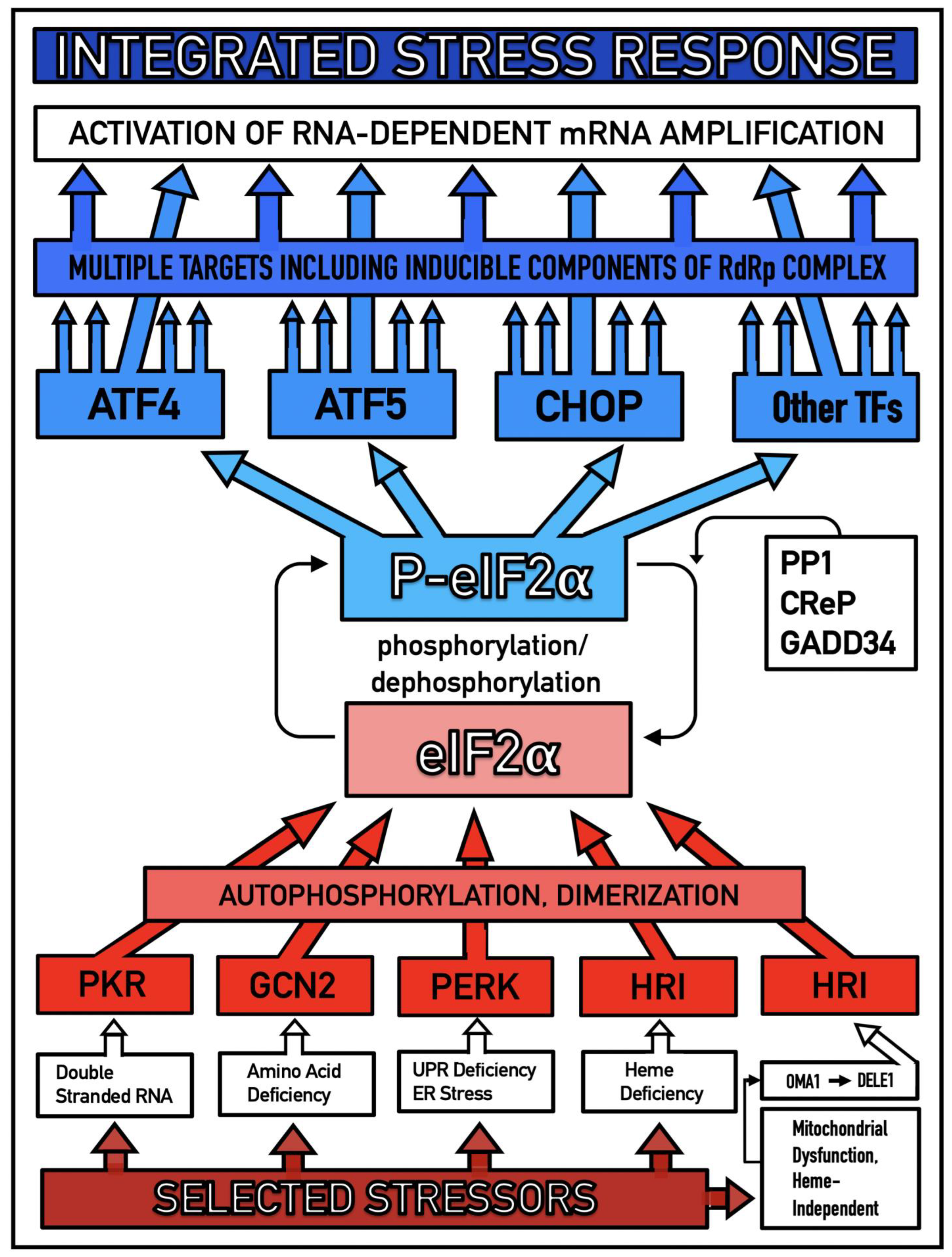
Figure 4.
Mammalian RNA-dependent mRNA amplification is enabled by the integrated stress response. Distinct stressors selectively named in the Figure activate, in a stressor-specific manner, a subset (one or more) of eIF2α kinases: PKR, GCN2, PERK, and HRI; the activation process entails autophosphorylation and dimerization of the kinases. When activated, kinases phosphorylate eIF2α at its Ser51 residue; this is the event, which integrates a multitude of signaling pathways into a single outcome; it is the essence of the integrated stress response pathway. When elicited, the ISR radically rearranges the transcriptional and translational landscapes of the cell. Global cellular protein production is severely inhibited, primarily through suppression of cap-dependent initiation of translation. Concurrently, the ISR activates cap-independent translation of a small set of selected mRNA species, including those that encode various transcription factors. Among the proteins whose production was enabled by the ISR are essential components of RdRp, not present under regular, non-ISR, conditions. Thus, ISR enables the operation of the mammalian RNA-dependent RNA amplification pathway.
Figure 4.
Mammalian RNA-dependent mRNA amplification is enabled by the integrated stress response. Distinct stressors selectively named in the Figure activate, in a stressor-specific manner, a subset (one or more) of eIF2α kinases: PKR, GCN2, PERK, and HRI; the activation process entails autophosphorylation and dimerization of the kinases. When activated, kinases phosphorylate eIF2α at its Ser51 residue; this is the event, which integrates a multitude of signaling pathways into a single outcome; it is the essence of the integrated stress response pathway. When elicited, the ISR radically rearranges the transcriptional and translational landscapes of the cell. Global cellular protein production is severely inhibited, primarily through suppression of cap-dependent initiation of translation. Concurrently, the ISR activates cap-independent translation of a small set of selected mRNA species, including those that encode various transcription factors. Among the proteins whose production was enabled by the ISR are essential components of RdRp, not present under regular, non-ISR, conditions. Thus, ISR enables the operation of the mammalian RNA-dependent RNA amplification pathway.
![Ijms 26 04252 g004]()
Figure 5.
Mouse AβPP mRNA is not a legitimate template for RNA-dependent mRNA amplification “Human”: human antisense AβPP RNA folded into the self-primed configuration. “Mouse”: The relationship between segments of the mouse antisense AβPP RNA molecule analogous to that of the human TCE and ICE elements. Asterisk: The nucleotide of the antisense RNA molecule corresponding to the transcription-initiating nucleotide of either human or mouse AβPP mRNA; in both human and mouse transcription of 149 upstream from the AUG translation initiation codon. There is no better than random complementarity between segments of mouse antisense AβPP RNA corresponding to the TCE and ICE elements of its human counterpart, and the 3′ overhang would effectively prevent the priming/extension. The possibility that the ICE of mouse antisense AβPP RNA is positioned somewhere else in the molecule was excluded by the blast analysis of its 3′-terminal portion, with the rest of the molecule showing no significant complementarity anywhere. Mouse AβPP mRNA is, therefore, not a legitimate template for RNA-dependent amplification.
Figure 5.
Mouse AβPP mRNA is not a legitimate template for RNA-dependent mRNA amplification “Human”: human antisense AβPP RNA folded into the self-primed configuration. “Mouse”: The relationship between segments of the mouse antisense AβPP RNA molecule analogous to that of the human TCE and ICE elements. Asterisk: The nucleotide of the antisense RNA molecule corresponding to the transcription-initiating nucleotide of either human or mouse AβPP mRNA; in both human and mouse transcription of 149 upstream from the AUG translation initiation codon. There is no better than random complementarity between segments of mouse antisense AβPP RNA corresponding to the TCE and ICE elements of its human counterpart, and the 3′ overhang would effectively prevent the priming/extension. The possibility that the ICE of mouse antisense AβPP RNA is positioned somewhere else in the molecule was excluded by the blast analysis of its 3′-terminal portion, with the rest of the molecule showing no significant complementarity anywhere. Mouse AβPP mRNA is, therefore, not a legitimate template for RNA-dependent amplification.
![Ijms 26 04252 g005]()
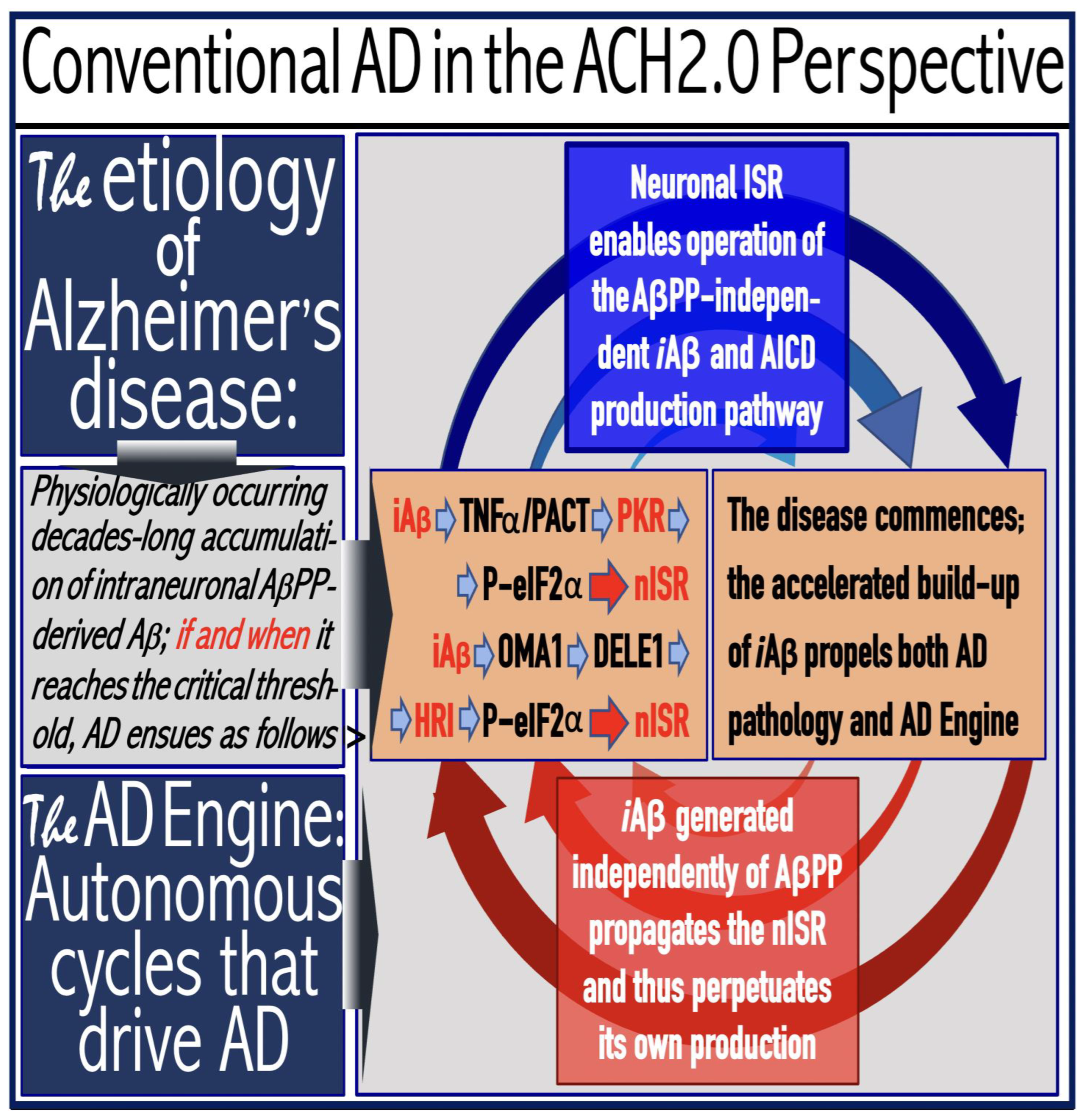
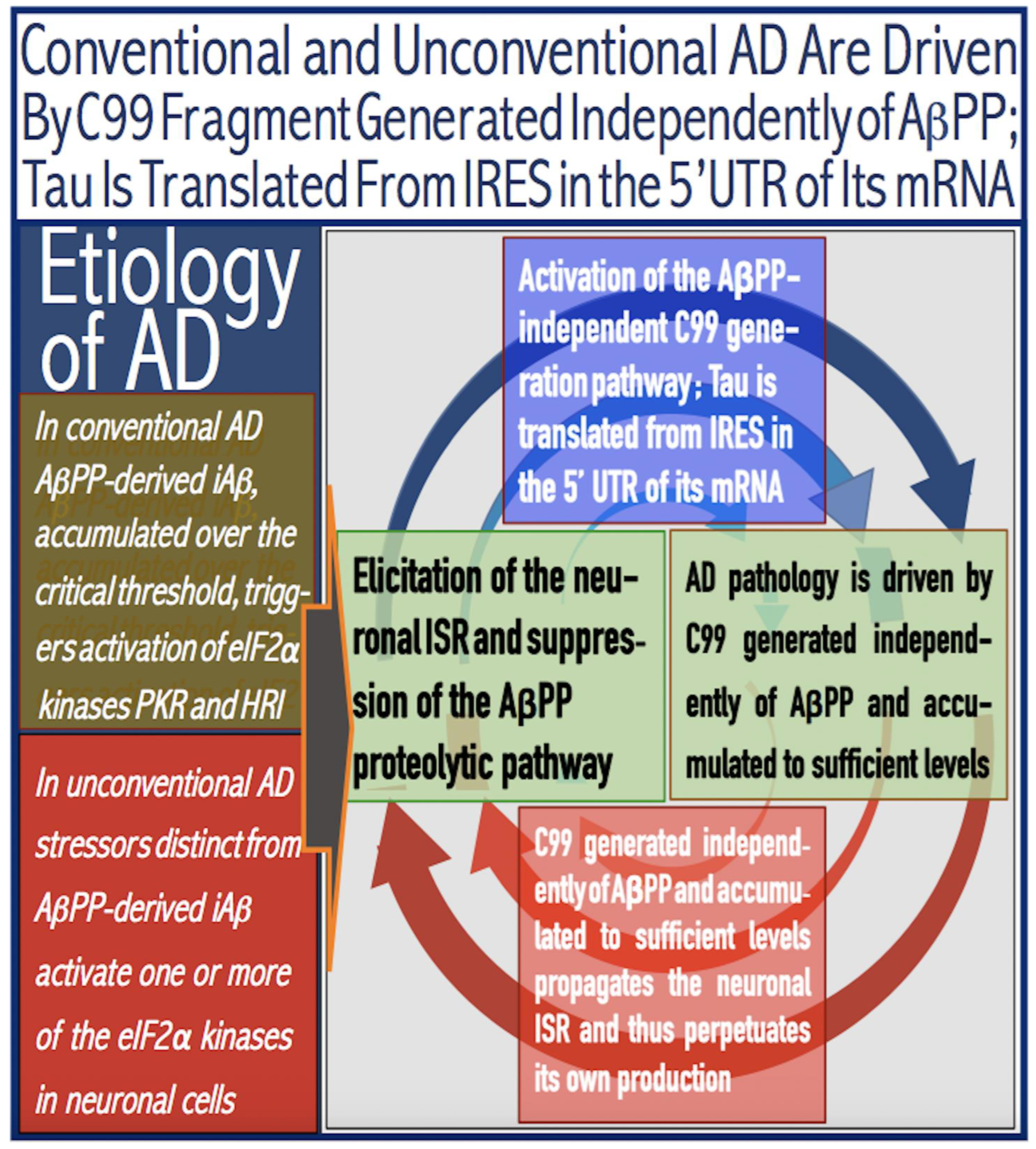
Figure 6.
AD is driven by the self-sustainable AβPP-independent C99 production pathway enabled by the neuronal ISR.
eIF2α: eukaryotic translation initiation factor 2
α.
PKR and HRI: kinases that phosphorylate eIF2α at its Ser51 residue.
PACT: PKR activator.
TNFα: tumor necrosis factor
α.
OMA1: mitochondrial protease activated during mitochondrial dysfunction.
DELE1: substrate of OMA1 protease. Cleavage of DELE1 leads to the activation of HRI. Phosphorylation of eIF2α elicits the integrated stress response, which, in turn, enables the generation of components essential for the activity of the AβPP-independent pathway of C99 and, subsequently, iAβ production. iAβ: intraneuronal Aβ. AβPP-derived iAβ accumulates physiologically via the importation of extracellular Aβ and the retention of iAβ produced by gamma-cleavages on intracellular membranes in an exceedingly slow process. In most individuals it does not reach the PKR- and/or HRI-activating, the neuronal ISR-eliciting threshold within their lifetimes and no conventional AD occurs. When this threshold is crossed, the neuronal ISR is elicited and the AβPP-independent C99 (and, subsequently, iAβ) production pathway activated. The iAβ product of the latter drives AD pathology. It also propagates the neuronal ISR and thus perpetuates its own production in the AβPP-independent pathway. It should be noted that the accumulation of iAβ produced in both the AβPP proteolytic and AβPP-independent pathways is accompanied by a proportional accumulation of AICD (AβPP Intracellular Domain). AICD was shown to be capable of interfering with numerous processes involved in AD [
191,
192,
193,
194,
195,
196,
197,
198,
199,
200,
201,
202,
203,
204,
205,
206] but its contribution to the disease, if any, remains to be elucidated.
Figure 6.
AD is driven by the self-sustainable AβPP-independent C99 production pathway enabled by the neuronal ISR.
eIF2α: eukaryotic translation initiation factor 2
α.
PKR and HRI: kinases that phosphorylate eIF2α at its Ser51 residue.
PACT: PKR activator.
TNFα: tumor necrosis factor
α.
OMA1: mitochondrial protease activated during mitochondrial dysfunction.
DELE1: substrate of OMA1 protease. Cleavage of DELE1 leads to the activation of HRI. Phosphorylation of eIF2α elicits the integrated stress response, which, in turn, enables the generation of components essential for the activity of the AβPP-independent pathway of C99 and, subsequently, iAβ production. iAβ: intraneuronal Aβ. AβPP-derived iAβ accumulates physiologically via the importation of extracellular Aβ and the retention of iAβ produced by gamma-cleavages on intracellular membranes in an exceedingly slow process. In most individuals it does not reach the PKR- and/or HRI-activating, the neuronal ISR-eliciting threshold within their lifetimes and no conventional AD occurs. When this threshold is crossed, the neuronal ISR is elicited and the AβPP-independent C99 (and, subsequently, iAβ) production pathway activated. The iAβ product of the latter drives AD pathology. It also propagates the neuronal ISR and thus perpetuates its own production in the AβPP-independent pathway. It should be noted that the accumulation of iAβ produced in both the AβPP proteolytic and AβPP-independent pathways is accompanied by a proportional accumulation of AICD (AβPP Intracellular Domain). AICD was shown to be capable of interfering with numerous processes involved in AD [
191,
192,
193,
194,
195,
196,
197,
198,
199,
200,
201,
202,
203,
204,
205,
206] but its contribution to the disease, if any, remains to be elucidated.
![Ijms 26 04252 g006]()
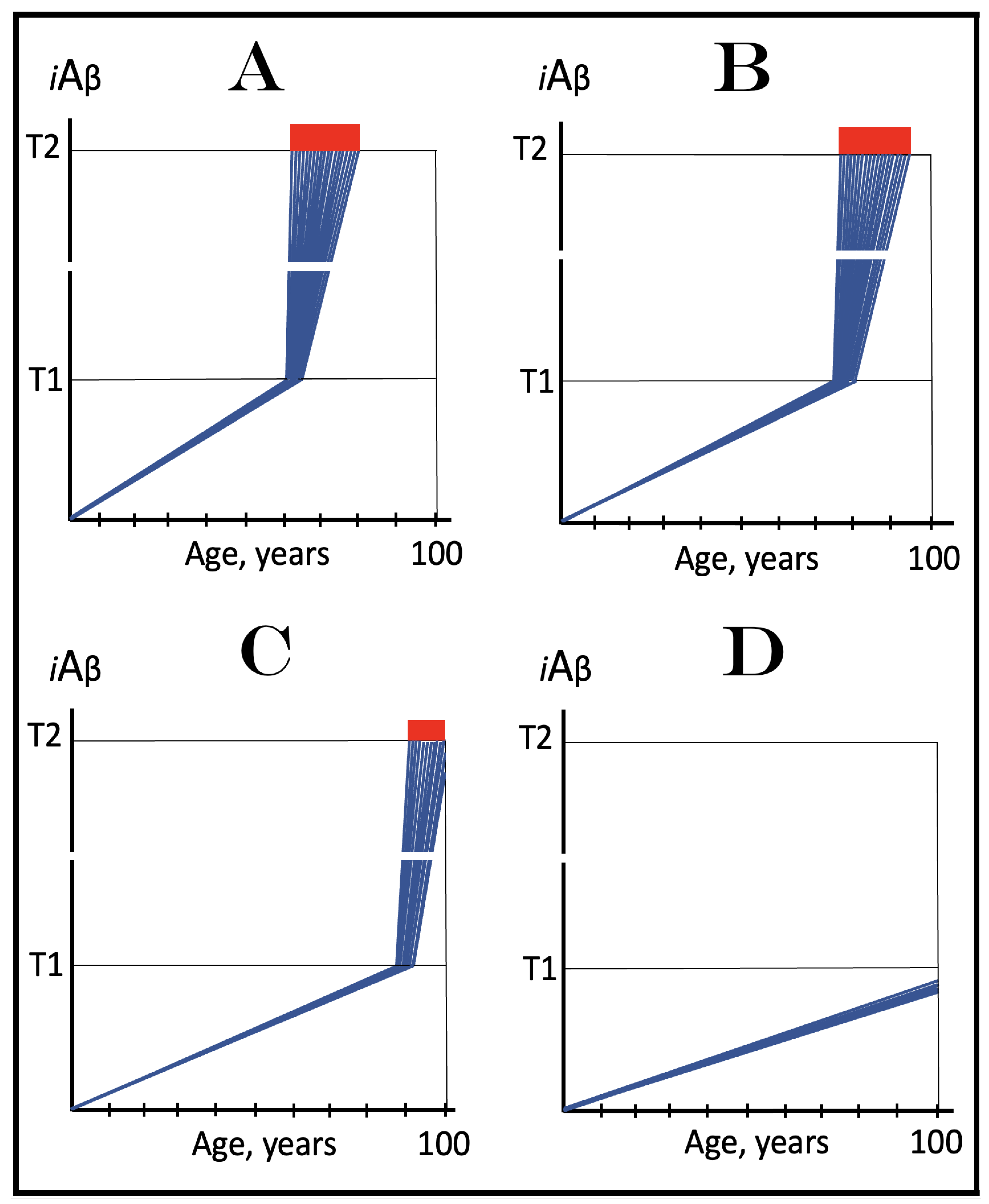
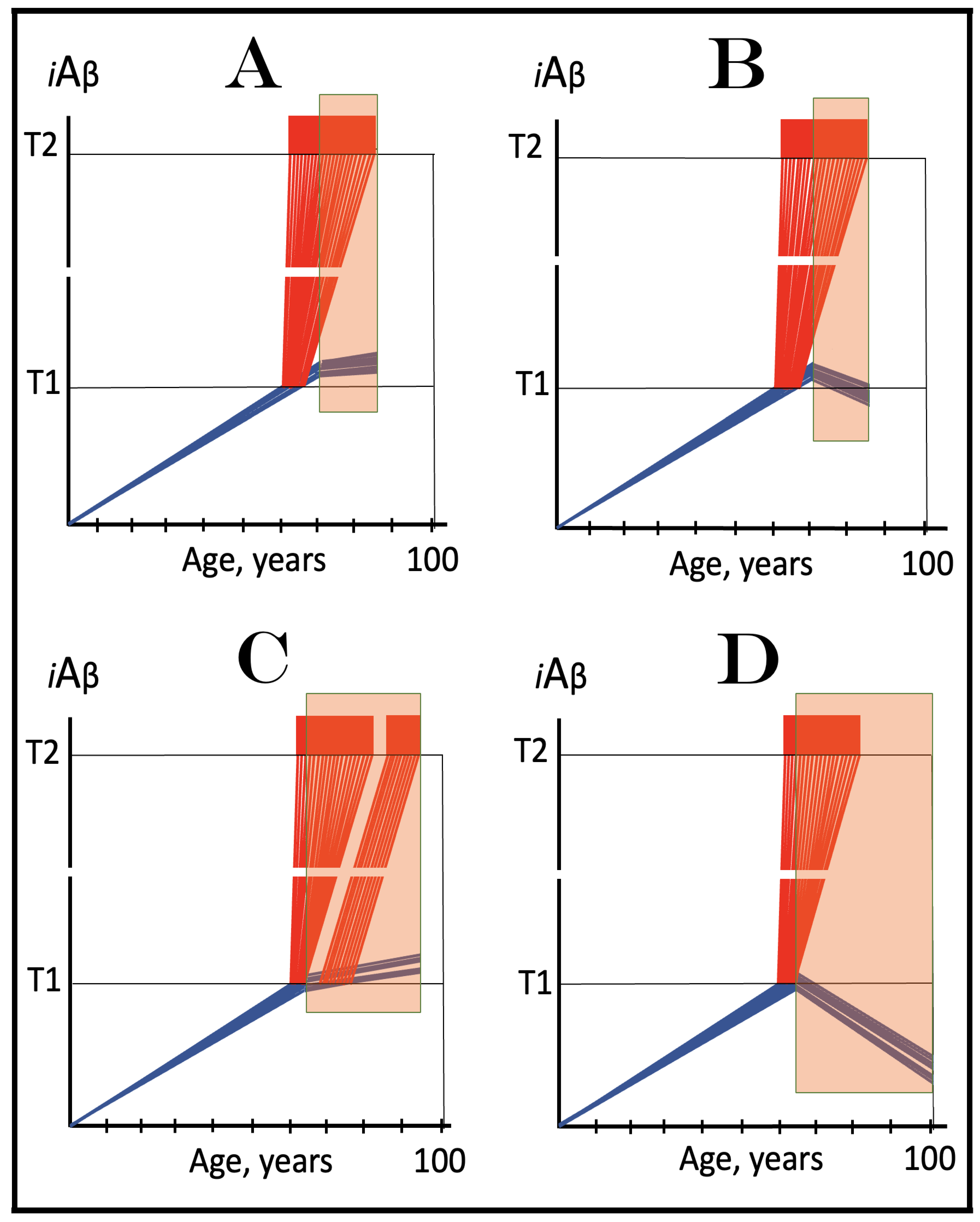
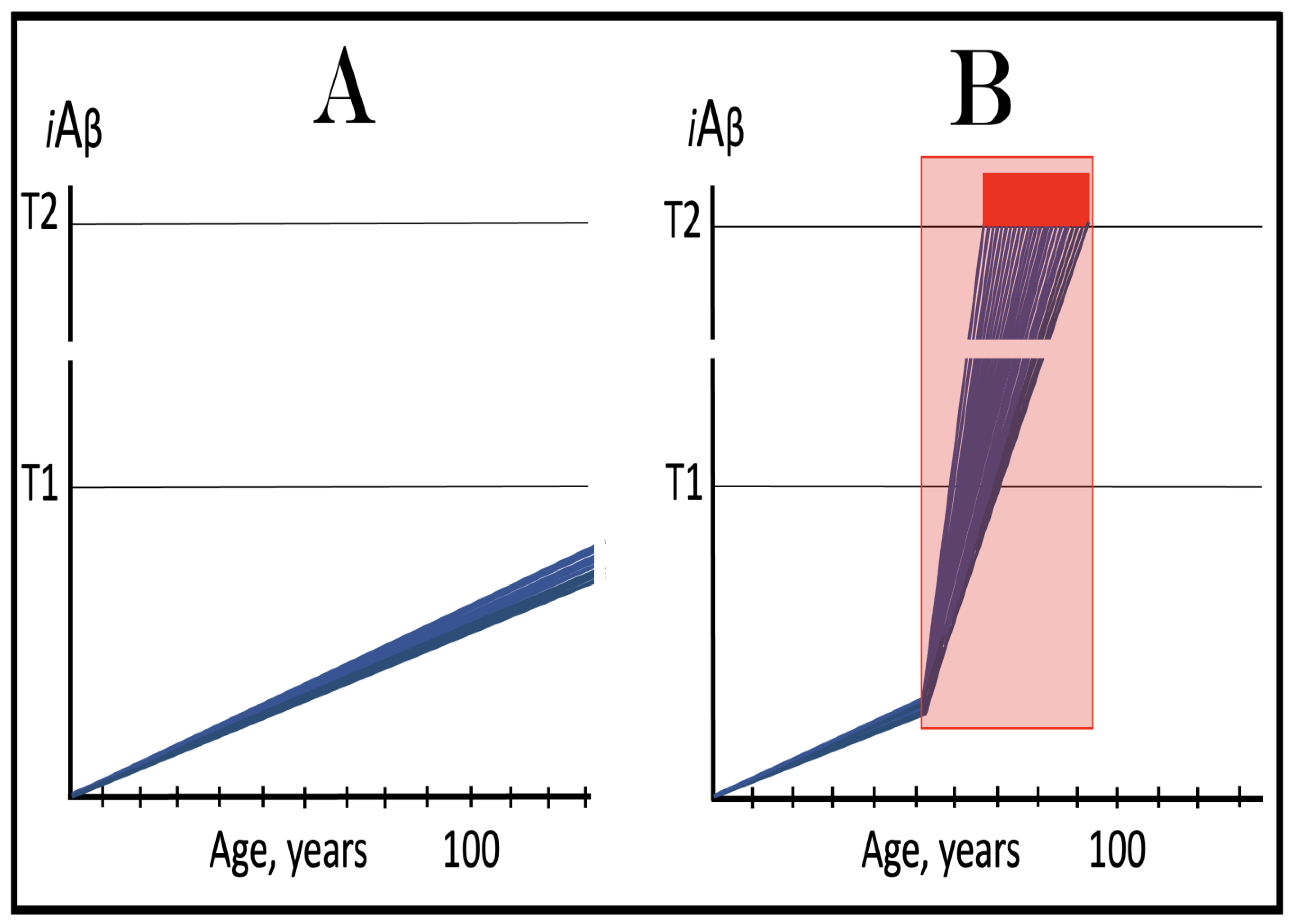
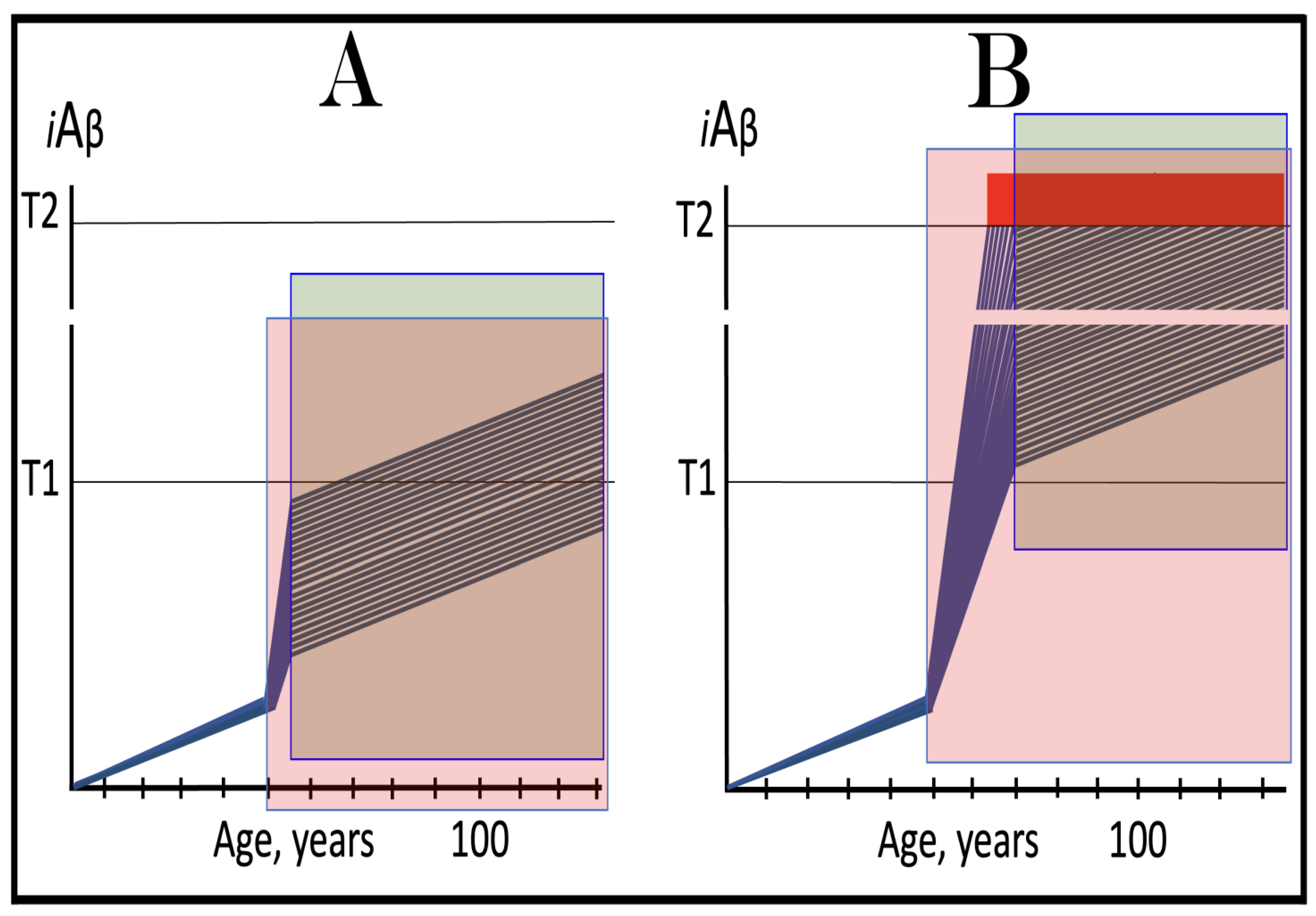
Figure 7.
The occurrence and timing of conventional AD is determined by the dynamics of accumulation of AβPP-derived iAβ.
Blue lines: Intraneuronal Aβ, iAβ.
T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD.
T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis [
207].
Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. The only variable parameter in the present figure is the rate of accumulation of AβPP-derived iAβ.
Panel (
A): The rate of accumulation of AβPP-derived iAβ is relatively rapid. It crosses the T1 threshold at mid-sixties. PKR and/or HRI are activated, eIF2α phosphorylated, the neuronal ISR elicited, AβPP-independent C99 generation pathway initiated, and AD commences.
Panel (
B): The rate of accumulation of AβPP-derived iAβ is reduced. The T1 threshold is reached and crossed, and the disease commences at around the age of eighty.
Panel (
C): The rate of accumulation of AβPP-derived iAβ is reduced further. The T1 threshold is reached, and the disease commences only in the nineties.
Panel (
D): AβPP-derived iAβ does not reach the T1 threshold within the lifespan of the individual; the neuronal ISR is not elicited, the AβPP-independent C99 generation pathway is not activated, and conventional AD does not occur. Such an outcome is typical for the majority of the human population.
Figure 7.
The occurrence and timing of conventional AD is determined by the dynamics of accumulation of AβPP-derived iAβ.
Blue lines: Intraneuronal Aβ, iAβ.
T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD.
T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis [
207].
Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. The only variable parameter in the present figure is the rate of accumulation of AβPP-derived iAβ.
Panel (
A): The rate of accumulation of AβPP-derived iAβ is relatively rapid. It crosses the T1 threshold at mid-sixties. PKR and/or HRI are activated, eIF2α phosphorylated, the neuronal ISR elicited, AβPP-independent C99 generation pathway initiated, and AD commences.
Panel (
B): The rate of accumulation of AβPP-derived iAβ is reduced. The T1 threshold is reached and crossed, and the disease commences at around the age of eighty.
Panel (
C): The rate of accumulation of AβPP-derived iAβ is reduced further. The T1 threshold is reached, and the disease commences only in the nineties.
Panel (
D): AβPP-derived iAβ does not reach the T1 threshold within the lifespan of the individual; the neuronal ISR is not elicited, the AβPP-independent C99 generation pathway is not activated, and conventional AD does not occur. Such an outcome is typical for the majority of the human population.
![Ijms 26 04252 g007]()
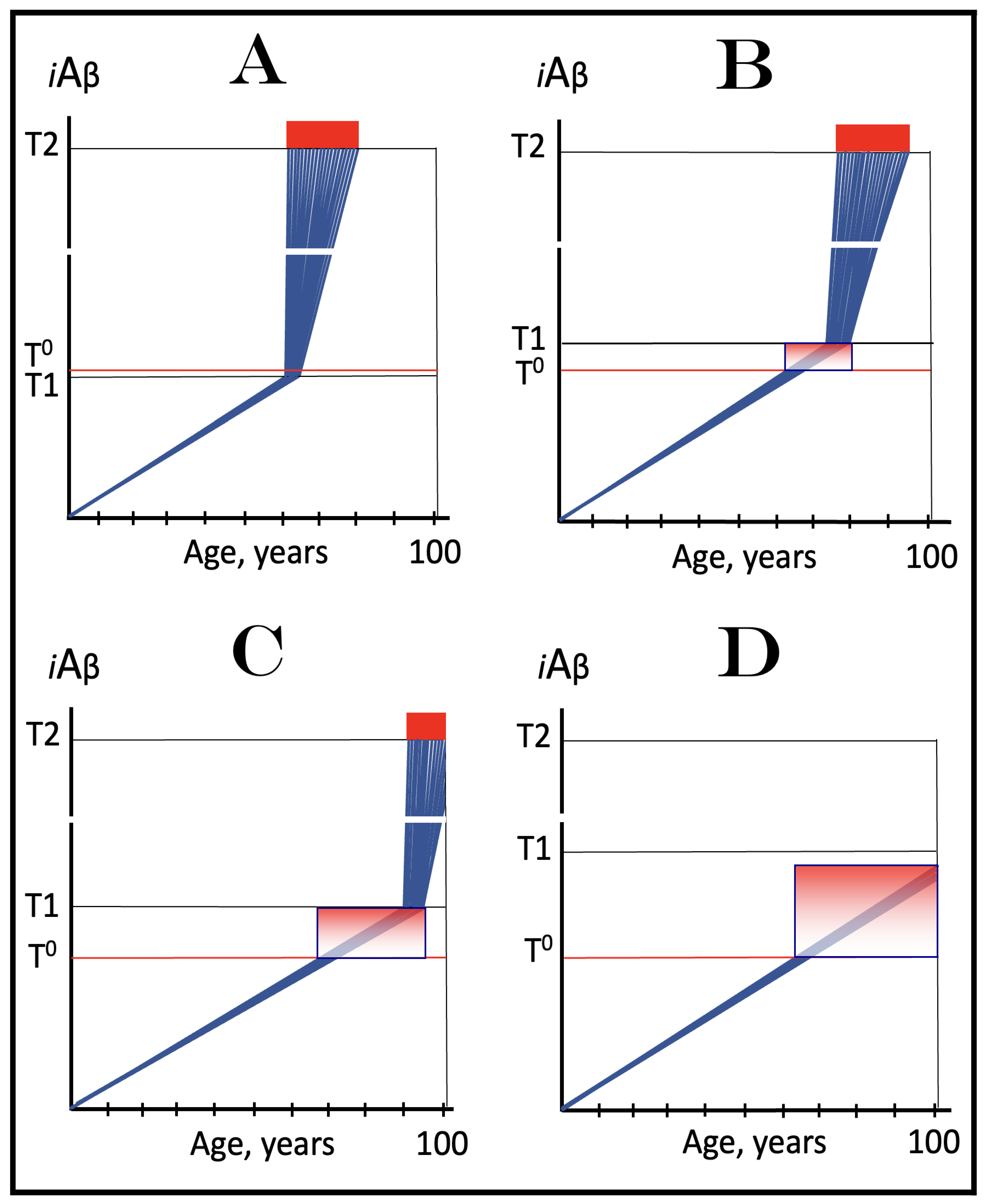
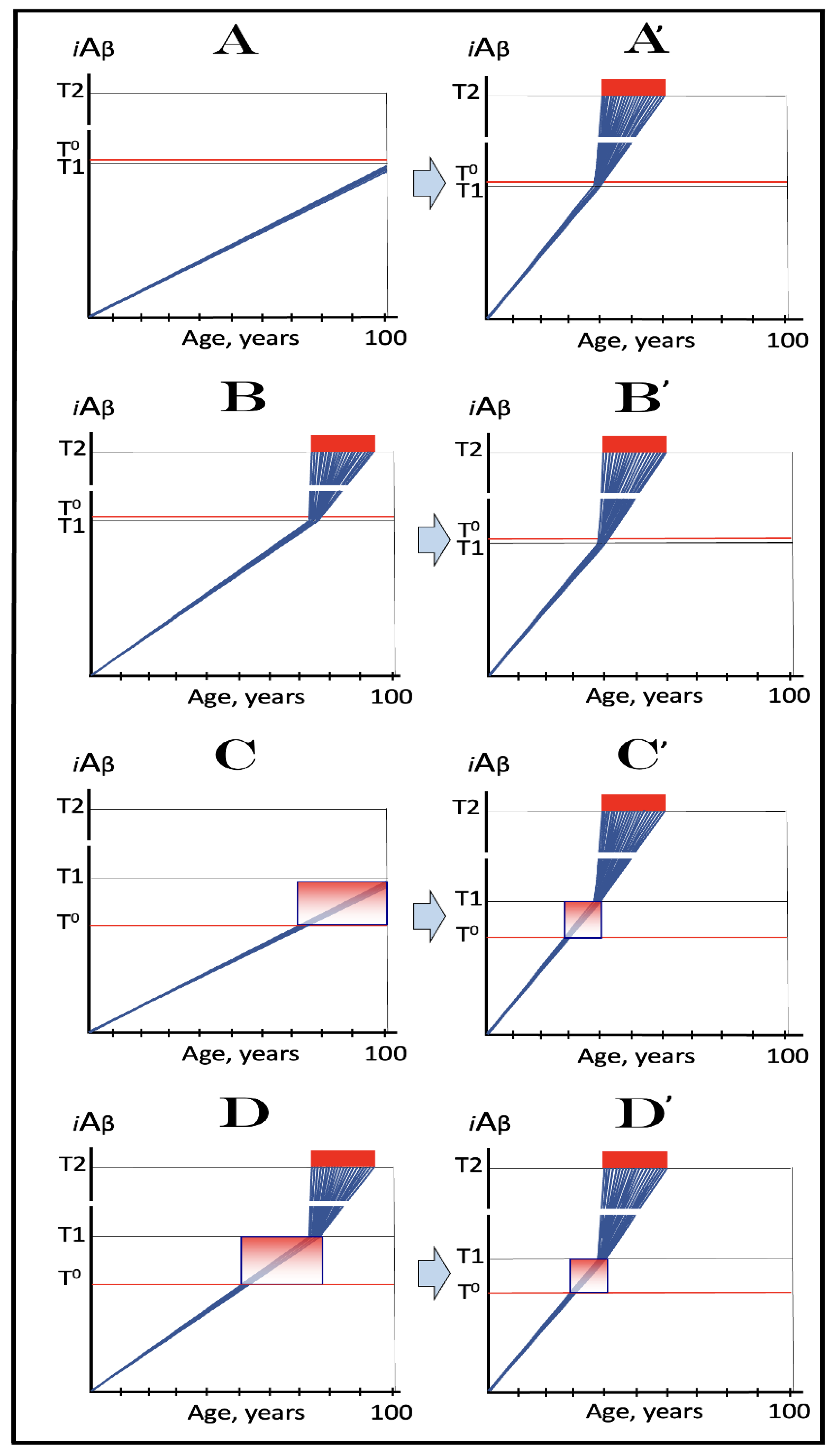
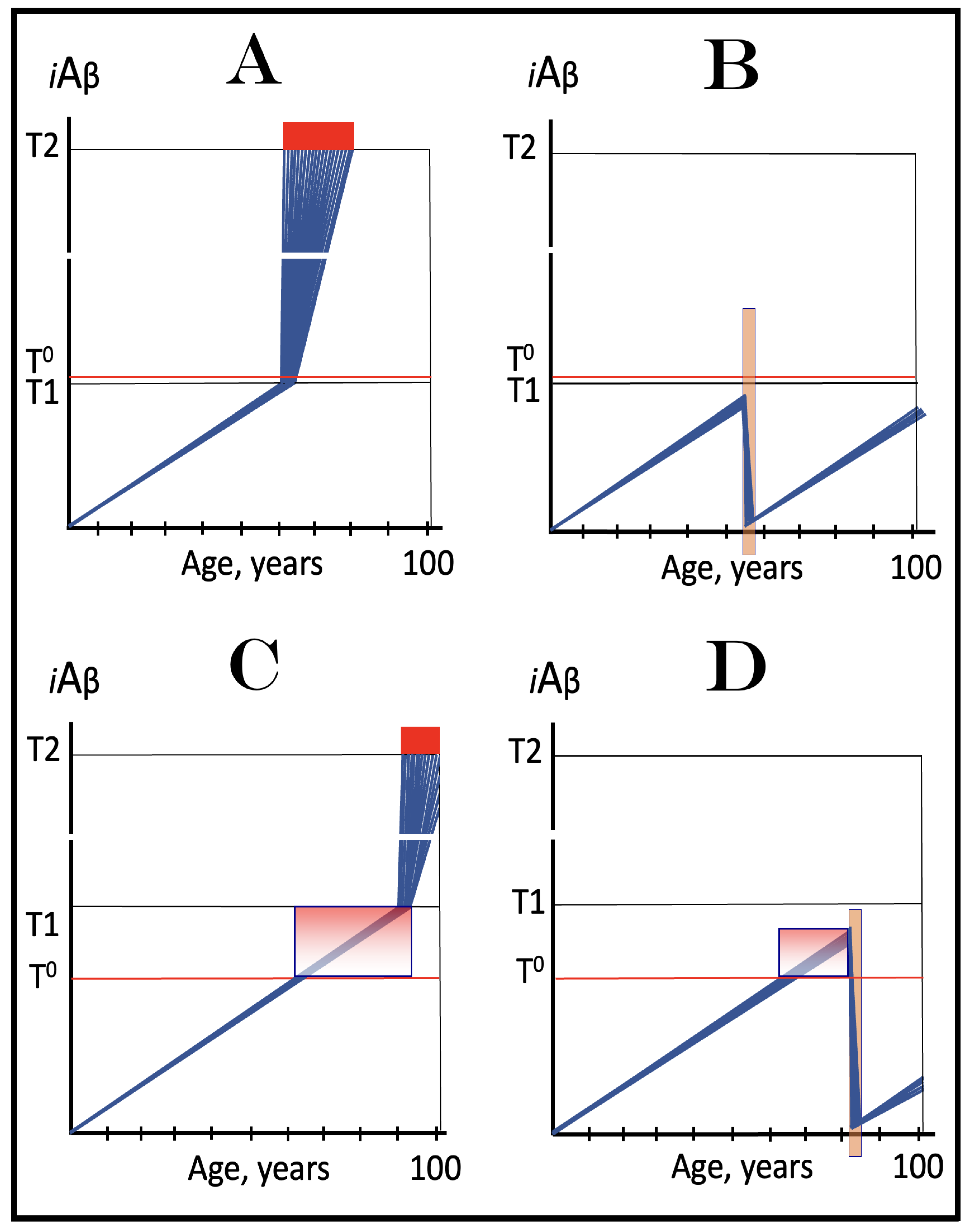
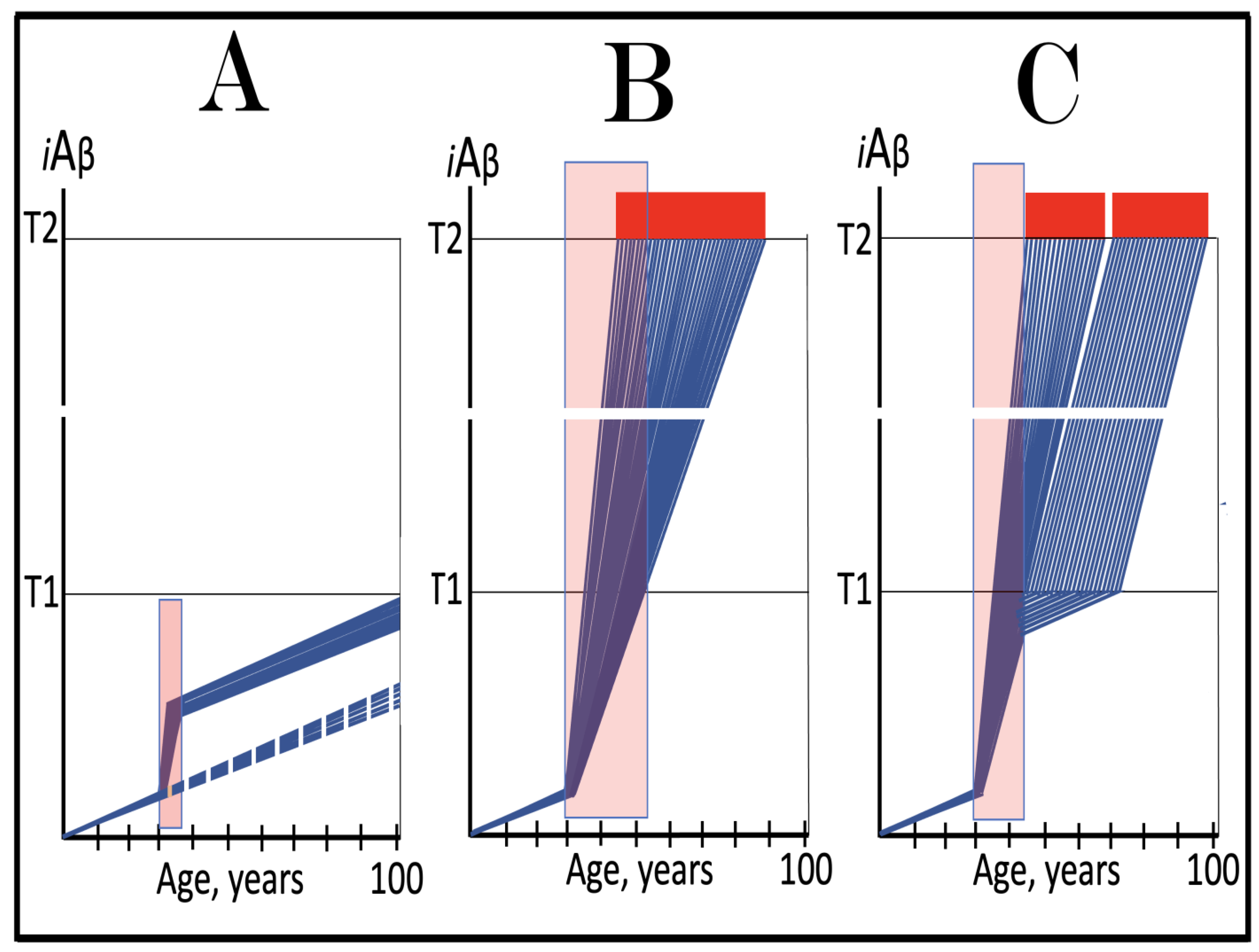
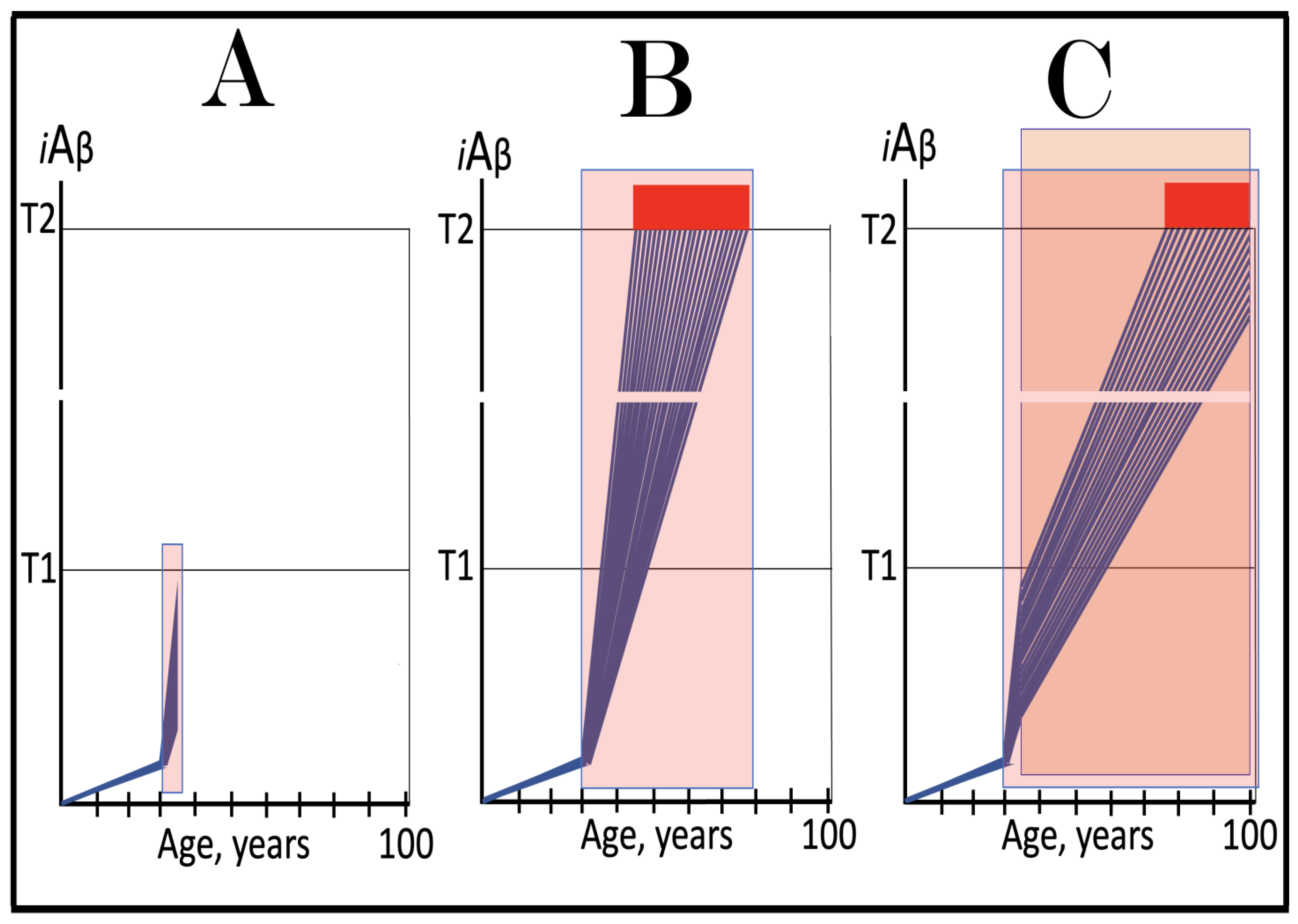
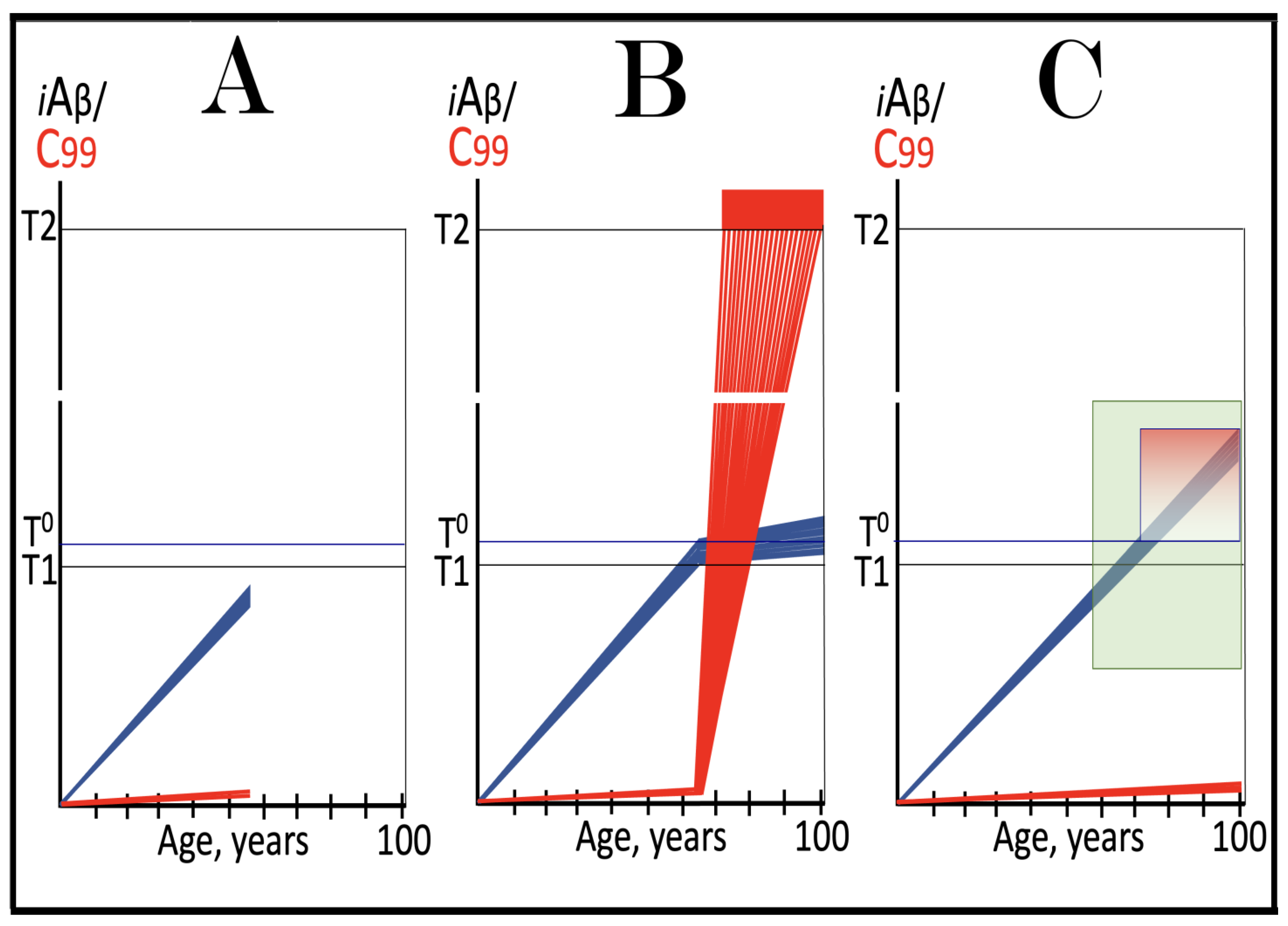
Figure 8.
Role of the extent of the T1 threshold in the incidence and timing of AD and of aging-associated cognitive decline, AACD. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing (if no T1 occurs, AACD persists for the remaining portion of the lifetime). It follows that AACD can occur only if the extent of the T0 threshold is smaller than that of the T1. Panel (A): The T1 threshold is crossed, the neuronal ISR is elicited, the AβPP-independent C99 and iAβ production pathway is activated, and AD commences at about sixty years of age. The extent of the T0 threshold is greater than that of the T1; consequently, no AACD occurs. Panel (B): The extent of the T1 threshold is above that of the T0 threshold. When the T0 is crossed by AβPP-derived iAβ, AACD commences and morphs into AD following the T1 crossing at about seventy-five years of age. Panel (C): The T1 threshold is increased further and is crossed at about ninety years of age. Since the extent of the T0 does not change, AACD commences at the same time as the preceding panel, but its duration increases. Panel (D): The extent of the T1 threshold is such that it is not crossed by AβPP-derived iAβ within the lifetime of the individual; no conventional AD occurs. On the other hand, AACD commences with the T0 crossing and persists for the remaining lifetime.
Figure 8.
Role of the extent of the T1 threshold in the incidence and timing of AD and of aging-associated cognitive decline, AACD. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing (if no T1 occurs, AACD persists for the remaining portion of the lifetime). It follows that AACD can occur only if the extent of the T0 threshold is smaller than that of the T1. Panel (A): The T1 threshold is crossed, the neuronal ISR is elicited, the AβPP-independent C99 and iAβ production pathway is activated, and AD commences at about sixty years of age. The extent of the T0 threshold is greater than that of the T1; consequently, no AACD occurs. Panel (B): The extent of the T1 threshold is above that of the T0 threshold. When the T0 is crossed by AβPP-derived iAβ, AACD commences and morphs into AD following the T1 crossing at about seventy-five years of age. Panel (C): The T1 threshold is increased further and is crossed at about ninety years of age. Since the extent of the T0 does not change, AACD commences at the same time as the preceding panel, but its duration increases. Panel (D): The extent of the T1 threshold is such that it is not crossed by AβPP-derived iAβ within the lifetime of the individual; no conventional AD occurs. On the other hand, AACD commences with the T0 crossing and persists for the remaining lifetime.
![Ijms 26 04252 g008]()
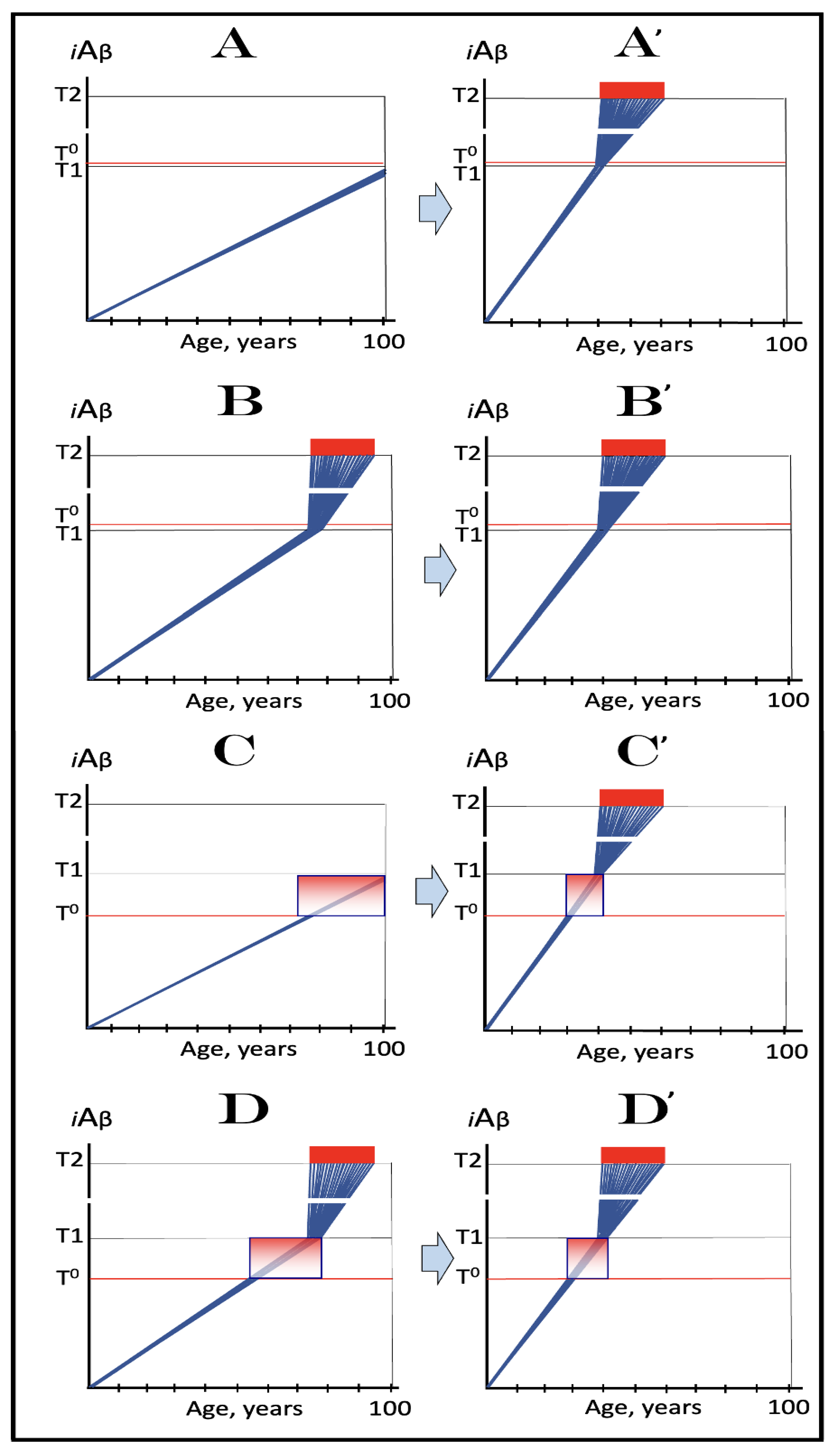
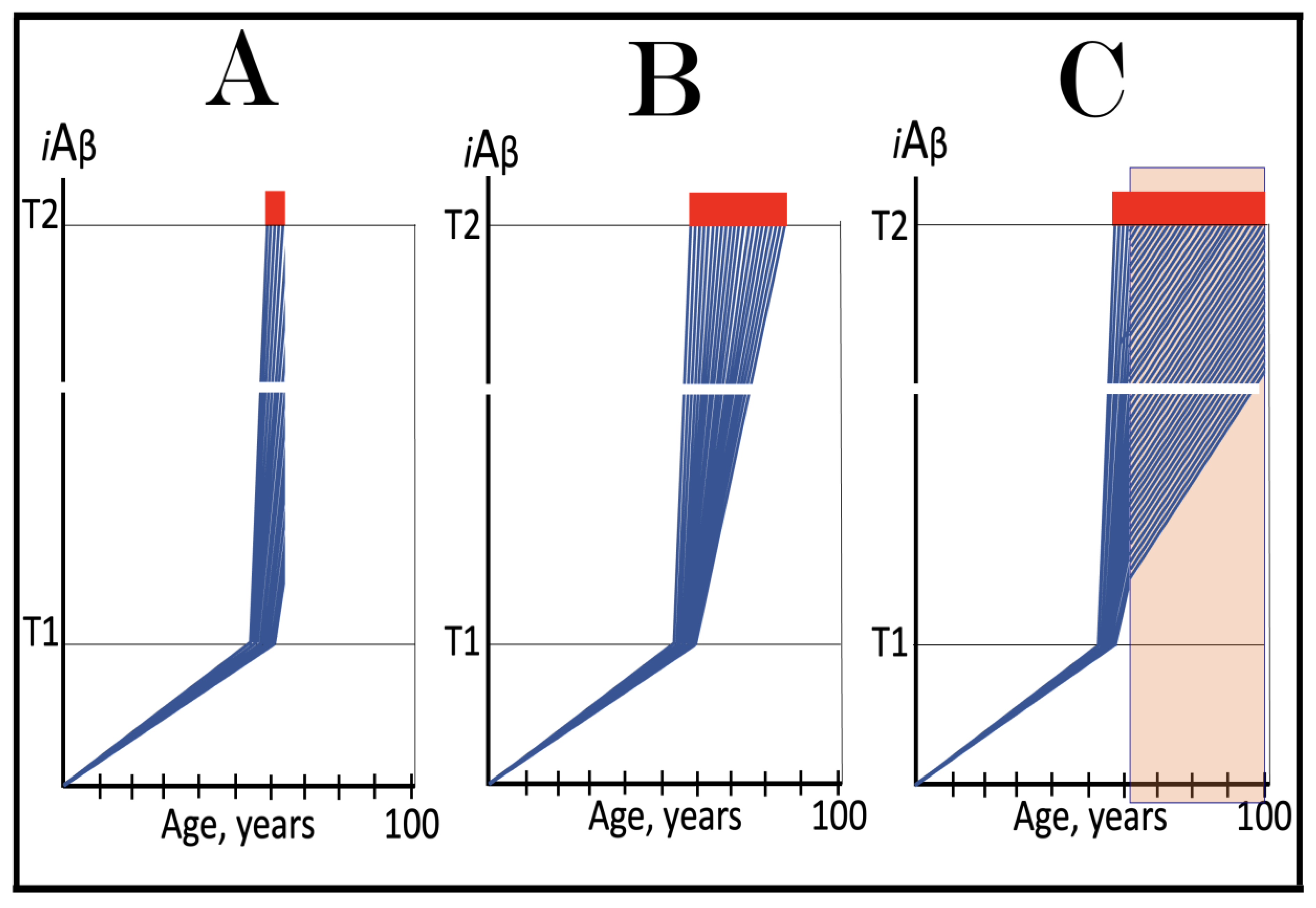
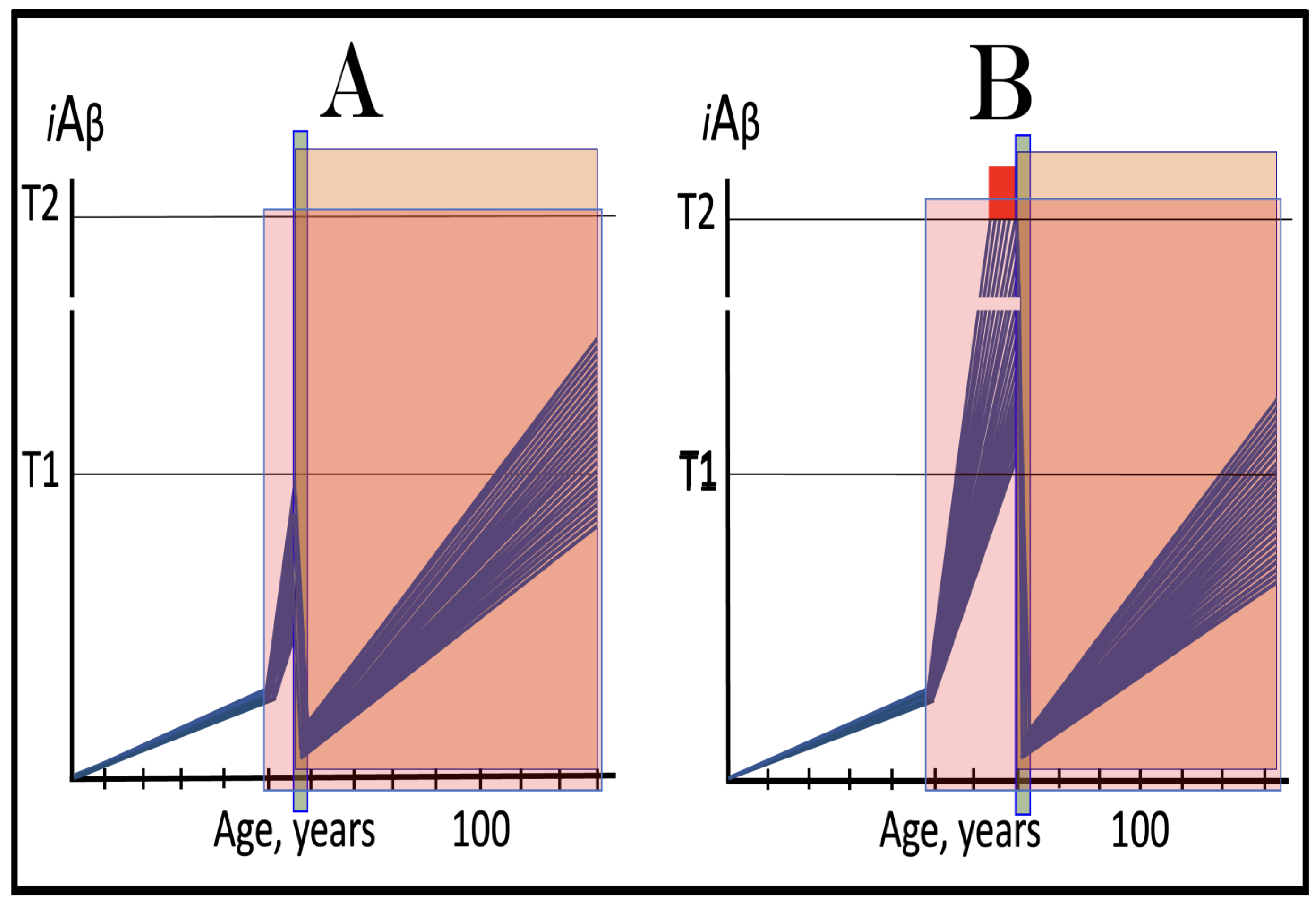
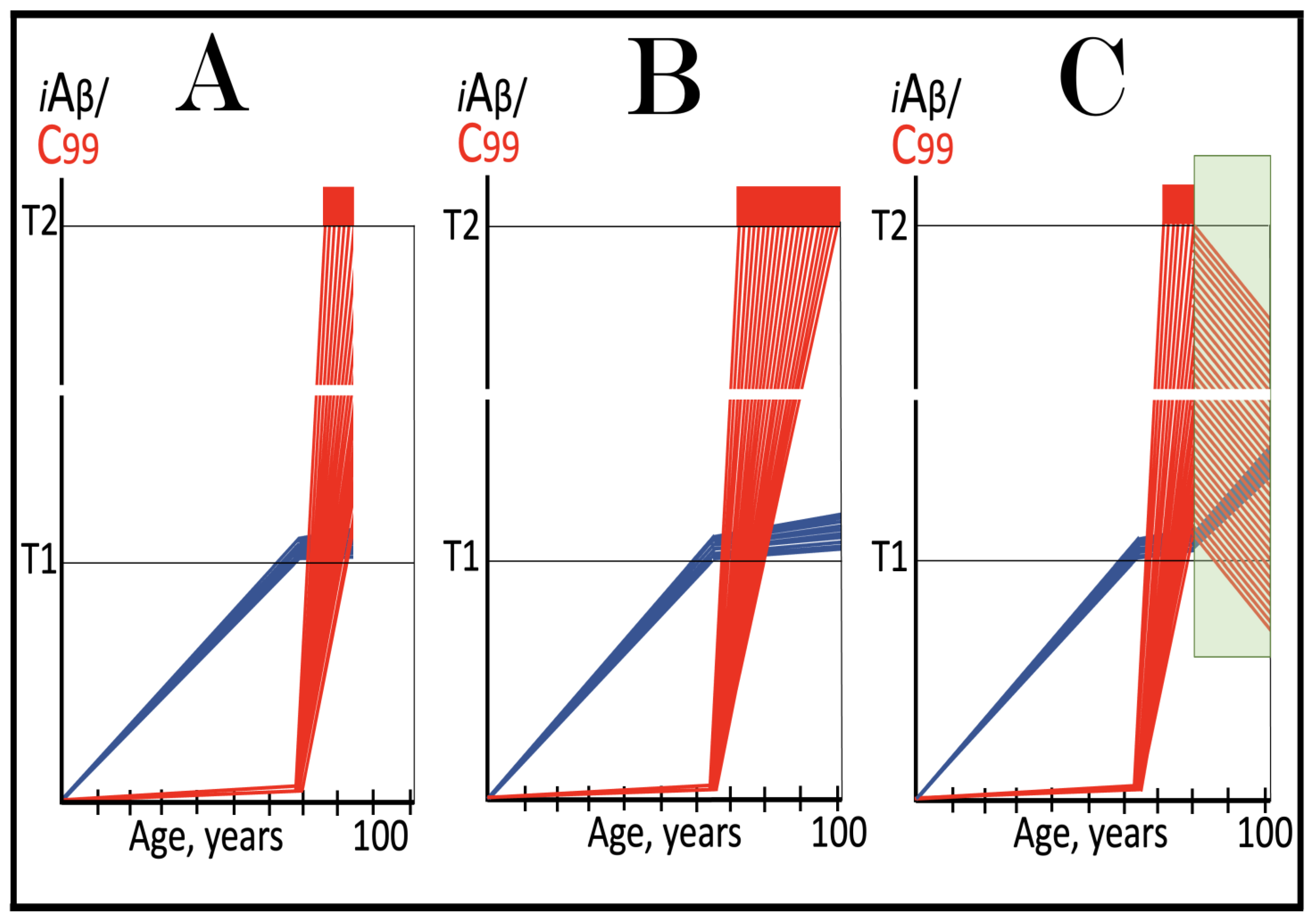
Figure 9.
Familial AD mutations that exert their effect by accelerating the rate of accumulation of AβPP-derived iAβ. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Panels (A–D): dynamics of iAβ accumulation in wild-type AβPP carriers. Panels (A,B): the T0 levels are above those of T1; no AACD occurs. Panel (A): the T1 threshold is not crossed; no disease occurs. Panel (B): the T1 threshold is crossed; this is followed by late onset AD. Panels (C,D): the T0 threshold is lower than T1. Panel (C): the T1 threshold is not reached; when the T0 threshold is crossed, AACD commences and persists for the remaining lifespan. Panel (D): AACD commences when the T0 is crossed and morphs into late onset AD upon the T1 crossing. Panels (A’–D’): Dynamics of iAβ accumulation in carriers of FAD mutations, which elevate the rate of accumulation of AβPP-derived iAβ. As a result, the T1 threshold is crossed sooner, and early-onset AD follows. Panels (A’,B’): the T0 levels are above those of T1; no AACD occurs. Panels (C’,D’): the extent of the T0 threshold is smaller than that of the T1; AACD commences with the T0 crossing and morphs into the early-onset AD when the T1 threshold is reached.
Figure 9.
Familial AD mutations that exert their effect by accelerating the rate of accumulation of AβPP-derived iAβ. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Panels (A–D): dynamics of iAβ accumulation in wild-type AβPP carriers. Panels (A,B): the T0 levels are above those of T1; no AACD occurs. Panel (A): the T1 threshold is not crossed; no disease occurs. Panel (B): the T1 threshold is crossed; this is followed by late onset AD. Panels (C,D): the T0 threshold is lower than T1. Panel (C): the T1 threshold is not reached; when the T0 threshold is crossed, AACD commences and persists for the remaining lifespan. Panel (D): AACD commences when the T0 is crossed and morphs into late onset AD upon the T1 crossing. Panels (A’–D’): Dynamics of iAβ accumulation in carriers of FAD mutations, which elevate the rate of accumulation of AβPP-derived iAβ. As a result, the T1 threshold is crossed sooner, and early-onset AD follows. Panels (A’,B’): the T0 levels are above those of T1; no AACD occurs. Panels (C’,D’): the extent of the T0 threshold is smaller than that of the T1; AACD commences with the T0 crossing and morphs into the early-onset AD when the T1 threshold is reached.
![Ijms 26 04252 g009]()
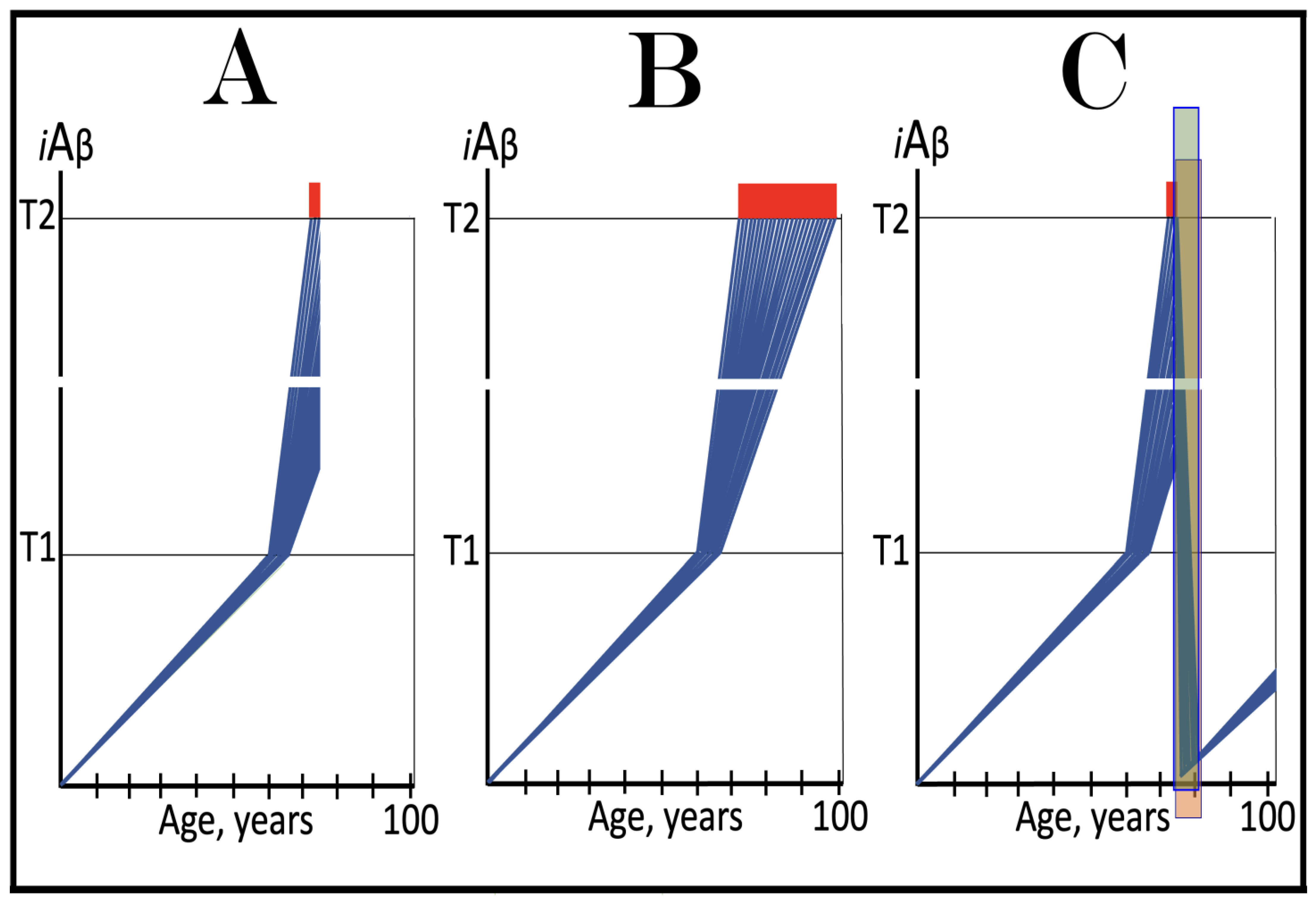
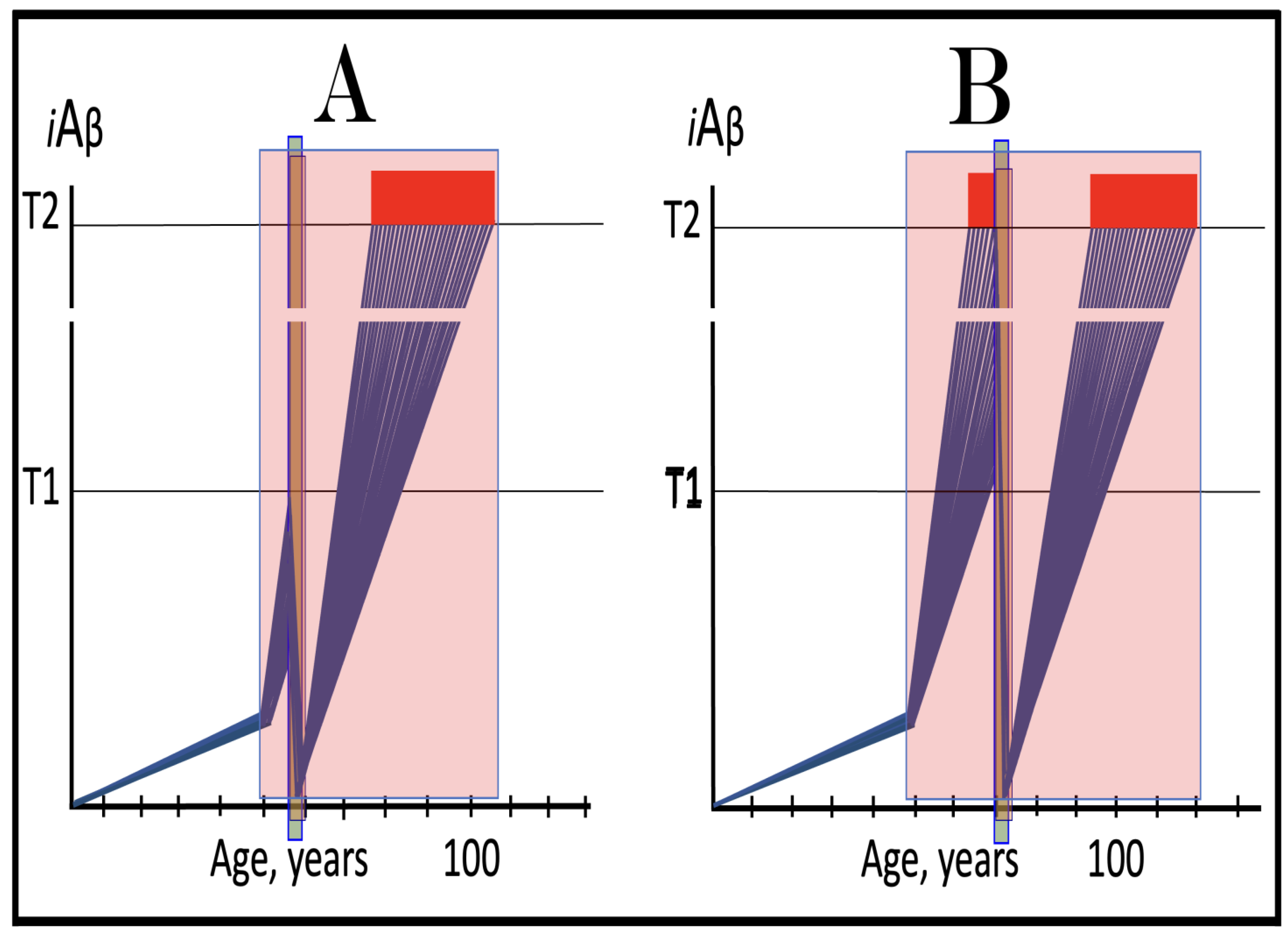
Figure 10.
FAD mutations that exert their effect by accelerating the rate of iAβ Accumulation and lowering the extent of T1. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Panels (A–D): dynamics of iAβ accumulation in wild-type AβPP carriers. Panels (A,B): the T0 levels are above those of T1; no AACD occurs. Panel (A): the T1 threshold is not crossed; no disease occurs. Panel (B): the T1 threshold is crossed; this is followed by late onset AD. Panels (C,D): the T0 threshold is lower than T1. Panel (C): the T1 threshold is not reached; when the T0 threshold is crossed, AACD commences and persists for the remaining lifespan. Panel (D): AACD commences when the T0 is crossed and morphs into late onset AD upon the T1 crossing. Panels (A’–D’): Dynamics of iAβ accumulation in carriers of FAD mutations, which not only elevate the rate of accumulation of AβPP-derived iAβ but also reduce the extent of the T1 threshold. As a result, the T1 threshold is crossed sooner, and early-onset AD follows. Panels (A’,B’): the T0 levels are above those of T1; no AACD occurs. Panels (C’,D’): the extent of the T0 threshold is smaller than that of the T1; AACD commences with the T0 crossing and morphs into the early-onset AD when the T1 is reached.
Figure 10.
FAD mutations that exert their effect by accelerating the rate of iAβ Accumulation and lowering the extent of T1. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Panels (A–D): dynamics of iAβ accumulation in wild-type AβPP carriers. Panels (A,B): the T0 levels are above those of T1; no AACD occurs. Panel (A): the T1 threshold is not crossed; no disease occurs. Panel (B): the T1 threshold is crossed; this is followed by late onset AD. Panels (C,D): the T0 threshold is lower than T1. Panel (C): the T1 threshold is not reached; when the T0 threshold is crossed, AACD commences and persists for the remaining lifespan. Panel (D): AACD commences when the T0 is crossed and morphs into late onset AD upon the T1 crossing. Panels (A’–D’): Dynamics of iAβ accumulation in carriers of FAD mutations, which not only elevate the rate of accumulation of AβPP-derived iAβ but also reduce the extent of the T1 threshold. As a result, the T1 threshold is crossed sooner, and early-onset AD follows. Panels (A’,B’): the T0 levels are above those of T1; no AACD occurs. Panels (C’,D’): the extent of the T0 threshold is smaller than that of the T1; AACD commences with the T0 crossing and morphs into the early-onset AD when the T1 is reached.
![Ijms 26 04252 g010]()
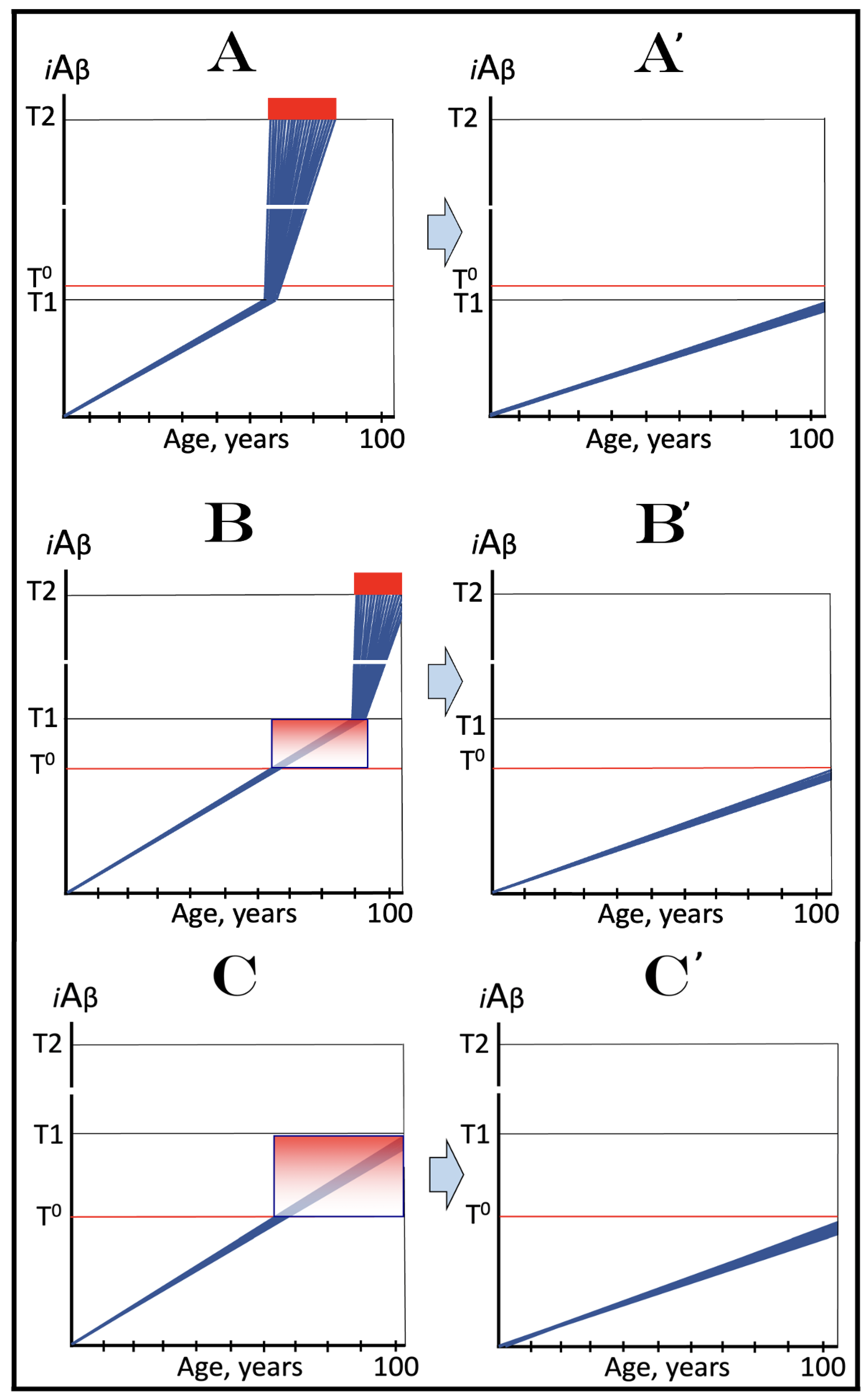
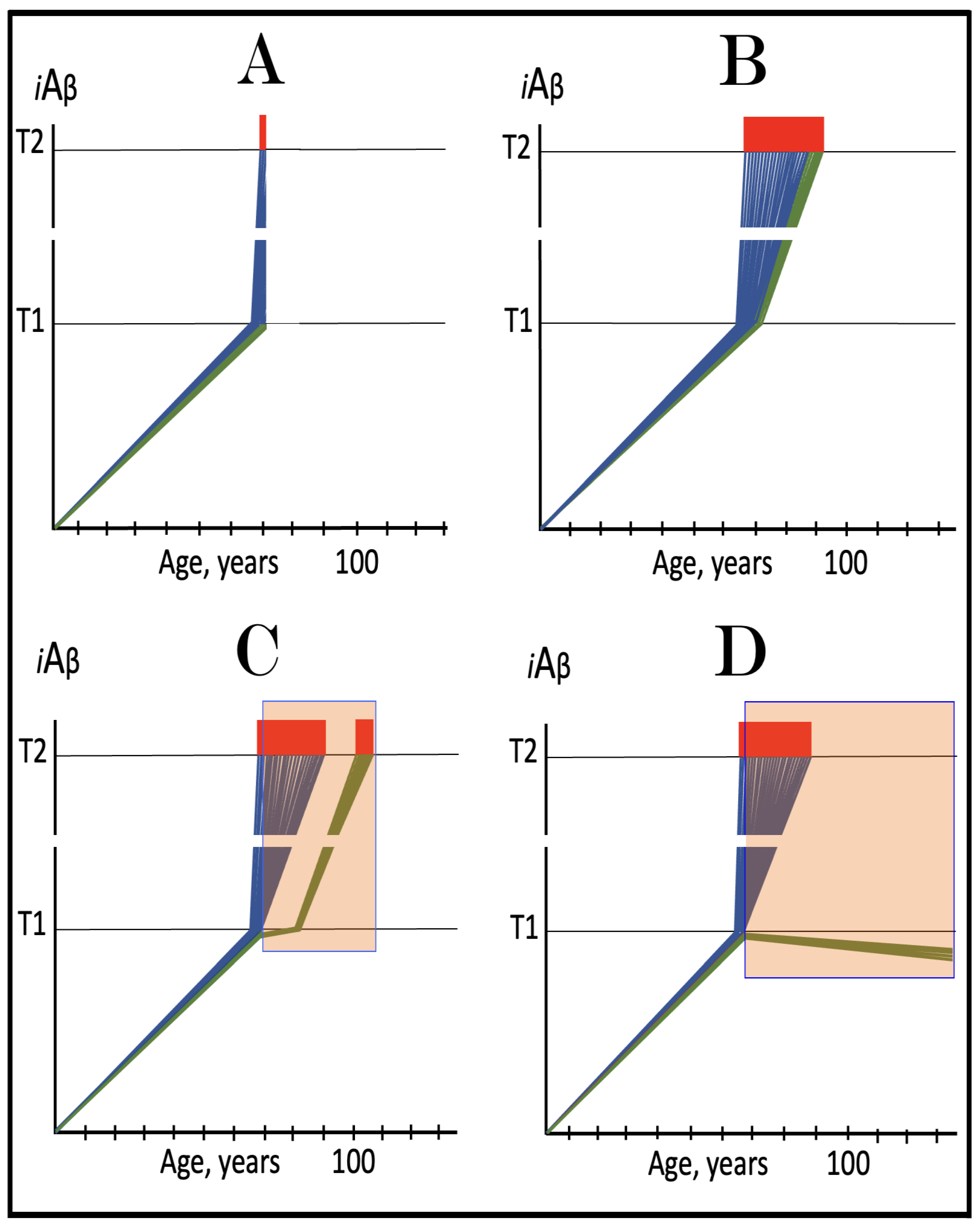
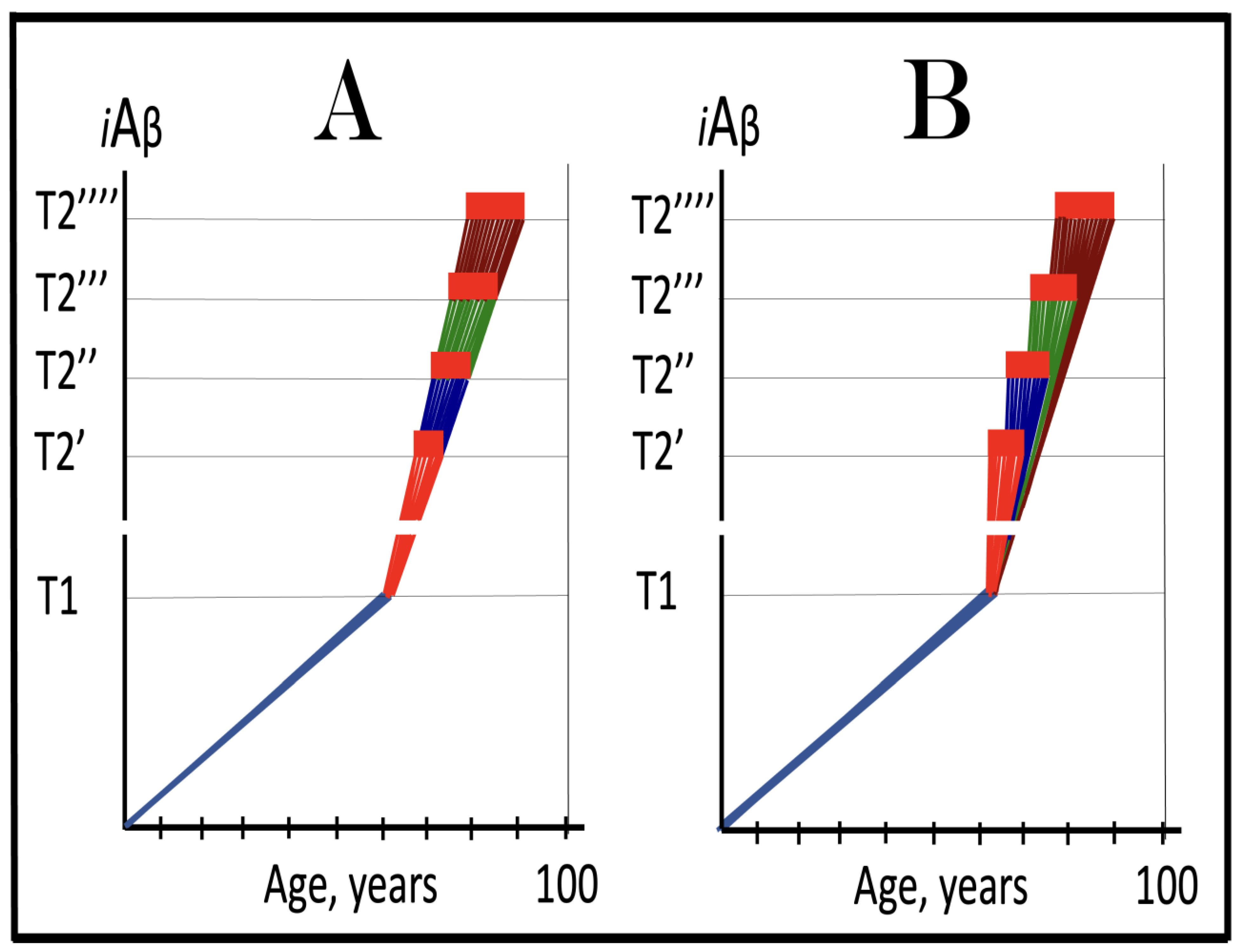
Figure 11.
The Icelandic AβPP mutation protects from AD and AACD by lowering the influx of AβPP-derived iAβ and decelerating its rate of accumulation. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Panels (A–C) illustrate principal forms of conventional AD and/or AACD that occur in wild-type AβPP carriers. Panel (A): The T1 threshold is below the AACD-triggering T0 threshold; no AACD occurs. Upon the T1 crossing the neuronal ISR is elicited, the AβPP-independent C99 and iAβ generation pathway activated, and conventional AD commences. Panel (B): The extent of the T0 threshold is smaller than that of the T1. When AβPP-derived iAβ crosses the T0, AACD commences and morphs into conventional AD upon the T1 crossing. Panel (C): The T1 threshold is not reached within the lifetime of the individual. When AβPP-derived iAβ crosses the T0 threshold, AACD commences and persists for the remaining lifetime. Panels (A’–C’) illustrate the protective effect of the Icelandic AβPP. In each panel the influx of AβPP-derived iAβ is reduced and its rate of accumulation lowered. Panel (A’): AβPP-derived iAβ does not reach the T1 threshold within the lifetime of the individual; no AD occurs. Panels (B’,C’): AβPP-derived iAβ reaches neither the T0 nor T1 threshold within the lifetime of the individual; neither AACD nor conventional AD occurs.
Figure 11.
The Icelandic AβPP mutation protects from AD and AACD by lowering the influx of AβPP-derived iAβ and decelerating its rate of accumulation. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Panels (A–C) illustrate principal forms of conventional AD and/or AACD that occur in wild-type AβPP carriers. Panel (A): The T1 threshold is below the AACD-triggering T0 threshold; no AACD occurs. Upon the T1 crossing the neuronal ISR is elicited, the AβPP-independent C99 and iAβ generation pathway activated, and conventional AD commences. Panel (B): The extent of the T0 threshold is smaller than that of the T1. When AβPP-derived iAβ crosses the T0, AACD commences and morphs into conventional AD upon the T1 crossing. Panel (C): The T1 threshold is not reached within the lifetime of the individual. When AβPP-derived iAβ crosses the T0 threshold, AACD commences and persists for the remaining lifetime. Panels (A’–C’) illustrate the protective effect of the Icelandic AβPP. In each panel the influx of AβPP-derived iAβ is reduced and its rate of accumulation lowered. Panel (A’): AβPP-derived iAβ does not reach the T1 threshold within the lifetime of the individual; no AD occurs. Panels (B’,C’): AβPP-derived iAβ reaches neither the T0 nor T1 threshold within the lifetime of the individual; neither AACD nor conventional AD occurs.
![Ijms 26 04252 g011]()
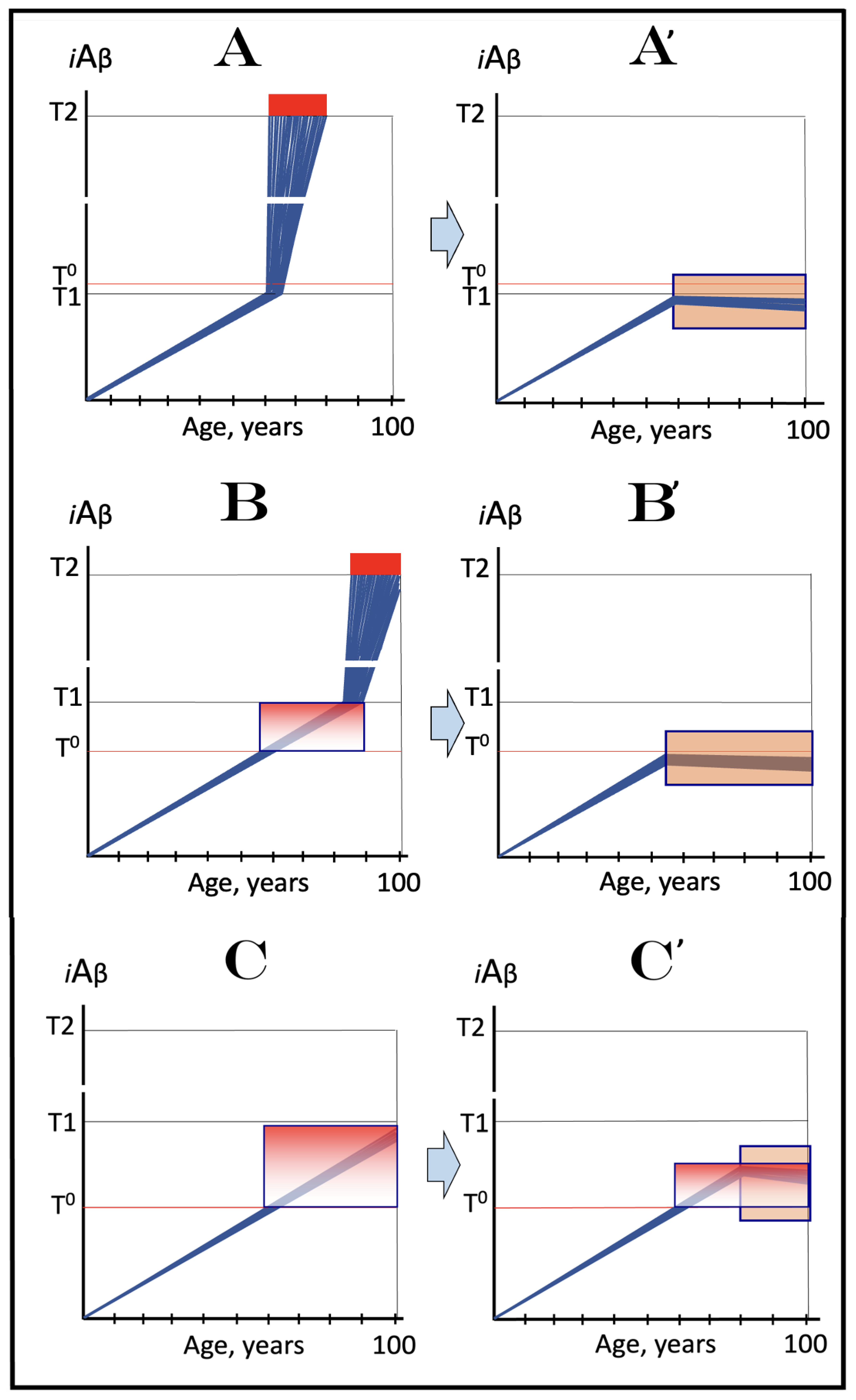
Figure 12.
Therapeutic strategies for conventional AD and AACD suggested by the Icelandic AβPP mutation. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Orange boxes: Duration of the treatment with the drug that reverses the rate of accumulation of AβPP-derived iAβ. Panels (A–C): Dynamics of iAβ accumulation in AD-affected neurons of the untreated AD and/or AACD patients. Panel (A): The T1 threshold is below the AACD-triggering T0 threshold; no AACD occurs. Upon the T1 crossing, the neuronal ISR is elicited, the AβPP-independent C99 and iAβ generation pathway activated, and conventional AD commences. Panel (B): The extent of the T0 threshold is smaller than that of the T1. When AβPP-derived iAβ crosses the T0, AACD commences and morphs into conventional AD upon the T1 crossing. Panel (C): The T1 threshold is not reached within the lifetime of the individual. When AβPP-derived iAβ crosses the T0 threshold, AACD commences and persists for the remaining lifetime. Panels (A’–C’): Dynamics of AβPP-derived iAβ in individuals treated with a drug that suppresses its influx and reveres its rate of accumulation. In Panels (A’,B’), the drug is administered prior to the crossing of the T0 and/or T1 thresholds. Neither threshold is reached for the duration of the treatment, nor does AD or AACD occur. In Panel (C’), the drug is administered after the T0 crossing but prior to the T1 crossing. The drug reverses the accumulation of AβPP-derived iAβ and stops the progression of AACD. The T1 threshold would not be reached, and conventional AD would not occur for the duration of the treatment.
Figure 12.
Therapeutic strategies for conventional AD and AACD suggested by the Icelandic AβPP mutation. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Orange boxes: Duration of the treatment with the drug that reverses the rate of accumulation of AβPP-derived iAβ. Panels (A–C): Dynamics of iAβ accumulation in AD-affected neurons of the untreated AD and/or AACD patients. Panel (A): The T1 threshold is below the AACD-triggering T0 threshold; no AACD occurs. Upon the T1 crossing, the neuronal ISR is elicited, the AβPP-independent C99 and iAβ generation pathway activated, and conventional AD commences. Panel (B): The extent of the T0 threshold is smaller than that of the T1. When AβPP-derived iAβ crosses the T0, AACD commences and morphs into conventional AD upon the T1 crossing. Panel (C): The T1 threshold is not reached within the lifetime of the individual. When AβPP-derived iAβ crosses the T0 threshold, AACD commences and persists for the remaining lifetime. Panels (A’–C’): Dynamics of AβPP-derived iAβ in individuals treated with a drug that suppresses its influx and reveres its rate of accumulation. In Panels (A’,B’), the drug is administered prior to the crossing of the T0 and/or T1 thresholds. Neither threshold is reached for the duration of the treatment, nor does AD or AACD occur. In Panel (C’), the drug is administered after the T0 crossing but prior to the T1 crossing. The drug reverses the accumulation of AβPP-derived iAβ and stops the progression of AACD. The T1 threshold would not be reached, and conventional AD would not occur for the duration of the treatment.
![Ijms 26 04252 g012]()
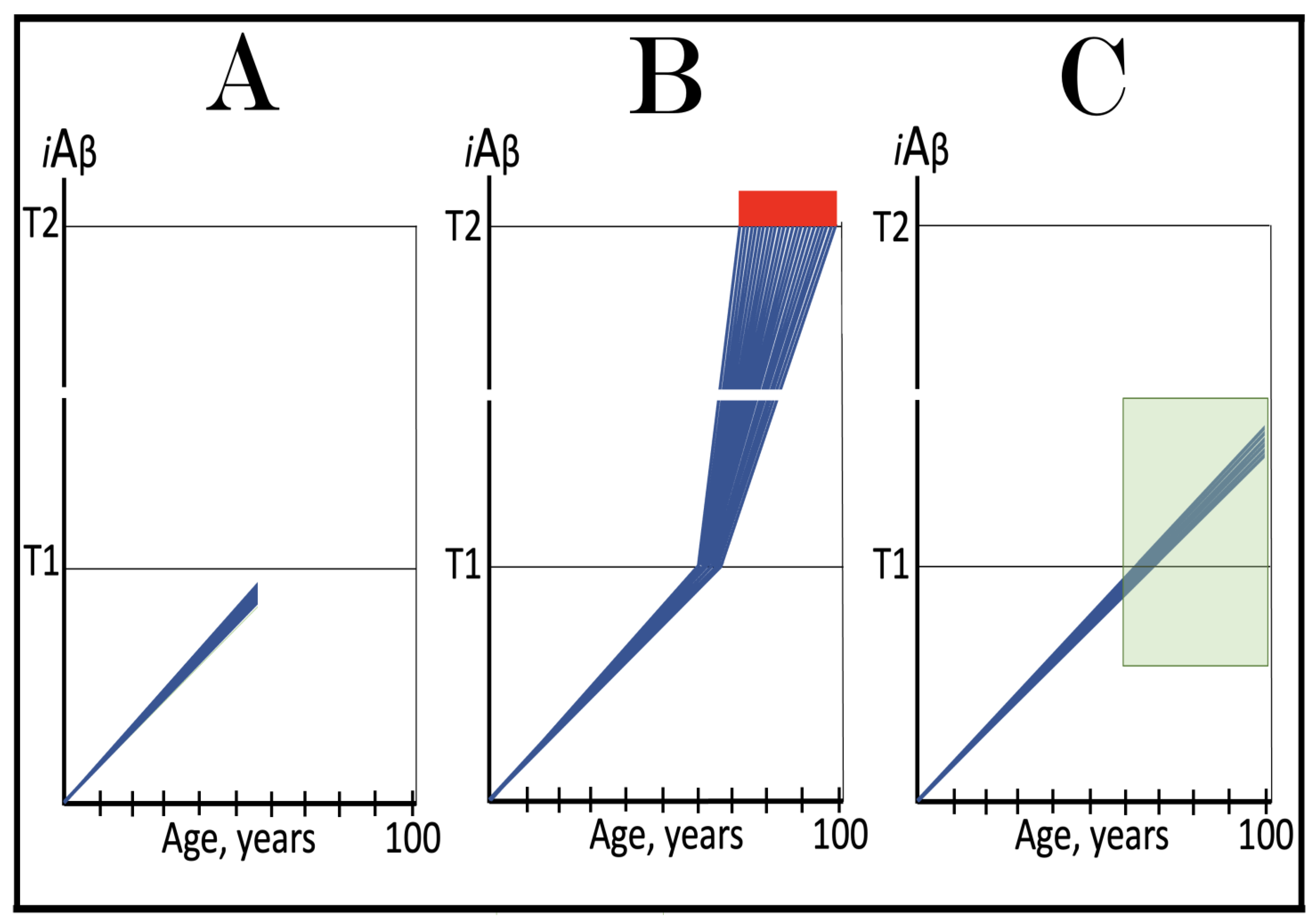
Figure 13.
ACH-based AD drugs could be effective if administered preventively. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange boxes: Duration of the treatment with the ACH-based drug that suppresses or reverses the rate of accumulation of AβPP-derived iAβ. Panels (A–C): Dynamics of iAβ accumulation in AD-affected neurons of the untreated and treated AD and/or AACD patients. Panel (A): Dynamics of the accumulation of iAβ in the untreated individual. AβPP-derived iAβ accumulates via two physiological mechanisms described in the main text. When it crosses the T1 threshold, PKR and/or HRI kinases are activated, eIF2α phosphorylated, the neuronal ISR elicited, the AβPP-independent C99 production pathway initiated, and conventional AD commences. Panel (B): The drug suppresses the influx of AβPP-derived iAβ, but its accumulation continues albeit at a reduced rate. Eventually, subject to sufficient longevity, it would cross the T1 threshold. The neuronal ISR would be elicited, the AβPP-independent C99 production pathway would be activated, and conventional AD would commence. In this scenario the drug delays the occurrence of AD. Panel (C): The drug suppresses the influx of AβPP-derived iAβ and reverses its rate of accumulation. The T1 threshold would not be reached, and conventional AD would not occur for the duration of the treatment. In this scenario the drug prevents the occurrence of conventional AD.
Figure 13.
ACH-based AD drugs could be effective if administered preventively. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange boxes: Duration of the treatment with the ACH-based drug that suppresses or reverses the rate of accumulation of AβPP-derived iAβ. Panels (A–C): Dynamics of iAβ accumulation in AD-affected neurons of the untreated and treated AD and/or AACD patients. Panel (A): Dynamics of the accumulation of iAβ in the untreated individual. AβPP-derived iAβ accumulates via two physiological mechanisms described in the main text. When it crosses the T1 threshold, PKR and/or HRI kinases are activated, eIF2α phosphorylated, the neuronal ISR elicited, the AβPP-independent C99 production pathway initiated, and conventional AD commences. Panel (B): The drug suppresses the influx of AβPP-derived iAβ, but its accumulation continues albeit at a reduced rate. Eventually, subject to sufficient longevity, it would cross the T1 threshold. The neuronal ISR would be elicited, the AβPP-independent C99 production pathway would be activated, and conventional AD would commence. In this scenario the drug delays the occurrence of AD. Panel (C): The drug suppresses the influx of AβPP-derived iAβ and reverses its rate of accumulation. The T1 threshold would not be reached, and conventional AD would not occur for the duration of the treatment. In this scenario the drug prevents the occurrence of conventional AD.
![Ijms 26 04252 g013]()
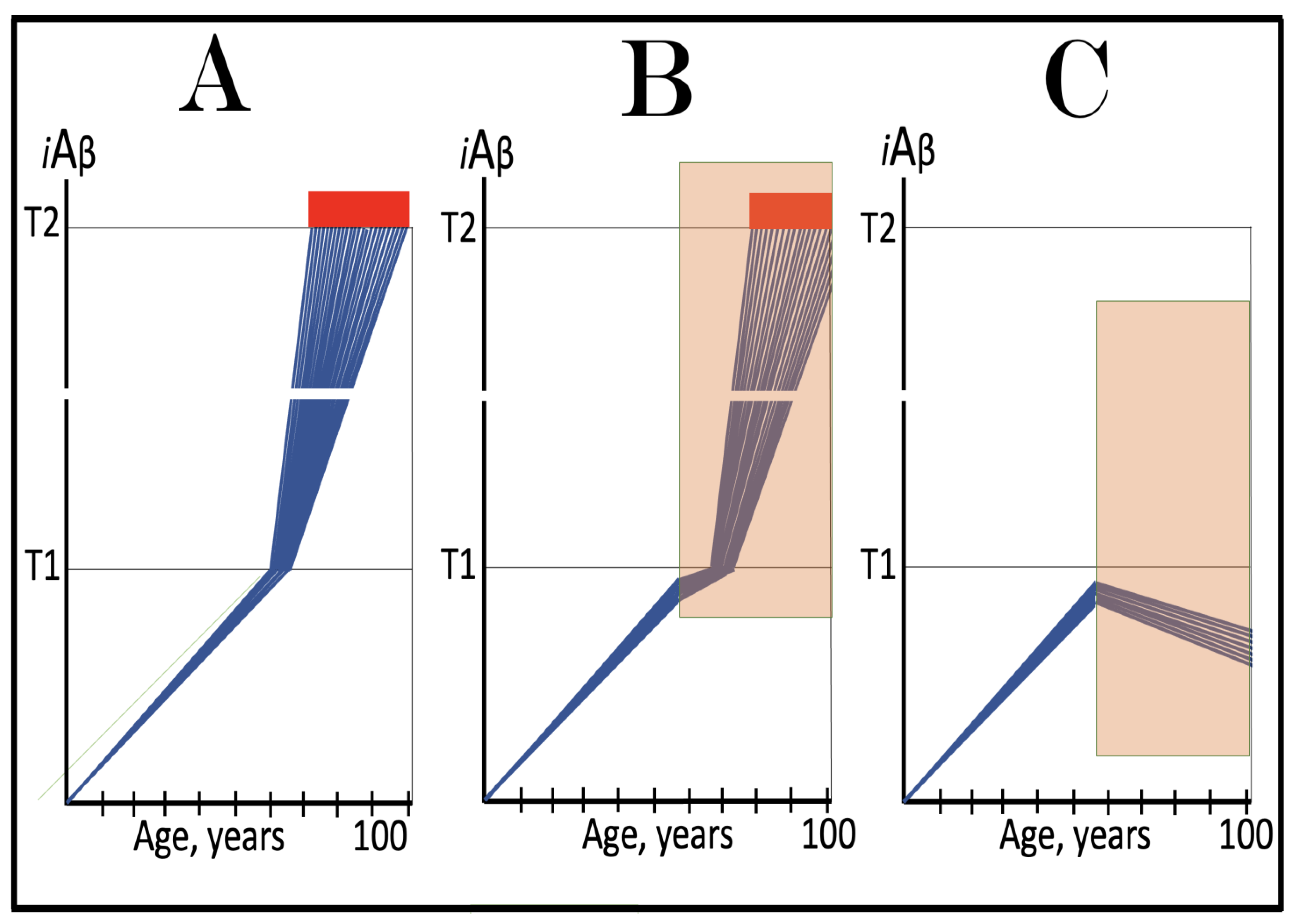
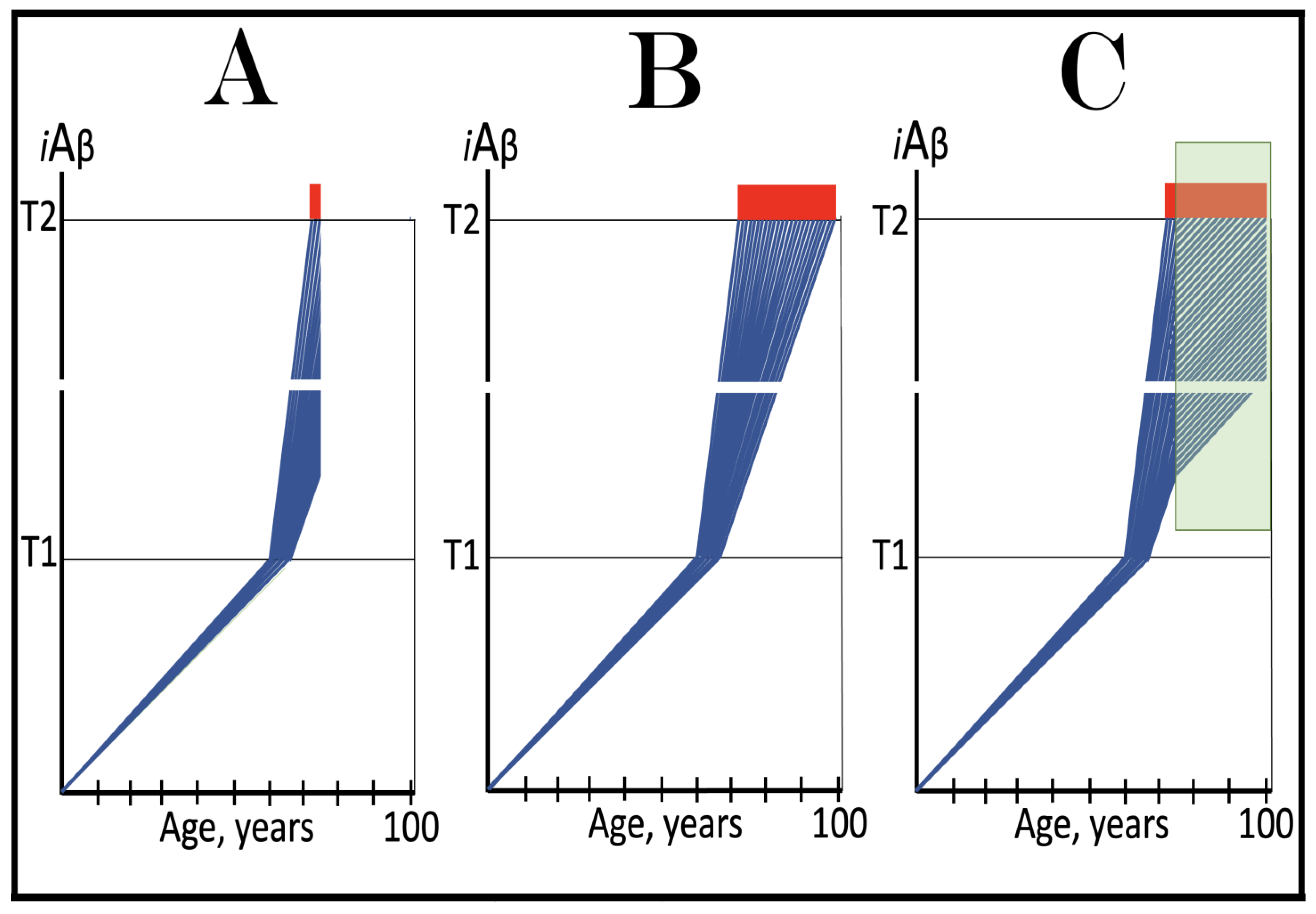
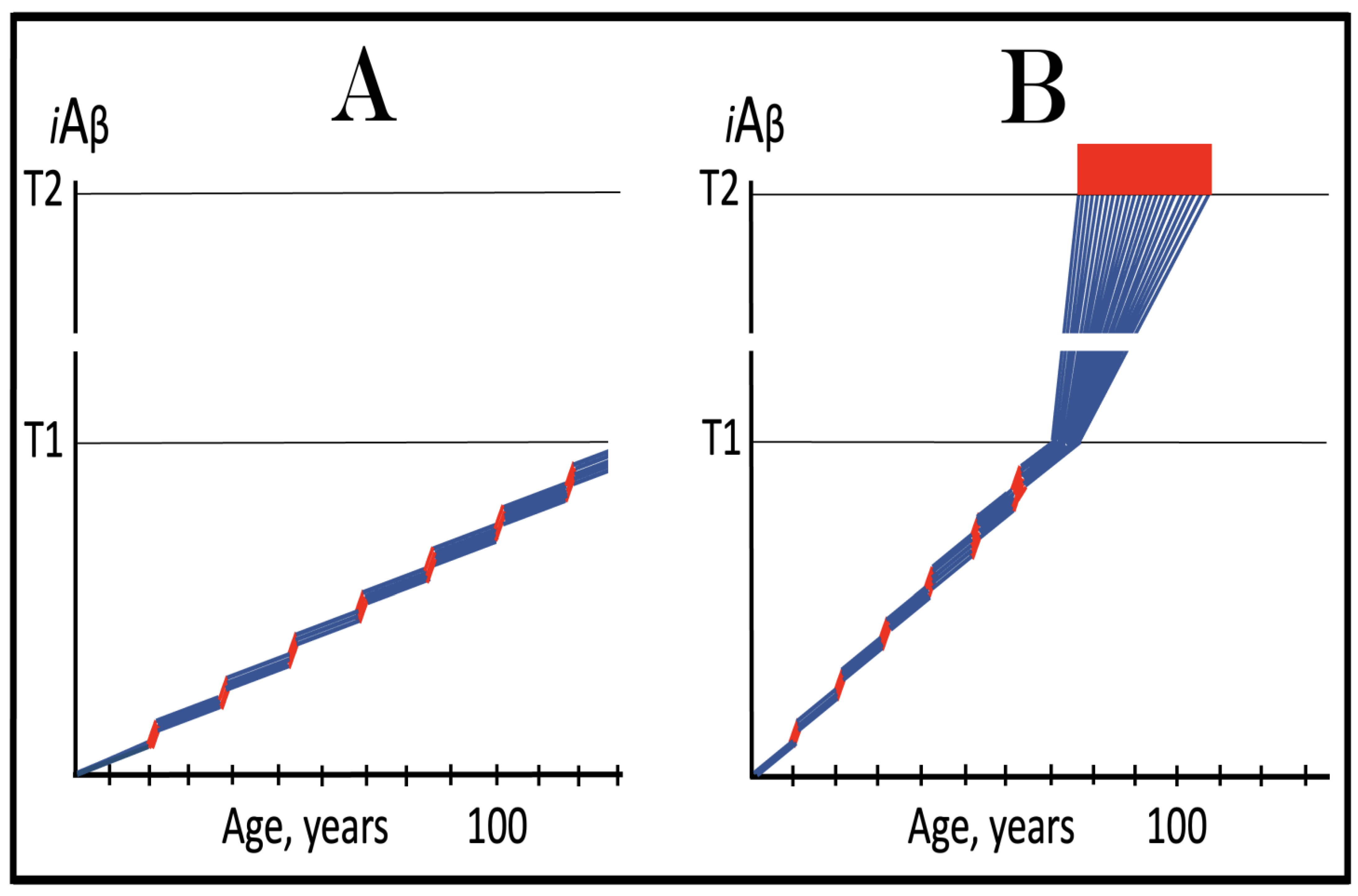
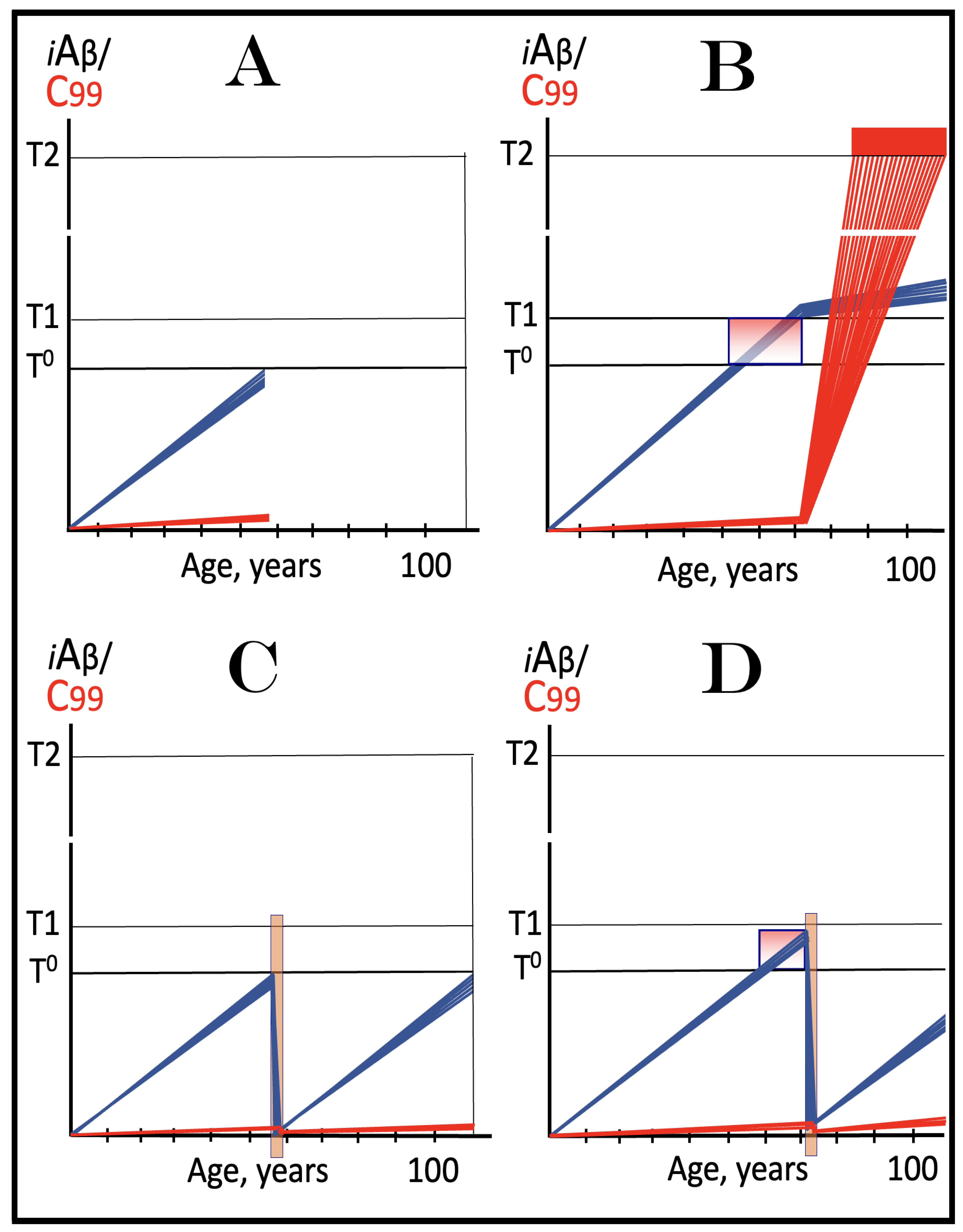
Figure 14.
Modulators of gamma-secretase may potentially protect from conventional AD via the elevation of the T1 threshold. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange boxes: Duration of the treatment with the gamma-secretase modulating drug that shifts the production of Aβ toward shorter, more benign species. Panel (A): Dynamics of the accumulation of iAβ in the untreated individual. AβPP-derived iAβ accumulates via two physiological mechanisms described in the main text. When it crosses the T1 threshold, PKR and/or HRI kinases are activated, eIF2α phosphorylated, the neuronal ISR elicited, the AβPP-independent C99 production pathway initiated, and conventional AD commences. Panel (B): In the presence of the drug shorter, more benign species of Aβ are produced and accumulated as AβPP-derived iAβ. More of it is needed to attain the same level of cellular toxicity and cellular stress than with regular Aβ species. Consequently, the extent of the T1 threshold and the timing of its crossing by AβPP-derived iAβ increase. In this scenario the treatment delays the commencement of conventional AD. Panel (C): The treatment-caused increase in the extent of the T1 threshold is such that it is not reached, and conventional AD does not occur for the duration of the treatment. In this scenario, the drug prevents the occurrence of AD.
Figure 14.
Modulators of gamma-secretase may potentially protect from conventional AD via the elevation of the T1 threshold. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange boxes: Duration of the treatment with the gamma-secretase modulating drug that shifts the production of Aβ toward shorter, more benign species. Panel (A): Dynamics of the accumulation of iAβ in the untreated individual. AβPP-derived iAβ accumulates via two physiological mechanisms described in the main text. When it crosses the T1 threshold, PKR and/or HRI kinases are activated, eIF2α phosphorylated, the neuronal ISR elicited, the AβPP-independent C99 production pathway initiated, and conventional AD commences. Panel (B): In the presence of the drug shorter, more benign species of Aβ are produced and accumulated as AβPP-derived iAβ. More of it is needed to attain the same level of cellular toxicity and cellular stress than with regular Aβ species. Consequently, the extent of the T1 threshold and the timing of its crossing by AβPP-derived iAβ increase. In this scenario the treatment delays the commencement of conventional AD. Panel (C): The treatment-caused increase in the extent of the T1 threshold is such that it is not reached, and conventional AD does not occur for the duration of the treatment. In this scenario, the drug prevents the occurrence of AD.
![Ijms 26 04252 g014]()
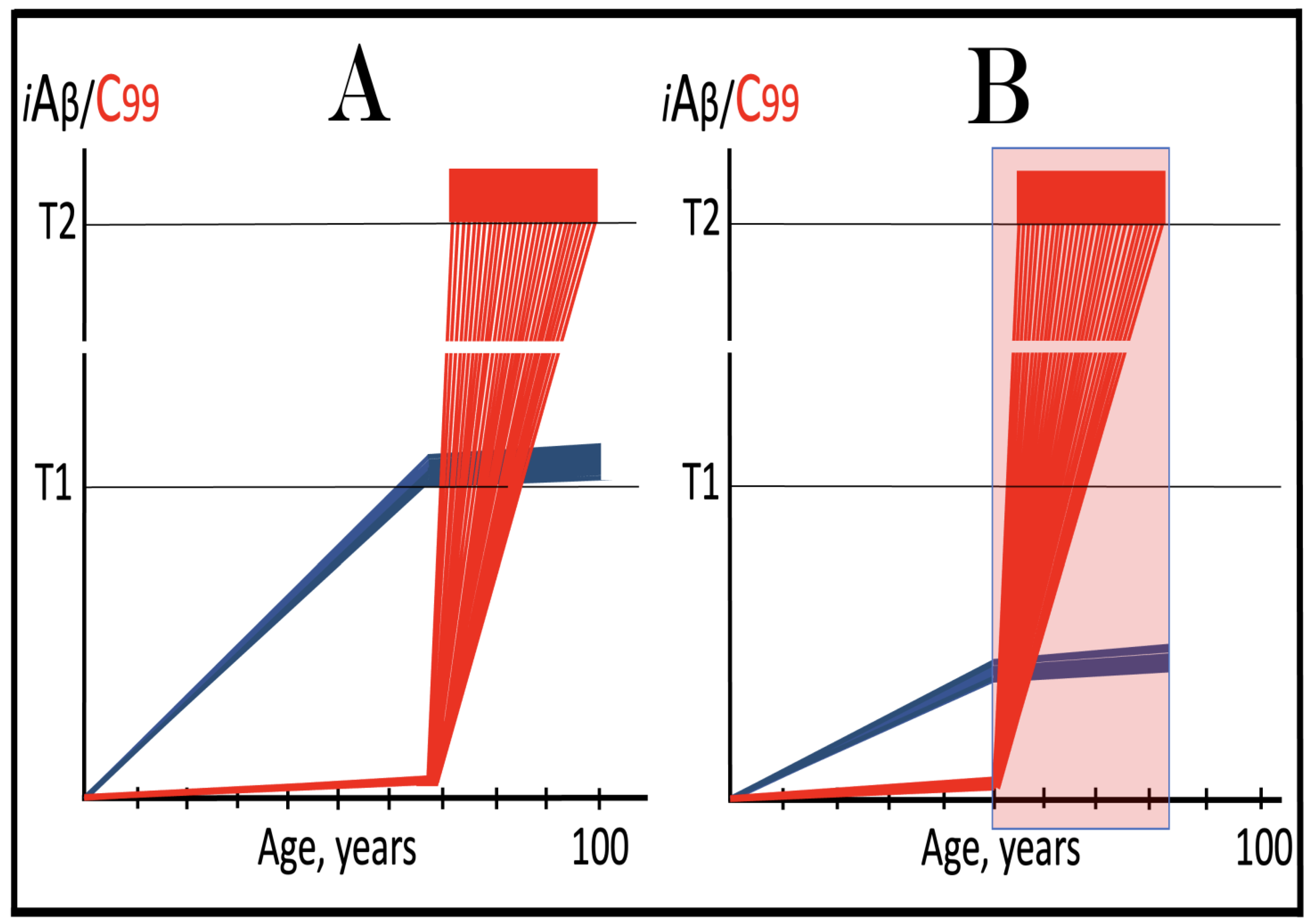
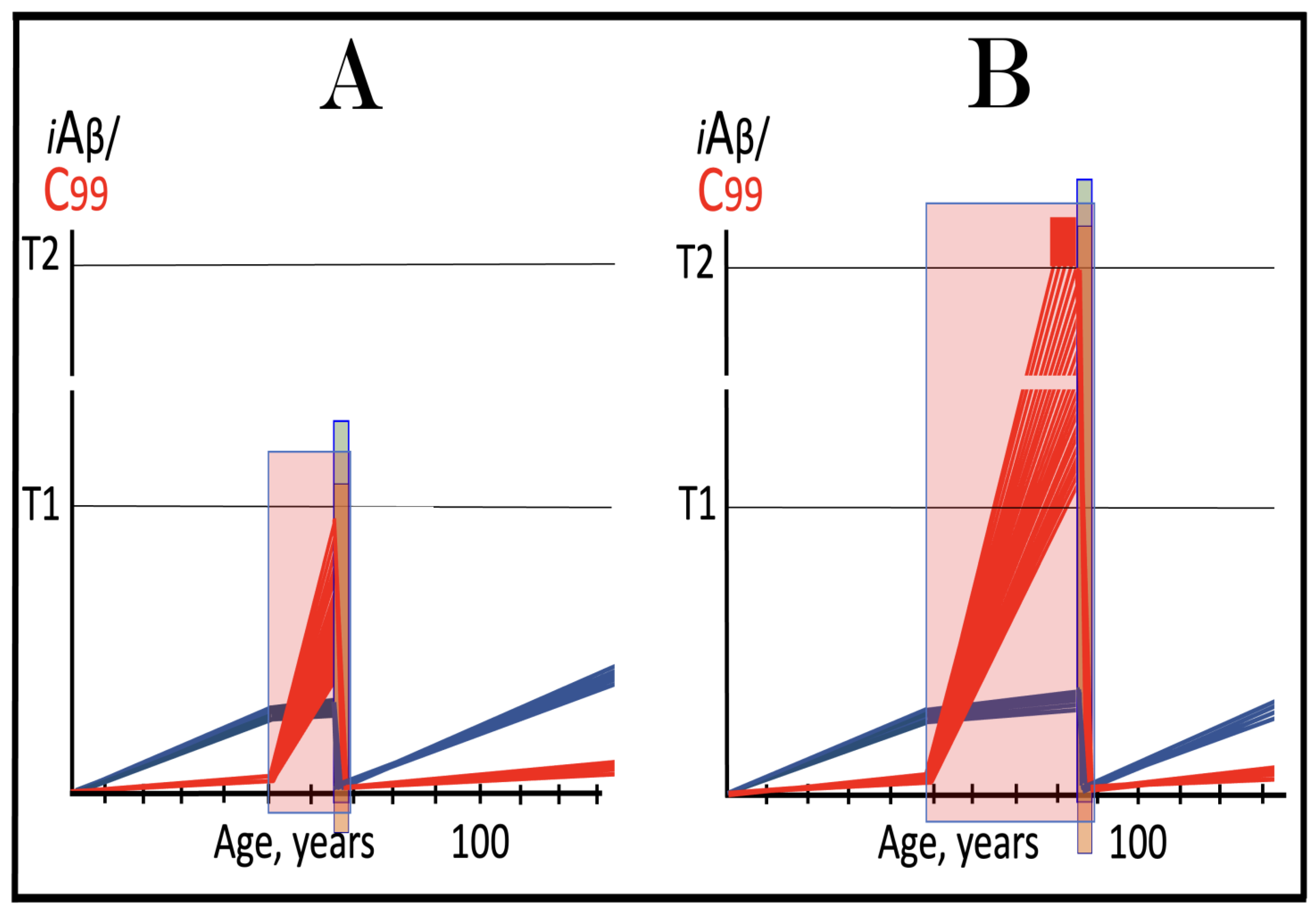
Figure 15.
ACH-based drugs are ineffective when the AβPP-independent C99/iAβ production pathway is operational. Blue lines: AβPP-derived iAβ. Red lines: iAβ generated independently of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange boxes: Duration of the treatment with the ACH-based drug that suppresses or reverses the rate of accumulation of AβPP-derived iAβ. Panel (A): AβPP-derived iAβ accumulates via two physiological mechanisms described in the main text. When it crosses the T1 threshold PKR and/or HRI kinases are activated, eIF2α phosphorylated, the neuronal ISR elicited, the AβPP-independent C99 production pathway initiated, and conventional AD commences. ACH-based drugs administered at this point reduce significantly the rate of accumulation of AβPP-derived iAβ. They have, however, little, if any, effect on both the production of iAβ in the AβPP-independent pathway and the progression of the disease. Panel (B): As in the preceding panel, the ACH-based drug is administered when all affected neurons have already crossed the T1 threshold, and the AβPP-independent C99 production pathway has already been activated. The reduction in the influx of AβPP-derived iAβ is such that the rate of its accumulation is reversed. Since the operation of the self-sustainable AβPP-independent C99 production pathway is not affected, this reversal is inconsequential for the progression of the disease. Panels (C,D): The ACH-based drug is administered when a fraction of the affected neurons has not yet crossed the T1 threshold. In over-T1 neurons, the AβPP-independent C99/iAβ production pathway has been activated, and the drug does not affect its operation. In Panel (C), the drug reduces the influx of AβPP-derived iAβ, but its accumulation continues. The initially under-T1 neurons eventually cross the T1 threshold, the AβPP-independent C99/iAβ production pathway is activated, and cellular AD pathology commences. In Panel (D), the drug reverses the rate of accumulation of AβPP-derived iAβ. The initially under-T1 neurons do not cross the T1 threshold and remain AD pathology-free for the duration of the treatment.
Figure 15.
ACH-based drugs are ineffective when the AβPP-independent C99/iAβ production pathway is operational. Blue lines: AβPP-derived iAβ. Red lines: iAβ generated independently of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange boxes: Duration of the treatment with the ACH-based drug that suppresses or reverses the rate of accumulation of AβPP-derived iAβ. Panel (A): AβPP-derived iAβ accumulates via two physiological mechanisms described in the main text. When it crosses the T1 threshold PKR and/or HRI kinases are activated, eIF2α phosphorylated, the neuronal ISR elicited, the AβPP-independent C99 production pathway initiated, and conventional AD commences. ACH-based drugs administered at this point reduce significantly the rate of accumulation of AβPP-derived iAβ. They have, however, little, if any, effect on both the production of iAβ in the AβPP-independent pathway and the progression of the disease. Panel (B): As in the preceding panel, the ACH-based drug is administered when all affected neurons have already crossed the T1 threshold, and the AβPP-independent C99 production pathway has already been activated. The reduction in the influx of AβPP-derived iAβ is such that the rate of its accumulation is reversed. Since the operation of the self-sustainable AβPP-independent C99 production pathway is not affected, this reversal is inconsequential for the progression of the disease. Panels (C,D): The ACH-based drug is administered when a fraction of the affected neurons has not yet crossed the T1 threshold. In over-T1 neurons, the AβPP-independent C99/iAβ production pathway has been activated, and the drug does not affect its operation. In Panel (C), the drug reduces the influx of AβPP-derived iAβ, but its accumulation continues. The initially under-T1 neurons eventually cross the T1 threshold, the AβPP-independent C99/iAβ production pathway is activated, and cellular AD pathology commences. In Panel (D), the drug reverses the rate of accumulation of AβPP-derived iAβ. The initially under-T1 neurons do not cross the T1 threshold and remain AD pathology-free for the duration of the treatment.
![Ijms 26 04252 g015]()
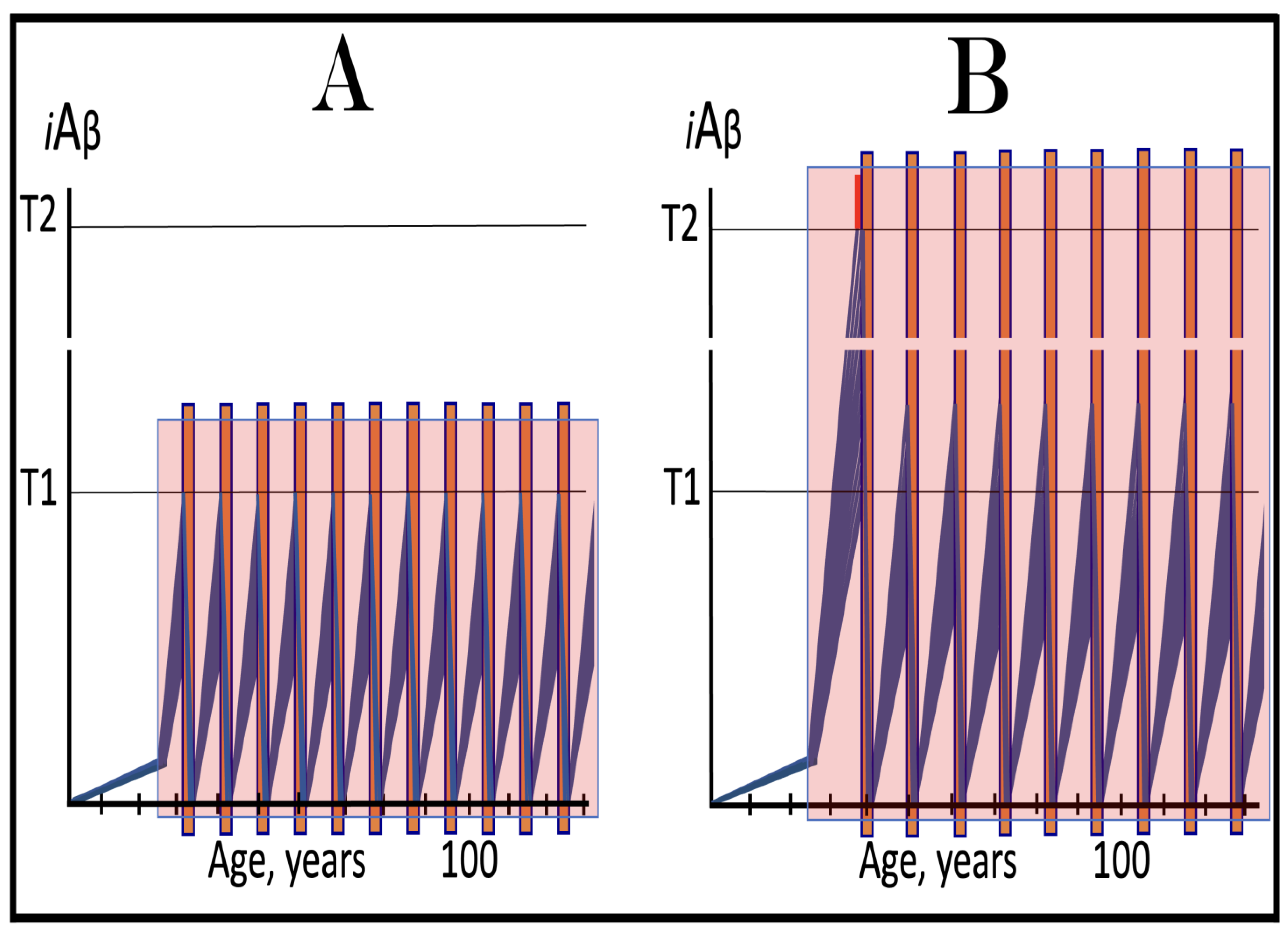
Figure 16.
Effect of Lecanemab and Donanemab in early AD: only a marginal fraction of neurons that have not yet crossed the T1 threshold is affected. Blue lines: Intraneuronal Aβ, iAβ. Green lines: Levels of iAβ in the neurons that have not yet crossed the T1 threshold at the commencement of the treatment. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange boxes: Duration of the treatment with lecanemab or donanemab. Panel (A): The initial state of levels of iAβ at the commencement of the drug’s administration. The bulk of the affected neurons have crossed the T1 threshold. The neuronal ISR has been elicited and the AβPP-independent C99/iAβ production pathway has been activated. Some neurons have reached the T2 threshold and AD symptoms have manifested. Panel (B): The evolution of the initial state in the untreated AD patient. All affected neurons cross the T1 threshold. The disease progresses and reaches the end stage. Panels (C,D): The evolution of the initial state in treated patients. The drug has no effect whatsoever on the operation of the self-sustainable AβPP-independent C99/iAβ production pathway; it affects only the rate of accumulation of AβPP-derived iAβ. Panel (C): The drug suppresses the influx of AβPP-derived iAβ, but its accumulation continues, albeit at a reduced rate. Eventually, subject to sufficient longevity, it would cross the T1 threshold in the initially under-T1 neurons. The neuronal ISR would be elicited, the AβPP-independent C99 production pathway would be activated, and cellular AD pathology would commence. Panel (D): The drug suppresses the influx of AβPP-derived iAβ and reverses its rate of accumulation. The T1 threshold would not be reached, and AD pathology would not occur in the initially under-T1 neurons for the duration of the treatment. In both scenarios, however, the effect is only marginal because the fraction of the drug-affected neurons is.
Figure 16.
Effect of Lecanemab and Donanemab in early AD: only a marginal fraction of neurons that have not yet crossed the T1 threshold is affected. Blue lines: Intraneuronal Aβ, iAβ. Green lines: Levels of iAβ in the neurons that have not yet crossed the T1 threshold at the commencement of the treatment. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange boxes: Duration of the treatment with lecanemab or donanemab. Panel (A): The initial state of levels of iAβ at the commencement of the drug’s administration. The bulk of the affected neurons have crossed the T1 threshold. The neuronal ISR has been elicited and the AβPP-independent C99/iAβ production pathway has been activated. Some neurons have reached the T2 threshold and AD symptoms have manifested. Panel (B): The evolution of the initial state in the untreated AD patient. All affected neurons cross the T1 threshold. The disease progresses and reaches the end stage. Panels (C,D): The evolution of the initial state in treated patients. The drug has no effect whatsoever on the operation of the self-sustainable AβPP-independent C99/iAβ production pathway; it affects only the rate of accumulation of AβPP-derived iAβ. Panel (C): The drug suppresses the influx of AβPP-derived iAβ, but its accumulation continues, albeit at a reduced rate. Eventually, subject to sufficient longevity, it would cross the T1 threshold in the initially under-T1 neurons. The neuronal ISR would be elicited, the AβPP-independent C99 production pathway would be activated, and cellular AD pathology would commence. Panel (D): The drug suppresses the influx of AβPP-derived iAβ and reverses its rate of accumulation. The T1 threshold would not be reached, and AD pathology would not occur in the initially under-T1 neurons for the duration of the treatment. In both scenarios, however, the effect is only marginal because the fraction of the drug-affected neurons is.
![Ijms 26 04252 g016]()
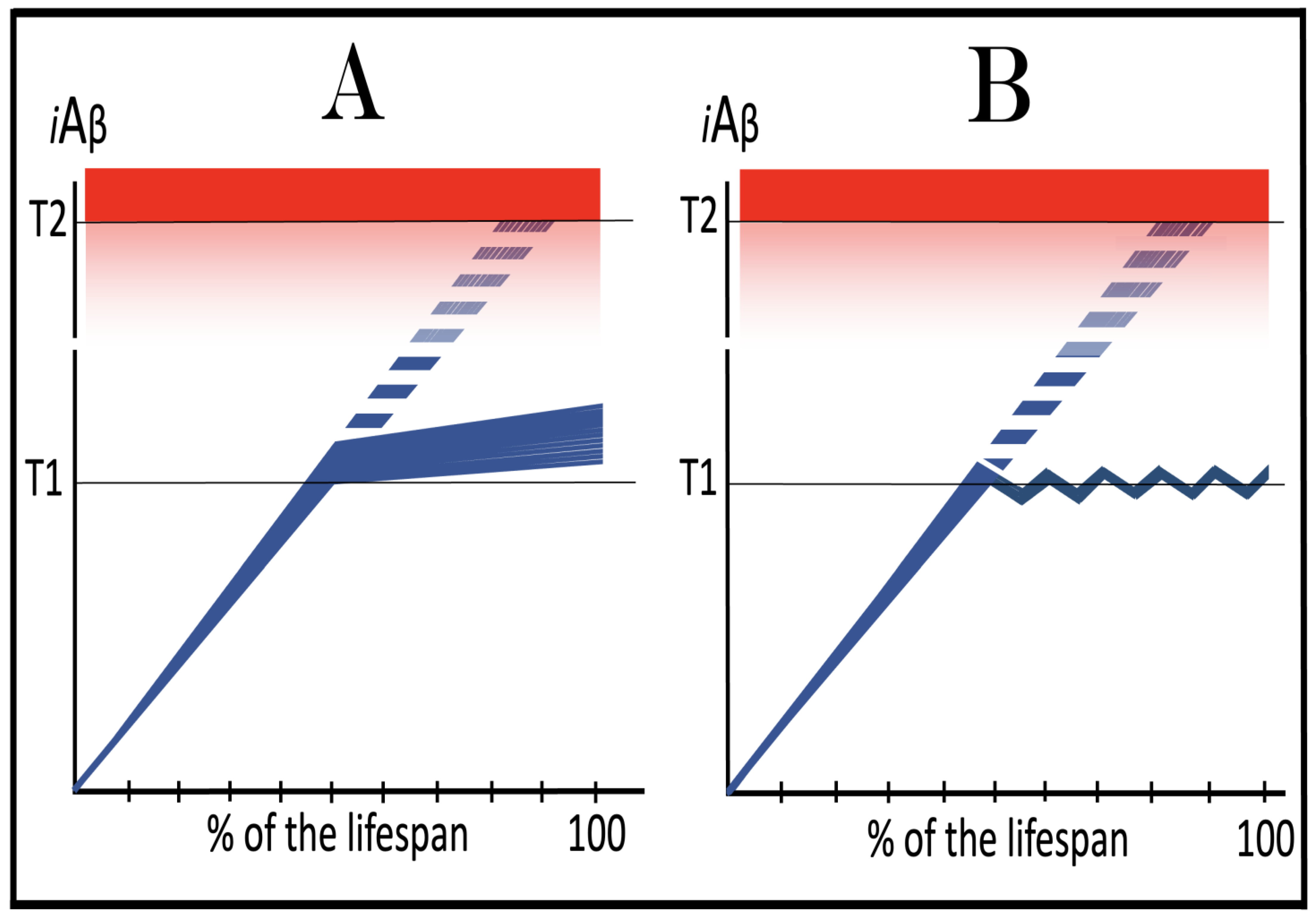
Figure 17.
Suppression of the neuronal integrated stress response in the prevention of conventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the healthy individual who would develop AD if untreated. AβPP-derived iAβ has been accumulating but its levels are below the T1 threshold Panel (B): The evolution of the initial state in the untreated individual. AβPP-derived iAβ accumulates unimpeded and crosses the T1 threshold. This triggers activation of PKR and/or HRI, phosphorylation of eIF2α, elicitation of the neuronal ISR, and initiation of the AβPP-independent C99/iAβ production pathway. AD commences and progresses until it reaches its end stage. Panel (C): The evolution of the initial state in the individual treated with the ISR-inhibiting drug. AβPP-derived iAβ reaches and crosses the T1 threshold but the neuronal ISR cannot be elicited. The influx of iAβ is supported solely by the AβPP proteolysis and it continues to accumulate at a slow pre-T1 crossing rate. Its level would not reach the AD pathology-causing range for the duration of the treatment.
Figure 17.
Suppression of the neuronal integrated stress response in the prevention of conventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the healthy individual who would develop AD if untreated. AβPP-derived iAβ has been accumulating but its levels are below the T1 threshold Panel (B): The evolution of the initial state in the untreated individual. AβPP-derived iAβ accumulates unimpeded and crosses the T1 threshold. This triggers activation of PKR and/or HRI, phosphorylation of eIF2α, elicitation of the neuronal ISR, and initiation of the AβPP-independent C99/iAβ production pathway. AD commences and progresses until it reaches its end stage. Panel (C): The evolution of the initial state in the individual treated with the ISR-inhibiting drug. AβPP-derived iAβ reaches and crosses the T1 threshold but the neuronal ISR cannot be elicited. The influx of iAβ is supported solely by the AβPP proteolysis and it continues to accumulate at a slow pre-T1 crossing rate. Its level would not reach the AD pathology-causing range for the duration of the treatment.
![Ijms 26 04252 g017]()
Figure 18.
Suppression of the neuronal integrated stress response in the treatment of conventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the AD patient. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons, the neuronal ISR has been elicited, the AβPP-independent C99/iAβ production pathway activated, the T2 threshold crossed in a neuronal fraction, and AD symptoms have manifested. Panel (B): The disease progresses and reaches its end stage. Panel (C): The evolution of the initial state in the individual treated with the ISR-inhibiting drug. The supply of components essential for the activity of the AβPP-independent C99/iAβ production pathway stops and it operation ceases. The accumulation of iAβ, however, continues but at a slow pre-T1 crossing rate. The rate of the progression of the disease is reduced for the duration of the treatment.
Figure 18.
Suppression of the neuronal integrated stress response in the treatment of conventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the AD patient. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons, the neuronal ISR has been elicited, the AβPP-independent C99/iAβ production pathway activated, the T2 threshold crossed in a neuronal fraction, and AD symptoms have manifested. Panel (B): The disease progresses and reaches its end stage. Panel (C): The evolution of the initial state in the individual treated with the ISR-inhibiting drug. The supply of components essential for the activity of the AβPP-independent C99/iAβ production pathway stops and it operation ceases. The accumulation of iAβ, however, continues but at a slow pre-T1 crossing rate. The rate of the progression of the disease is reduced for the duration of the treatment.
![Ijms 26 04252 g018]()
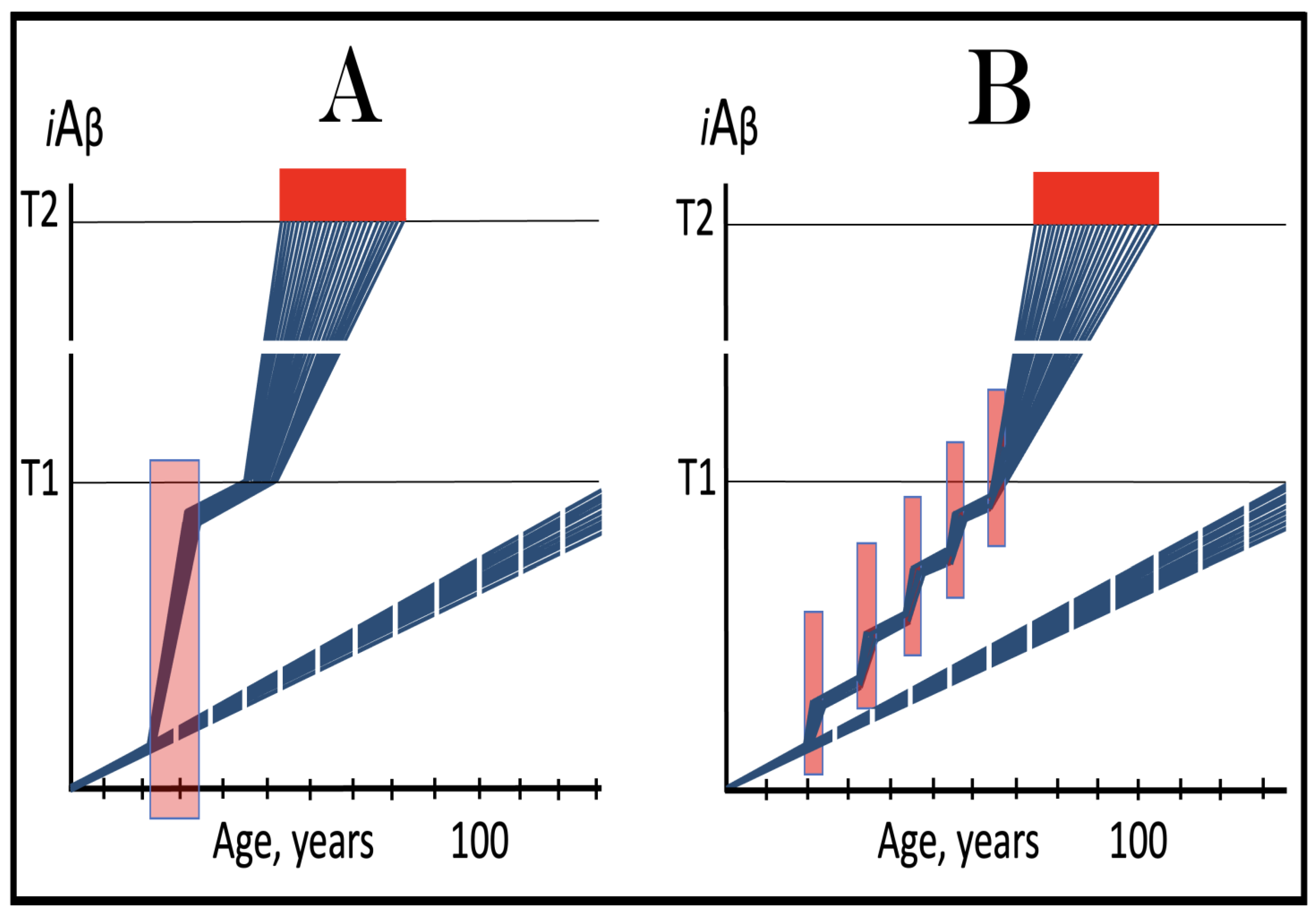
Figure 19.
Activators of BACE1 and/or BACE2 in the prevention of conventional AD and the prevention and treatment of AACD. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Orange boxes: Duration of the treatment with the BACE1- and/or BAEC2-activating drug. Panel (A): Dynamics of the accumulation of iAβ in the untreated AD patient. AβPP-derived iAβ accumulates via two physiological mechanisms described in the main text. When it crosses the T1 threshold PKR and/or HRI kinases are activated, eIF2α phosphorylated, the neuronal ISR elicited, the AβPP-independent C99 production pathway initiated, and conventional AD commences. The T0 threshold is above the T1; there is no AACD. Panel (B): Activators of BACE1 and/or BACE2 are administered transiently prior to the T1 crossing. As a result, AβPP-derived iAβ is substantially depleted. Following the withdrawal of the drug, the de novo accumulation commences from low baseline and proceeds at the pre-treatment rate. Neither the T1 threshold would be crossed, nor AD would occur within the lifetime of the treated individual. Panel (C): Dynamics of the accumulation of iAβ in the untreated AACD/AD patient. When AβPP-derived iAβ reaches the T0 threshold, AACD commences and persists until the T1 crossing, the activation of the AβPP-independent C99/iAβ production pathway, and the commencement of AD. At this point AACD morphs into AD. Panel (D): Activators of BACE1 and/or BACE2 are administered transiently following the T0 crossing and the commencement of AACD but prior to the T1 crossing. As a result, iAβ is substantially depleted well below the T0 threshold. At this point AACD is cured. The accumulation of AβPP-derived iAβ starts de novo from low baseline and proceeds at the pre-treatment rate. It would not reach the T0 threshold within the lifetime of the treated individual and neither AACD would recur nor AD would occur.
Figure 19.
Activators of BACE1 and/or BACE2 in the prevention of conventional AD and the prevention and treatment of AACD. Blue lines: Intraneuronal Aβ, iAβ. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Orange boxes: Duration of the treatment with the BACE1- and/or BAEC2-activating drug. Panel (A): Dynamics of the accumulation of iAβ in the untreated AD patient. AβPP-derived iAβ accumulates via two physiological mechanisms described in the main text. When it crosses the T1 threshold PKR and/or HRI kinases are activated, eIF2α phosphorylated, the neuronal ISR elicited, the AβPP-independent C99 production pathway initiated, and conventional AD commences. The T0 threshold is above the T1; there is no AACD. Panel (B): Activators of BACE1 and/or BACE2 are administered transiently prior to the T1 crossing. As a result, AβPP-derived iAβ is substantially depleted. Following the withdrawal of the drug, the de novo accumulation commences from low baseline and proceeds at the pre-treatment rate. Neither the T1 threshold would be crossed, nor AD would occur within the lifetime of the treated individual. Panel (C): Dynamics of the accumulation of iAβ in the untreated AACD/AD patient. When AβPP-derived iAβ reaches the T0 threshold, AACD commences and persists until the T1 crossing, the activation of the AβPP-independent C99/iAβ production pathway, and the commencement of AD. At this point AACD morphs into AD. Panel (D): Activators of BACE1 and/or BACE2 are administered transiently following the T0 crossing and the commencement of AACD but prior to the T1 crossing. As a result, iAβ is substantially depleted well below the T0 threshold. At this point AACD is cured. The accumulation of AβPP-derived iAβ starts de novo from low baseline and proceeds at the pre-treatment rate. It would not reach the T0 threshold within the lifetime of the treated individual and neither AACD would recur nor AD would occur.
![Ijms 26 04252 g019]()
Figure 20.
With the AβPP-independent C99/iAβ production pathway operational, the rate of degradation of iAβ by activated BACE1 and/or BACE2 cannot match or exceed that of its influx. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange box: Duration of the treatment with the BACE1- and/or BACE2-activating drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the AD patient. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons, the neuronal ISR has been elicited, the AβPP-independent C99/iAβ production pathway activated, the T2 threshold crossed in a neuronal fraction, and AD symptoms have manifested. Panel (B): The disease progresses and reaches its end stage. Panel (C): The evolution of the initial state in the individual treated with the BACE1- and/or BACE2-activating drug. The rate of degradation of iAβ via its internal cleavages increases but it cannot match the rate of the influx of iAβ produced independently of AβPP. The rate of the accumulation of iAβ decreases and the progression of AD slows down for the duration of the treatment, but the disease persists.
Figure 20.
With the AβPP-independent C99/iAβ production pathway operational, the rate of degradation of iAβ by activated BACE1 and/or BACE2 cannot match or exceed that of its influx. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Orange box: Duration of the treatment with the BACE1- and/or BACE2-activating drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the AD patient. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons, the neuronal ISR has been elicited, the AβPP-independent C99/iAβ production pathway activated, the T2 threshold crossed in a neuronal fraction, and AD symptoms have manifested. Panel (B): The disease progresses and reaches its end stage. Panel (C): The evolution of the initial state in the individual treated with the BACE1- and/or BACE2-activating drug. The rate of degradation of iAβ via its internal cleavages increases but it cannot match the rate of the influx of iAβ produced independently of AβPP. The rate of the accumulation of iAβ decreases and the progression of AD slows down for the duration of the treatment, but the disease persists.
![Ijms 26 04252 g020]()
Figure 21.
Concurrent transient BACE1 and/or BACE2 activation and ISR inhibition in the treatment of conventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Orange box: Duration of the treatment with the BACE1- and/or BACE2-activating drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the AD patient. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons, the neuronal ISR has been elicited, the AβPP-independent C99/iAβ production pathway activated, the T2 threshold crossed in a neuronal fraction, and AD symptoms have manifested. Panel (B): The disease progresses and reaches its end stage. Panel (C): The evolution of the initial state in the treated patient. The ISR-inhibiting drug is administered transiently and concurrently with the activator of BACE1 and/or BACE2. Suppression of the neuronal ISR disables the operation of the AβPP-independent C99/iAβ production pathway and the influx of its product stops. This allows activated BACE1 and/or BACE2 to efficiently deplete iAβ. Following the withdrawal of both ISR inhibitors and BACE activators, the AβPP-independent C99/iAβ production pathway remains inoperative, and the progression of AD ceases. De novo accumulation of iAβ commences from a low baseline supported solely by the AβPP proteolysis. It would not reach the T1 threshold, the AβPP-independent C99/iAβ production pathway would not be reactivated, and the disease would not recur within the remaining lifetime of the treated AD patient.
Figure 21.
Concurrent transient BACE1 and/or BACE2 activation and ISR inhibition in the treatment of conventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Orange box: Duration of the treatment with the BACE1- and/or BACE2-activating drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the AD patient. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons, the neuronal ISR has been elicited, the AβPP-independent C99/iAβ production pathway activated, the T2 threshold crossed in a neuronal fraction, and AD symptoms have manifested. Panel (B): The disease progresses and reaches its end stage. Panel (C): The evolution of the initial state in the treated patient. The ISR-inhibiting drug is administered transiently and concurrently with the activator of BACE1 and/or BACE2. Suppression of the neuronal ISR disables the operation of the AβPP-independent C99/iAβ production pathway and the influx of its product stops. This allows activated BACE1 and/or BACE2 to efficiently deplete iAβ. Following the withdrawal of both ISR inhibitors and BACE activators, the AβPP-independent C99/iAβ production pathway remains inoperative, and the progression of AD ceases. De novo accumulation of iAβ commences from a low baseline supported solely by the AβPP proteolysis. It would not reach the T1 threshold, the AβPP-independent C99/iAβ production pathway would not be reactivated, and the disease would not recur within the remaining lifetime of the treated AD patient.
![Ijms 26 04252 g021]()
Figure 22.
Sequential occurrence of AD pathology in the defined brain compartments as the consequence of the variability in the rates of AβPP-independent iAβ accumulation and the extents of the T2 threshold. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. T2′, T2’’, T2’’’, T’’’’: Variable T2 thresholds in the defined regions of the brain. Red, blue, green, and brown lines above the T1 threshold: iAβ accumulation in the AD-affected neurons in various regions of the brain. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Panel (A): Effects of the variable extent of the T2 threshold at the background of the given rate of accumulation of iAβ produced independently of AβPP on the sequential occurrence of AD pathology in the defined regions of the brain. The greater the former, the later the neurodegeneration manifests. Panel (B): the combined effect of modulation of both the extent of the T1 threshold and the rate of accumulation of iAβ produced independently of AβPP on the sequential occurrence of AD pathology in the defined brain compartment.
Figure 22.
Sequential occurrence of AD pathology in the defined brain compartments as the consequence of the variability in the rates of AβPP-independent iAβ accumulation and the extents of the T2 threshold. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. T2′, T2’’, T2’’’, T’’’’: Variable T2 thresholds in the defined regions of the brain. Red, blue, green, and brown lines above the T1 threshold: iAβ accumulation in the AD-affected neurons in various regions of the brain. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Panel (A): Effects of the variable extent of the T2 threshold at the background of the given rate of accumulation of iAβ produced independently of AβPP on the sequential occurrence of AD pathology in the defined regions of the brain. The greater the former, the later the neurodegeneration manifests. Panel (B): the combined effect of modulation of both the extent of the T1 threshold and the rate of accumulation of iAβ produced independently of AβPP on the sequential occurrence of AD pathology in the defined brain compartment.
![Ijms 26 04252 g022]()
Figure 23.
Alzheimer’s is a multiform disease of the neuronal ISR: Conventional AD is triggered by AβPP-derived iAβ whereas neuronal ISR-eliciting stressors distinct from AβPP-derived iAβ initiate unconventional AD. eIF2α: eukaryotic translation initiation factor 2α. PKR and HRI: kinases that phosphorylate eIF2α at its Ser51 residue. PACT: PKR activator. TNFα: tumor necrosis factor α. OMA1: mitochondrial protease activated during mitochondrial dysfunction. DELE1: substrate of OMA1 protease. Cleavage of DELE1 leads to the activation of HRI. Phosphorylation of eIF2α elicits the integrated stress response, which, in turn, enables the generation of components essential for the activity of the AβPP-independent pathway of C99 and iAβ production. iAβ: intraneuronal Aβ. The accumulation of iAβ produced in both the AβPP proteolytic and AβPP-independent pathways is accompanied by a proportional accumulation of AICD, AβPP Intracellular Domain. AICD was shown to be capable of interfering with numerous processes involved in AD but its contribution to the disease remains to be elucidated. AβPP-derived iAβ accumulates physiologically via the importation of extracellular Aβ and the retention of iAβ produced by gamma-cleavages on intracellular membranes in an exceedingly slow process. In most individuals it does not reach the PKR- and/or HRI-activating, the neuronal ISR-eliciting threshold within their lifetimes and no conventional AD occurs. When this threshold is crossed, the neuronal ISR is elicited and the AβPP-independent C99 (and, subsequently, iAβ) production pathway activated, and conventional AD ensues. In unconventional AD stressors that are distinct from AβPP-derived iAβ and capable of activating one or more of eIF2α kinases trigger the elicitation of the neuronal ISR. In both conventional and unconventional forms of AD the iAβ product of the AβPP-independent pathway of C99 and iAβ production drives AD pathology. It also propagates the neuronal ISR and thus perpetuates its own production in the AβPP-independent pathway. Thus, conventional and unconventional AD differ only in the manner of the elicitation of the neuronal integrated stress response.
Figure 23.
Alzheimer’s is a multiform disease of the neuronal ISR: Conventional AD is triggered by AβPP-derived iAβ whereas neuronal ISR-eliciting stressors distinct from AβPP-derived iAβ initiate unconventional AD. eIF2α: eukaryotic translation initiation factor 2α. PKR and HRI: kinases that phosphorylate eIF2α at its Ser51 residue. PACT: PKR activator. TNFα: tumor necrosis factor α. OMA1: mitochondrial protease activated during mitochondrial dysfunction. DELE1: substrate of OMA1 protease. Cleavage of DELE1 leads to the activation of HRI. Phosphorylation of eIF2α elicits the integrated stress response, which, in turn, enables the generation of components essential for the activity of the AβPP-independent pathway of C99 and iAβ production. iAβ: intraneuronal Aβ. The accumulation of iAβ produced in both the AβPP proteolytic and AβPP-independent pathways is accompanied by a proportional accumulation of AICD, AβPP Intracellular Domain. AICD was shown to be capable of interfering with numerous processes involved in AD but its contribution to the disease remains to be elucidated. AβPP-derived iAβ accumulates physiologically via the importation of extracellular Aβ and the retention of iAβ produced by gamma-cleavages on intracellular membranes in an exceedingly slow process. In most individuals it does not reach the PKR- and/or HRI-activating, the neuronal ISR-eliciting threshold within their lifetimes and no conventional AD occurs. When this threshold is crossed, the neuronal ISR is elicited and the AβPP-independent C99 (and, subsequently, iAβ) production pathway activated, and conventional AD ensues. In unconventional AD stressors that are distinct from AβPP-derived iAβ and capable of activating one or more of eIF2α kinases trigger the elicitation of the neuronal ISR. In both conventional and unconventional forms of AD the iAβ product of the AβPP-independent pathway of C99 and iAβ production drives AD pathology. It also propagates the neuronal ISR and thus perpetuates its own production in the AβPP-independent pathway. Thus, conventional and unconventional AD differ only in the manner of the elicitation of the neuronal integrated stress response.
![Ijms 26 04252 g023]()
Figure 24.
Unconventional activation of the AβPP-independent C99/iAβ production pathway: Effect of the long-term presence of unconventional ISR-eliciting stressors. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: Duration of the occurrence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Panel (A): Kinetics of the accumulation of AβPP-derived iAβ in the healthy individual. The rate of accumulation of iAβ is such that it does not reach the T1 threshold. eIF2α kinases are not activated, eIF2α is not phosphorylated, the neuronal ISR is not elicited, the AβPP-independent C99/iAβ production pathway is not initiated, and AD does not occur. Panel (B): Kinetics of the iAβ accumulation in the presence of the unconventional stressor. The unconventional (i.e., distinct from AβPP-derived iAβ) stressor occurs when the levels of AβPP-derived iAβ are well below the T1 threshold and persists for the remaining portion of the lifetime. The neuronal ISR is unconventionally elicited, and the AβPP-independent C99/iAβ production pathway is unconventionally activated. iAβ generated independently of AβPP rapidly accumulates. When its levels cross the T1 threshold, the AβPP-independent C99/iAβ production pathway is rendered self-sustainable and unconventional AD commences.
Figure 24.
Unconventional activation of the AβPP-independent C99/iAβ production pathway: Effect of the long-term presence of unconventional ISR-eliciting stressors. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: Duration of the occurrence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Panel (A): Kinetics of the accumulation of AβPP-derived iAβ in the healthy individual. The rate of accumulation of iAβ is such that it does not reach the T1 threshold. eIF2α kinases are not activated, eIF2α is not phosphorylated, the neuronal ISR is not elicited, the AβPP-independent C99/iAβ production pathway is not initiated, and AD does not occur. Panel (B): Kinetics of the iAβ accumulation in the presence of the unconventional stressor. The unconventional (i.e., distinct from AβPP-derived iAβ) stressor occurs when the levels of AβPP-derived iAβ are well below the T1 threshold and persists for the remaining portion of the lifetime. The neuronal ISR is unconventionally elicited, and the AβPP-independent C99/iAβ production pathway is unconventionally activated. iAβ generated independently of AβPP rapidly accumulates. When its levels cross the T1 threshold, the AβPP-independent C99/iAβ production pathway is rendered self-sustainable and unconventional AD commences.
![Ijms 26 04252 g024]()
Figure 25.
Unconventional activation of the AβPP-independent C99/iAβ production pathway: effects of the transient presence of unconventional ISR-eliciting stressors. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: Duration of the occurrence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Broken lines: The projected dynamics of the accumulation of AβPP-derived iAβ in the absence of the episodes of unconventional activity of the AβPP-independent C99 production pathway. Panel (A): The unconventional stressor occurs when AβPP-derived iAβ is well below the T1 threshold and accumulates at such a rate that it would not reach the T1 threshold within the individual’s lifetime (projected accumulation of AβPP-derived iAβ in the absence of the unconventional stressor is shown as broken lines). Following the occurrence of the unconventional stressor, the neuronal ISR is unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated. The levels of iAβ produced independently of AβPP rapidly increase. But before they reach the T1 threshold, the unconventional stressor is withdrawn. The ISR conditions are reversed, and the operation of the AβPP-independent C99/iAβ production pathway ceases. The accumulation of AβPP-derived iAβ resumes from an elevated baseline but proceeds at a low rate, supported only by the AβPP proteolysis. It does not reach the T1 threshold within the lifetime of the individual and no AD occurs. Panel (B): The unconventional stressor persists for a considerable duration. The neuronal ISR is unconventionally elicited, the AβPP-independent C99/iAβ production pathway is unconventionally activated, and iAβ produced independently of AβPP rapidly accumulates. When the unconventional stressor is withdrawn, iAβ has crossed the T1 threshold in all affected neurons. The AβPP-independent C99/iAβ production pathway has attained self-sustainability and the withdrawal of the initial unconventional stressor has no effect whatsoever on its continuous operation. Panel (C): The unconventional stressor is withdrawn when iAβ produced independently of AβPP has crossed the T1 threshold in only a fraction of affected neurons. In these neurons the AβPP-independent C99/iAβ production pathway has attained self-sustainability and its continuous operation is not affected by the withdrawal of the initial unconventional stressor. In the neurons that have not yet crossed the T1 threshold, the withdrawal of the unconventional stressor terminates the operation of the AβPP-independent C99/iAβ production pathway. The accumulation of AβPP-derived iAβ resumes and when it crosses the T1 threshold the neuronal ISR is elicited, the AβPP-independent C99/iAβ production pathway is activated, and the progression of AD pathology in these neurons commences.
Figure 25.
Unconventional activation of the AβPP-independent C99/iAβ production pathway: effects of the transient presence of unconventional ISR-eliciting stressors. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: Duration of the occurrence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Broken lines: The projected dynamics of the accumulation of AβPP-derived iAβ in the absence of the episodes of unconventional activity of the AβPP-independent C99 production pathway. Panel (A): The unconventional stressor occurs when AβPP-derived iAβ is well below the T1 threshold and accumulates at such a rate that it would not reach the T1 threshold within the individual’s lifetime (projected accumulation of AβPP-derived iAβ in the absence of the unconventional stressor is shown as broken lines). Following the occurrence of the unconventional stressor, the neuronal ISR is unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated. The levels of iAβ produced independently of AβPP rapidly increase. But before they reach the T1 threshold, the unconventional stressor is withdrawn. The ISR conditions are reversed, and the operation of the AβPP-independent C99/iAβ production pathway ceases. The accumulation of AβPP-derived iAβ resumes from an elevated baseline but proceeds at a low rate, supported only by the AβPP proteolysis. It does not reach the T1 threshold within the lifetime of the individual and no AD occurs. Panel (B): The unconventional stressor persists for a considerable duration. The neuronal ISR is unconventionally elicited, the AβPP-independent C99/iAβ production pathway is unconventionally activated, and iAβ produced independently of AβPP rapidly accumulates. When the unconventional stressor is withdrawn, iAβ has crossed the T1 threshold in all affected neurons. The AβPP-independent C99/iAβ production pathway has attained self-sustainability and the withdrawal of the initial unconventional stressor has no effect whatsoever on its continuous operation. Panel (C): The unconventional stressor is withdrawn when iAβ produced independently of AβPP has crossed the T1 threshold in only a fraction of affected neurons. In these neurons the AβPP-independent C99/iAβ production pathway has attained self-sustainability and its continuous operation is not affected by the withdrawal of the initial unconventional stressor. In the neurons that have not yet crossed the T1 threshold, the withdrawal of the unconventional stressor terminates the operation of the AβPP-independent C99/iAβ production pathway. The accumulation of AβPP-derived iAβ resumes and when it crosses the T1 threshold the neuronal ISR is elicited, the AβPP-independent C99/iAβ production pathway is activated, and the progression of AD pathology in these neurons commences.
![Ijms 26 04252 g025]()
Figure 26.
Microbursts of unconventional activity of the AβPP-independent C99/iAβ production pathway contribute to the normal Under-T1 accumulation of iAβ. Blue lines: Intraneuronal Aβ, iAβ. iAβ produced in the microbursts of the unconventional activity of the AβPP-independent C99/iAβ generation pathway. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Panel (A): The accumulation of iAβ in neurons of the healthy individual. Unconventional neuronal ISR-eliciting stressors appear for only a short duration but on numerous occasions. Every time their occurrence causes a microburst of the activity of the AβPP-independent C99/iAβ generation pathway and the levels of iAβ rapidly increase. Each burst concludes with the withdrawal of the unconventional stressor and the resumption of the slow accumulation of AβPP-derived iAβ from the elevated baseline. Despite numerous microbursts, the levels of iAβ do not reach the T1 threshold within the lifetime of the individual and no AD occurs. Panel (B): The rate of accumulation of AβPP-derived iAβ combined with the extents and number of microbursts generating iAβ independently of AβPP are such that the T1 threshold is reached and crossed. The neuronal ISR is sustainably elicited, the self-sustainable AβPP-independent C99/iAβ generation pathway is activated, and conventional AD commences and progresses. Thus, numerous microbursts of the unconventional activity of the AβPP-independent C99/iAβ production pathway potentially occur “normally”, without adverse effects, in healthy individuals. On the other hand, conventional AD potentially always contains an unconventional component in the form of the microbursts of the unconventional activity of the AβPP-independent C99/iAβ generation pathway.
Figure 26.
Microbursts of unconventional activity of the AβPP-independent C99/iAβ production pathway contribute to the normal Under-T1 accumulation of iAβ. Blue lines: Intraneuronal Aβ, iAβ. iAβ produced in the microbursts of the unconventional activity of the AβPP-independent C99/iAβ generation pathway. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Panel (A): The accumulation of iAβ in neurons of the healthy individual. Unconventional neuronal ISR-eliciting stressors appear for only a short duration but on numerous occasions. Every time their occurrence causes a microburst of the activity of the AβPP-independent C99/iAβ generation pathway and the levels of iAβ rapidly increase. Each burst concludes with the withdrawal of the unconventional stressor and the resumption of the slow accumulation of AβPP-derived iAβ from the elevated baseline. Despite numerous microbursts, the levels of iAβ do not reach the T1 threshold within the lifetime of the individual and no AD occurs. Panel (B): The rate of accumulation of AβPP-derived iAβ combined with the extents and number of microbursts generating iAβ independently of AβPP are such that the T1 threshold is reached and crossed. The neuronal ISR is sustainably elicited, the self-sustainable AβPP-independent C99/iAβ generation pathway is activated, and conventional AD commences and progresses. Thus, numerous microbursts of the unconventional activity of the AβPP-independent C99/iAβ production pathway potentially occur “normally”, without adverse effects, in healthy individuals. On the other hand, conventional AD potentially always contains an unconventional component in the form of the microbursts of the unconventional activity of the AβPP-independent C99/iAβ generation pathway.
![Ijms 26 04252 g026]()
Figure 27.
TBI and CTE cause unconventional AD via the unconventional transient activation of the AβPP-independent C99/iAβ generation pathway. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: Duration of the occurrence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Broken lines: The projected dynamics of the accumulation of AβPP-derived iAβ in the absence of the episodes of unconventional activity of the AβPP-independent C99 production pathway; no T1 would be crossed within the lifetime of the individual, no AD would occur. Panel (A): TBI provides, probably via the suppression of CBF, the unconventional neuronal ISR-eliciting stressor and thus enables the unconventional activation and transient operation of the AβPP-independent C99/iAβ generation pathway. Upon withdrawal of the unconventional stressor, AβPP-derived iAβ resumes its accumulation from the elevated (but still under-T1) baseline. When the T1 is crossed, PKR and/or HRI are activated, eIF2α is phosphorylated, the neuronal ISR is elicited, the self-sustainable AβPP-independent C99/iAβ generation pathway is initiated, and AD commences. The time period between the TBI event and the commencement of AD is defined by the severity of TBI and, consequently, by the duration of the presence of the unconventional neuronal ISR-eliciting stressor. Panel (B): In CTE, the severity of trauma is substantially smaller than in TBI, but it does provide, potentially through the suppression of CBF, an unconventional neuronal ISR-eliciting stressor. The duration of the transient operation of the unconventionally activated AβPP-independent C99/iAβ generation pathway is shorter and the extent of the elevation of the baseline of the resumed accumulation of AβPP-derived iAβ is smaller in CTE than in TBI. But in the former traumatic events occur repeatedly, and after each event the baseline of the resumed accumulation of AβPP-derived iAβ is elevated further until, eventually, the T1 threshold is crossed, the self-sustainable AβPP-independent C99/iAβ generation pathway is activated, and AD commences.
Figure 27.
TBI and CTE cause unconventional AD via the unconventional transient activation of the AβPP-independent C99/iAβ generation pathway. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: Duration of the occurrence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Broken lines: The projected dynamics of the accumulation of AβPP-derived iAβ in the absence of the episodes of unconventional activity of the AβPP-independent C99 production pathway; no T1 would be crossed within the lifetime of the individual, no AD would occur. Panel (A): TBI provides, probably via the suppression of CBF, the unconventional neuronal ISR-eliciting stressor and thus enables the unconventional activation and transient operation of the AβPP-independent C99/iAβ generation pathway. Upon withdrawal of the unconventional stressor, AβPP-derived iAβ resumes its accumulation from the elevated (but still under-T1) baseline. When the T1 is crossed, PKR and/or HRI are activated, eIF2α is phosphorylated, the neuronal ISR is elicited, the self-sustainable AβPP-independent C99/iAβ generation pathway is initiated, and AD commences. The time period between the TBI event and the commencement of AD is defined by the severity of TBI and, consequently, by the duration of the presence of the unconventional neuronal ISR-eliciting stressor. Panel (B): In CTE, the severity of trauma is substantially smaller than in TBI, but it does provide, potentially through the suppression of CBF, an unconventional neuronal ISR-eliciting stressor. The duration of the transient operation of the unconventionally activated AβPP-independent C99/iAβ generation pathway is shorter and the extent of the elevation of the baseline of the resumed accumulation of AβPP-derived iAβ is smaller in CTE than in TBI. But in the former traumatic events occur repeatedly, and after each event the baseline of the resumed accumulation of AβPP-derived iAβ is elevated further until, eventually, the T1 threshold is crossed, the self-sustainable AβPP-independent C99/iAβ generation pathway is activated, and AD commences.
![Ijms 26 04252 g027]()
Figure 28.
Inhibitors of the neuronal integrated stress response in the prevention and treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Green box: The duration of the treatment with the ISR-inhibiting drug. Panel (A): ISR inhibitors in the prevention of unconventional AD. Following the occurrence of the unconventional stressor, the neuronal ISR has been unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated. iAβ levels have rapidly increased but have not yet reached the T1 threshold. The administration of ISR inhibitors at this point reverse the ISR conditions, deprive the AβPP-independent C99/iAβ production pathway of its essential components and render it inoperative. The accumulation of iAβ continues, but at a low rate, supported only by the AβPP proteolysis. The crossing of the T1 threshold would be inconsequential since no ISR can be elicited in the presence of the drug. The levels of iAβ would not reach the AD pathology-causing range and AD would not occur for the duration of the treatment. Panel (B): ISR inhibitors in the treatment of unconventional AD. Inhibition of the neuronal ISR deprives the AβPP-independent C99/iAβ production pathway of its essential components and its operation ceases. The rate of accumulation of iAβ, produced at this point only by the AβPP proteolysis, significantly decreases. The progression of the disease continues but at a much slower rate for the duration of the treatment.
Figure 28.
Inhibitors of the neuronal integrated stress response in the prevention and treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Green box: The duration of the treatment with the ISR-inhibiting drug. Panel (A): ISR inhibitors in the prevention of unconventional AD. Following the occurrence of the unconventional stressor, the neuronal ISR has been unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated. iAβ levels have rapidly increased but have not yet reached the T1 threshold. The administration of ISR inhibitors at this point reverse the ISR conditions, deprive the AβPP-independent C99/iAβ production pathway of its essential components and render it inoperative. The accumulation of iAβ continues, but at a low rate, supported only by the AβPP proteolysis. The crossing of the T1 threshold would be inconsequential since no ISR can be elicited in the presence of the drug. The levels of iAβ would not reach the AD pathology-causing range and AD would not occur for the duration of the treatment. Panel (B): ISR inhibitors in the treatment of unconventional AD. Inhibition of the neuronal ISR deprives the AβPP-independent C99/iAβ production pathway of its essential components and its operation ceases. The rate of accumulation of iAβ, produced at this point only by the AβPP proteolysis, significantly decreases. The progression of the disease continues but at a much slower rate for the duration of the treatment.
![Ijms 26 04252 g028]()
Figure 29.
Activation of BACE1 and/or BACE2 in the prevention of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Orange box: The duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The initial state of the levels of iAβ in individual neurons at the time of the commencement of the drug’s administration. In these neurons, the occurrence of the unconventional stressor elicited the neuronal ISR, which, in turn, provided components essential for the operation of the AβPP-independent C99/iAβ production pathway and thus activated it. iAβ generated independently of AβPP rapidly accumulated but is still below the T1 threshold. Panel (B): The evolution of the initial state in the untreated individual. The accumulation of iAβ produced independently of AβPP continues unchecked. It crosses the T1 threshold and the AβPP-independent C99/iAβ production pathway is rendered self-sustainable. AD commences and progresses until it reaches its end stage. Panel (C): The evolution of the initial state in the individual treated with the BACE1 and/or BACE2-activating drug. In the presence of the drug, the rate of the efflux of iAβ increases but it cannot match that of the influx of iAβ produced independently of AβPP. However, the rate of the accumulation of iAβ is reduced and the T1 crossing is delayed. The commencement of AD is also delayed, and its progression is slowed down for the duration of the treatment.
Figure 29.
Activation of BACE1 and/or BACE2 in the prevention of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Orange box: The duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The initial state of the levels of iAβ in individual neurons at the time of the commencement of the drug’s administration. In these neurons, the occurrence of the unconventional stressor elicited the neuronal ISR, which, in turn, provided components essential for the operation of the AβPP-independent C99/iAβ production pathway and thus activated it. iAβ generated independently of AβPP rapidly accumulated but is still below the T1 threshold. Panel (B): The evolution of the initial state in the untreated individual. The accumulation of iAβ produced independently of AβPP continues unchecked. It crosses the T1 threshold and the AβPP-independent C99/iAβ production pathway is rendered self-sustainable. AD commences and progresses until it reaches its end stage. Panel (C): The evolution of the initial state in the individual treated with the BACE1 and/or BACE2-activating drug. In the presence of the drug, the rate of the efflux of iAβ increases but it cannot match that of the influx of iAβ produced independently of AβPP. However, the rate of the accumulation of iAβ is reduced and the T1 crossing is delayed. The commencement of AD is also delayed, and its progression is slowed down for the duration of the treatment.
![Ijms 26 04252 g029]()
Figure 30.
Activation of BACE1 and/or BACE2 in the treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Orange box: The duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The initial state of the levels of iAβ in individual neurons at the time of the commencement of the drug’s administration. The neuronal ISR has been unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated at the levels of AβPP-derived iAβ well below the T1 threshold. iAβ produced independently of AβPP rapidly accumulated and crossed the T1 threshold in all affected neurons, and the AβPP-independent C99/iAβ production pathway was rendered self-sustainable. AD commenced and progressed; a fraction of the neurons crossed the T2 threshold, and AD symptoms manifested. Panel (B): The evolution of the initial state in the untreated individual. iAβ produced independently of AβPP continues to accumulate unhindered, additional neurons cross the T2 threshold, and the disease enters its end stage. Panel (C): The evolution of the initial state in the individual treated with the BACE1 and/or BACE2-activating drug. iAβ is being degraded by the intra-iAβ cleaving activities of BACE1 and/or BACE2 but the rate of its efflux is significantly smaller than that of its influx. Its accumulation continues and the disease progresses, although at a decreased rate, for the duration of the treatment.
Figure 30.
Activation of BACE1 and/or BACE2 in the treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Orange box: The duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The initial state of the levels of iAβ in individual neurons at the time of the commencement of the drug’s administration. The neuronal ISR has been unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated at the levels of AβPP-derived iAβ well below the T1 threshold. iAβ produced independently of AβPP rapidly accumulated and crossed the T1 threshold in all affected neurons, and the AβPP-independent C99/iAβ production pathway was rendered self-sustainable. AD commenced and progressed; a fraction of the neurons crossed the T2 threshold, and AD symptoms manifested. Panel (B): The evolution of the initial state in the untreated individual. iAβ produced independently of AβPP continues to accumulate unhindered, additional neurons cross the T2 threshold, and the disease enters its end stage. Panel (C): The evolution of the initial state in the individual treated with the BACE1 and/or BACE2-activating drug. iAβ is being degraded by the intra-iAβ cleaving activities of BACE1 and/or BACE2 but the rate of its efflux is significantly smaller than that of its influx. Its accumulation continues and the disease progresses, although at a decreased rate, for the duration of the treatment.
![Ijms 26 04252 g030]()
Figure 31.
Composite transient ISR inhibition/long-term BACE activation therapy in the prevention and treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Green box: The duration of the treatment with the ISR-inhibiting drug. Orange box: The duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The neuronal ISR is unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated at the levels of AβPP-derived iAβ well below the T1 threshold. The levels of iAβ generated independently of AβPP rapidly increase but at the time of the commencement of the composite therapy do not yet reach the T1 threshold. The ISR inhibitor disables the AβPP-independent C99/iAβ production pathway and stops the influx of iAβ generated independently of AβPP. The concurrent activation of BACE1 and/or BACE2 substantially depletes iAβ. Following the withdrawal of the ISR inhibitor, the neuronal ISR is re-elicited and the AβPP-independent C99/iAβ production pathway reactivated. The continuous presence of the BACE activator reduces the rate of accumulation of iAβ produced independently of AβPP and its levels increase relatively slowly. They do not reach the AD pathology-causing range and AD does not occur for the duration of the treatment. Panel (B): iAβ produced independently of AβPP has crossed the T1 threshold in all affected neurons and the AβPP-independent C99/iAβ production pathway has been rendered self-sustainable. At the time of the commencement of the composite therapy a fraction of the neurons had crossed the T2 threshold and AD symptoms manifested. The presence of the ISR inhibitor abrogates the influx of iAβ produced independently of AβPP and allows the activator of BACE1 and/or BACE2 to substantially reduce iAβ levels. Following the withdrawal of the ISR inhibitor, the neuronal ISR is unconventionally re-elicited and the AβPP-independent C99/iAβ production pathway unconventionally reactivated. However, the continuous presence of the BACE1 and/or BACE2 activator maintains a relatively low rate of accumulation of iAβ. It does not reach the AD pathology-causing levels and AD does not recur for the duration of the treatment.
Figure 31.
Composite transient ISR inhibition/long-term BACE activation therapy in the prevention and treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Green box: The duration of the treatment with the ISR-inhibiting drug. Orange box: The duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The neuronal ISR is unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated at the levels of AβPP-derived iAβ well below the T1 threshold. The levels of iAβ generated independently of AβPP rapidly increase but at the time of the commencement of the composite therapy do not yet reach the T1 threshold. The ISR inhibitor disables the AβPP-independent C99/iAβ production pathway and stops the influx of iAβ generated independently of AβPP. The concurrent activation of BACE1 and/or BACE2 substantially depletes iAβ. Following the withdrawal of the ISR inhibitor, the neuronal ISR is re-elicited and the AβPP-independent C99/iAβ production pathway reactivated. The continuous presence of the BACE activator reduces the rate of accumulation of iAβ produced independently of AβPP and its levels increase relatively slowly. They do not reach the AD pathology-causing range and AD does not occur for the duration of the treatment. Panel (B): iAβ produced independently of AβPP has crossed the T1 threshold in all affected neurons and the AβPP-independent C99/iAβ production pathway has been rendered self-sustainable. At the time of the commencement of the composite therapy a fraction of the neurons had crossed the T2 threshold and AD symptoms manifested. The presence of the ISR inhibitor abrogates the influx of iAβ produced independently of AβPP and allows the activator of BACE1 and/or BACE2 to substantially reduce iAβ levels. Following the withdrawal of the ISR inhibitor, the neuronal ISR is unconventionally re-elicited and the AβPP-independent C99/iAβ production pathway unconventionally reactivated. However, the continuous presence of the BACE1 and/or BACE2 activator maintains a relatively low rate of accumulation of iAβ. It does not reach the AD pathology-causing levels and AD does not recur for the duration of the treatment.
![Ijms 26 04252 g031]()
Figure 32.
Transient composite ISR inhibition/BACE activation therapy in the prevention and treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Green box: The duration of the treatment with the ISR-inhibiting drug. Orange box: The duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The effect of the transient concurrent ISR inhibition and BACE1 and/or BACE2 activation in the prevention of unconventional AD. The neuronal ISR has been unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated at the levels of AβPP-derived iAβ well below the T1 threshold. iAβ generated independently of AβPP has rapidly accumulated but at the time of the implementation of the composite therapy is still below the T1 threshold. The treatment with the ISR inhibitor disables the AβPP-independent C99/iAβ production pathway and terminates the influx of its iAβ product. This enables a substantial depletion of iAβ by concurrently activated BACE. When both drugs are withdrawn, the neuronal IST is unconventionally re-elicited and the AβPP-independent C99/iAβ production pathway unconventionally reactivated. The accumulation of iAβ resumes from a low baseline but proceeds rapidly. When it crosses the T1 threshold the AβPP-independent C99/iAβ production pathway is rendered self-sustainable and AD commences. The composite therapy does not prevent the disease but provides a short reprieve. Panel (B): The effect of the transient concurrent ISR inhibition and BACE1 and/or BACE2 activation in the treatment of unconventional AD. At the time of the implementation of the composite therapy all affected neurons have crossed the T1 threshold, a fraction of the neurons has crossed the T2 threshold, and AD symptoms manifested. Inhibition of the neuronal ISR deprives the AβPP-independent C99/iAβ production pathway of its essential components, disables it, and stops the influx of iAβ generated independently of AβPP. The concurrent activation of BACE1 and/or BACE2 substantially depletes iAβ. Upon the completion of the treatment and the withdrawal of the drugs the neuronal ISR is re-elicited and the AβPP-independent C99/iAβ production pathway reactivated. De novo accumulation of iAβ commences from a low baseline but proceeds rapidly. When it reaches the T1 threshold, the disease recurs.
Figure 32.
Transient composite ISR inhibition/BACE activation therapy in the prevention and treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Green box: The duration of the treatment with the ISR-inhibiting drug. Orange box: The duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The effect of the transient concurrent ISR inhibition and BACE1 and/or BACE2 activation in the prevention of unconventional AD. The neuronal ISR has been unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated at the levels of AβPP-derived iAβ well below the T1 threshold. iAβ generated independently of AβPP has rapidly accumulated but at the time of the implementation of the composite therapy is still below the T1 threshold. The treatment with the ISR inhibitor disables the AβPP-independent C99/iAβ production pathway and terminates the influx of its iAβ product. This enables a substantial depletion of iAβ by concurrently activated BACE. When both drugs are withdrawn, the neuronal IST is unconventionally re-elicited and the AβPP-independent C99/iAβ production pathway unconventionally reactivated. The accumulation of iAβ resumes from a low baseline but proceeds rapidly. When it crosses the T1 threshold the AβPP-independent C99/iAβ production pathway is rendered self-sustainable and AD commences. The composite therapy does not prevent the disease but provides a short reprieve. Panel (B): The effect of the transient concurrent ISR inhibition and BACE1 and/or BACE2 activation in the treatment of unconventional AD. At the time of the implementation of the composite therapy all affected neurons have crossed the T1 threshold, a fraction of the neurons has crossed the T2 threshold, and AD symptoms manifested. Inhibition of the neuronal ISR deprives the AβPP-independent C99/iAβ production pathway of its essential components, disables it, and stops the influx of iAβ generated independently of AβPP. The concurrent activation of BACE1 and/or BACE2 substantially depletes iAβ. Upon the completion of the treatment and the withdrawal of the drugs the neuronal ISR is re-elicited and the AβPP-independent C99/iAβ production pathway reactivated. De novo accumulation of iAβ commences from a low baseline but proceeds rapidly. When it reaches the T1 threshold, the disease recurs.
![Ijms 26 04252 g032]()
Figure 33.
Recurrent transient composite ISR inhibition/BACE activation therapy in the prevention and treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Brown boxes: The duration of the composite treatment with concurrently administered ISR inhibitors and BACE activators. Panel (A): Recurrent administration of the composite therapy in the prevention of unconventional AD. At the time of the implementation of the therapy the neuronal ISR has been unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated at the low levels of AβPP-derived iAβ. iAβ generated independently of AβPP has rapidly accumulated but has not yet reached the T1 threshold. The concurrent administration of ISR inhibitors and BACE activators disables the AβPP-independent C99/iAβ production pathway and substantially depletes iAβ. Upon the completion of the treatment, the neuronal ISR is unconventionally re-elicited and the AβPP-independent C99/iAβ production pathway unconventionally reactivated. The accumulation of iAβ produced independently of AβPP resumes but from a low baseline. Before it reaches the T1 threshold, at a point indicated by suitable biomarkers, the composite therapy is repeated recurrently. The T1 threshold is not crossed, and the disease does not occur for the duration of the treatments. Panel (B): Recurrent administration of the composite therapy in the prevention of unconventional AD. The first round of the transient composite therapy is administered when iAβ produced independently of AβPP have already crossed the T1 threshold in all affected neurons, the AβPP-independent C99/iAβ production pathway has been rendered self-sustainable, some neurons have crossed the T2 threshold, and AD symptoms have already manifested. The concurrent transient inhibition of the neuronal ISR and activation of BACE1 and/or BACE2 disable the AβPP-independent C99/iAβ production pathway, stop the influx of iAβ generated independently of AβPP, and substantially deplete iAβ. When drugs are withdrawn, the neuronal ISR is unconventionally re-elicited and the AβPP-independent C99/iAβ production pathway unconventionally reactivated. The accumulation of iAβ resumes from a low baseline. Before it reaches the AD pathology-causing levels, at a point defined by the appropriate biomarkers, the composite therapy is re-administered and repeated recurrently as needed. The AD pathology-causing range of iAβ concentrations is not reached and the disease does not recur for the duration of the treatment.
Figure 33.
Recurrent transient composite ISR inhibition/BACE activation therapy in the prevention and treatment of unconventional AD. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Brown boxes: The duration of the composite treatment with concurrently administered ISR inhibitors and BACE activators. Panel (A): Recurrent administration of the composite therapy in the prevention of unconventional AD. At the time of the implementation of the therapy the neuronal ISR has been unconventionally elicited and the AβPP-independent C99/iAβ production pathway unconventionally activated at the low levels of AβPP-derived iAβ. iAβ generated independently of AβPP has rapidly accumulated but has not yet reached the T1 threshold. The concurrent administration of ISR inhibitors and BACE activators disables the AβPP-independent C99/iAβ production pathway and substantially depletes iAβ. Upon the completion of the treatment, the neuronal ISR is unconventionally re-elicited and the AβPP-independent C99/iAβ production pathway unconventionally reactivated. The accumulation of iAβ produced independently of AβPP resumes but from a low baseline. Before it reaches the T1 threshold, at a point indicated by suitable biomarkers, the composite therapy is repeated recurrently. The T1 threshold is not crossed, and the disease does not occur for the duration of the treatments. Panel (B): Recurrent administration of the composite therapy in the prevention of unconventional AD. The first round of the transient composite therapy is administered when iAβ produced independently of AβPP have already crossed the T1 threshold in all affected neurons, the AβPP-independent C99/iAβ production pathway has been rendered self-sustainable, some neurons have crossed the T2 threshold, and AD symptoms have already manifested. The concurrent transient inhibition of the neuronal ISR and activation of BACE1 and/or BACE2 disable the AβPP-independent C99/iAβ production pathway, stop the influx of iAβ generated independently of AβPP, and substantially deplete iAβ. When drugs are withdrawn, the neuronal ISR is unconventionally re-elicited and the AβPP-independent C99/iAβ production pathway unconventionally reactivated. The accumulation of iAβ resumes from a low baseline. Before it reaches the AD pathology-causing levels, at a point defined by the appropriate biomarkers, the composite therapy is re-administered and repeated recurrently as needed. The AD pathology-causing range of iAβ concentrations is not reached and the disease does not recur for the duration of the treatment.
![Ijms 26 04252 g033]()
Figure 34.
The neuronal ISR inhibits the production of Aβ and suppresses the rate of iAβ accumulation: lessons from transgenic mouse models. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: Range of AD pathology-causing concentrations of iAβ. Broken blue lines: the projected dynamics of accumulation of AβPP-derived iAβ occurring at the pre-T1 crossing rate; it would cause massive neurodegeneration and neuronal loss. Panel (A): Exogenously produced AβPP-derived iAβ rapidly accumulates and crosses the T1 threshold. PKR and/or HRI are activated, eIF2α is phosphorylated at its Ser51 residue, and the neuronal ISR is elicited. ISR-mediated suppression of cellular protein synthesis causes mild neurodegeneration and cognitive impairment. Were the accumulation of exogenous AβPP-derived iAβ to continue at the pre-T1 crossing rate (shown by broken blue lines), it would inevitably cause massive neuronal loss. Instead, the production of constituents of the AβPP proteolytic pathway is suppressed in the framework of the neuronal ISR together with that of the decisive bulk of cellular proteins. Consequently, the rate of accumulation of AβPP-derived iAβ is substantially reduced and the degree of the neuronal damage and of cognitive impairment does not change significantly with time. Panel (B): The rate of accumulation of AβPP-derived iAβ is reversed under ISR conditions, and its levels are decreasing. When iAβ levels reverse-cross the T1 threshold the ISR state is also reversed. This restores cellular protein synthesis and enables the production of the components of the AβPP proteolytic pathway and, consequently, the generation of iAβ. With its rate of accumulation restored, levels of iAβ increase. When they cross the T1 threshold, the neuronal ISR is re-elicited, the AβPP proteolytic pathway and the production of iAβ are suppressed, and the rate of accumulation of the latter is again reversed. The cycle repeats, and the levels of iAβ oscillate around the T1 threshold.
Figure 34.
The neuronal ISR inhibits the production of Aβ and suppresses the rate of iAβ accumulation: lessons from transgenic mouse models. Blue lines: Intraneuronal Aβ, iAβ. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: Range of AD pathology-causing concentrations of iAβ. Broken blue lines: the projected dynamics of accumulation of AβPP-derived iAβ occurring at the pre-T1 crossing rate; it would cause massive neurodegeneration and neuronal loss. Panel (A): Exogenously produced AβPP-derived iAβ rapidly accumulates and crosses the T1 threshold. PKR and/or HRI are activated, eIF2α is phosphorylated at its Ser51 residue, and the neuronal ISR is elicited. ISR-mediated suppression of cellular protein synthesis causes mild neurodegeneration and cognitive impairment. Were the accumulation of exogenous AβPP-derived iAβ to continue at the pre-T1 crossing rate (shown by broken blue lines), it would inevitably cause massive neuronal loss. Instead, the production of constituents of the AβPP proteolytic pathway is suppressed in the framework of the neuronal ISR together with that of the decisive bulk of cellular proteins. Consequently, the rate of accumulation of AβPP-derived iAβ is substantially reduced and the degree of the neuronal damage and of cognitive impairment does not change significantly with time. Panel (B): The rate of accumulation of AβPP-derived iAβ is reversed under ISR conditions, and its levels are decreasing. When iAβ levels reverse-cross the T1 threshold the ISR state is also reversed. This restores cellular protein synthesis and enables the production of the components of the AβPP proteolytic pathway and, consequently, the generation of iAβ. With its rate of accumulation restored, levels of iAβ increase. When they cross the T1 threshold, the neuronal ISR is re-elicited, the AβPP proteolytic pathway and the production of iAβ are suppressed, and the rate of accumulation of the latter is again reversed. The cycle repeats, and the levels of iAβ oscillate around the T1 threshold.
![Ijms 26 04252 g034]()
Figure 35.
Conventional and unconventional AD is driven by C99 generated independently of AβPP; Tau is translated from IRES in the 5′UTR of its mRNA. eIF2α: eukaryotic translation initiation factor 2α. PKR and HRI: kinases that phosphorylate eIF2α at its Ser51 residue. PACT: PKR activator. TNFα: tumor necrosis factor α. OMA1: mitochondrial protease activated during mitochondrial dysfunction. DELE1: substrate of OMA1 protease. Cleavage of DELE1 leads to the activation of HRI. Phosphorylation of eIF2α elicits the integrated stress response, which, in turn, enables the generation of components essential for the activity of the AβPP-independent pathway of C99 and iAβ production. iAβ: intraneuronal Aβ. AβPP-derived iAβ accumulates physiologically via the importation of extracellular Aβ and the retention of iAβ produced by gamma-cleavages on intracellular membranes in an exceedingly slow process. In most individuals it does not reach the PKR- and/or HRI-activating, the neuronal ISR-eliciting threshold within their lifetimes and no conventional AD occurs. When this threshold is crossed, the neuronal ISR is elicited, and the AβPP-independent C99 production pathway activated, and conventional AD ensues. In unconventional AD stressors that are distinct from AβPP-derived iAβ and capable of activating one or more of eIF2α kinases trigger the elicitation of the neuronal ISR. In both cases the neuronal ISR provides components essential for the operation of the AβPP-independent C99 production pathway. Concurrently, under the neuronal ISR conditions the global cellular protein synthesis, including that of all components of the AβPP proteolytic pathway, is suppressed. Consequently, the accumulation of AβPP-derived iAβ is also suppressed. The production of C99 in the AβPP-independent pathway, on the other hand, proceeds unimpeded; due to the deficiency of gamma secretase it is processed no further. It rapidly accumulates, and it is C99 generated independently of AβPP, which drives AD pathology. Translation of tau protein, on the other hand, persists under ISR conditions because its mRNA contains an internal ribosomal entry site in the 5′UTR.
Figure 35.
Conventional and unconventional AD is driven by C99 generated independently of AβPP; Tau is translated from IRES in the 5′UTR of its mRNA. eIF2α: eukaryotic translation initiation factor 2α. PKR and HRI: kinases that phosphorylate eIF2α at its Ser51 residue. PACT: PKR activator. TNFα: tumor necrosis factor α. OMA1: mitochondrial protease activated during mitochondrial dysfunction. DELE1: substrate of OMA1 protease. Cleavage of DELE1 leads to the activation of HRI. Phosphorylation of eIF2α elicits the integrated stress response, which, in turn, enables the generation of components essential for the activity of the AβPP-independent pathway of C99 and iAβ production. iAβ: intraneuronal Aβ. AβPP-derived iAβ accumulates physiologically via the importation of extracellular Aβ and the retention of iAβ produced by gamma-cleavages on intracellular membranes in an exceedingly slow process. In most individuals it does not reach the PKR- and/or HRI-activating, the neuronal ISR-eliciting threshold within their lifetimes and no conventional AD occurs. When this threshold is crossed, the neuronal ISR is elicited, and the AβPP-independent C99 production pathway activated, and conventional AD ensues. In unconventional AD stressors that are distinct from AβPP-derived iAβ and capable of activating one or more of eIF2α kinases trigger the elicitation of the neuronal ISR. In both cases the neuronal ISR provides components essential for the operation of the AβPP-independent C99 production pathway. Concurrently, under the neuronal ISR conditions the global cellular protein synthesis, including that of all components of the AβPP proteolytic pathway, is suppressed. Consequently, the accumulation of AβPP-derived iAβ is also suppressed. The production of C99 in the AβPP-independent pathway, on the other hand, proceeds unimpeded; due to the deficiency of gamma secretase it is processed no further. It rapidly accumulates, and it is C99 generated independently of AβPP, which drives AD pathology. Translation of tau protein, on the other hand, persists under ISR conditions because its mRNA contains an internal ribosomal entry site in the 5′UTR.
![Ijms 26 04252 g035]()
Figure 36.
Dynamics of conventional and unconventional AD in the ACH2.0 Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Panel (A): Dynamics of conventional AD. AβPP-derived iAβ accumulates physiologically via mechanisms discussed in the main text. When it crosses the T1 threshold, PKR and/or HRI are activated, eIF2α is phosphorylated at its Ser51 residue, and the neuronal ISR is elicited. Global cellular protein synthesis, including all constituents of the AβPP proteolytic pathway: AβPP, BACE enzymes, components of the gamma-secretase complex, and, consequently Aβ, is severely suppressed. The influx of AβPP-derived iAβ is inhibited, and the rate of its accumulation is reduced. Concurrently, the neuronal ISR enables the production of components essential for the operation of the AβPP-independent C99 generation pathway. The pathway is self-sustainable from the instance of its activation because the ISR state is initially sustained by over-T1 AβPP-derived iAβ and eventually by C99 after it crosses the T1 threshold. C99 produced independently of AβPP drives AD pathology; when it crosses the T2 threshold the disease enters its end stage. Panel (B): Dynamics of unconventional AD. The neuronal ISR is unconventionally elicited, and the AβPP-independent C99 generation pathway is unconventionally activated at the levels of AβPP-derived iAβ well below the T1 threshold. Under the neuronal ISR conditions the AβPP proteolytic pathway is suppressed and the rate of accumulation of AβPP-derived iAβ is substantially reduced. On the other hand, C99 generated independently of AβPP rapidly accumulates and crosses the T1 threshold (provided the unconventional stressor persists for a sufficient duration). Following the T1 crossing, the AβPP-independent C99 production pathway is rendered self-sustainable, and AD commences and progresses. When the T2 threshold is reached in a sufficient neuronal fraction, the disease enters its end stage.
Figure 36.
Dynamics of conventional and unconventional AD in the ACH2.0 Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Panel (A): Dynamics of conventional AD. AβPP-derived iAβ accumulates physiologically via mechanisms discussed in the main text. When it crosses the T1 threshold, PKR and/or HRI are activated, eIF2α is phosphorylated at its Ser51 residue, and the neuronal ISR is elicited. Global cellular protein synthesis, including all constituents of the AβPP proteolytic pathway: AβPP, BACE enzymes, components of the gamma-secretase complex, and, consequently Aβ, is severely suppressed. The influx of AβPP-derived iAβ is inhibited, and the rate of its accumulation is reduced. Concurrently, the neuronal ISR enables the production of components essential for the operation of the AβPP-independent C99 generation pathway. The pathway is self-sustainable from the instance of its activation because the ISR state is initially sustained by over-T1 AβPP-derived iAβ and eventually by C99 after it crosses the T1 threshold. C99 produced independently of AβPP drives AD pathology; when it crosses the T2 threshold the disease enters its end stage. Panel (B): Dynamics of unconventional AD. The neuronal ISR is unconventionally elicited, and the AβPP-independent C99 generation pathway is unconventionally activated at the levels of AβPP-derived iAβ well below the T1 threshold. Under the neuronal ISR conditions the AβPP proteolytic pathway is suppressed and the rate of accumulation of AβPP-derived iAβ is substantially reduced. On the other hand, C99 generated independently of AβPP rapidly accumulates and crosses the T1 threshold (provided the unconventional stressor persists for a sufficient duration). Following the T1 crossing, the AβPP-independent C99 production pathway is rendered self-sustainable, and AD commences and progresses. When the T2 threshold is reached in a sufficient neuronal fraction, the disease enters its end stage.
![Ijms 26 04252 g036]()
Figure 37.
ISR inhibitors in the prevention of conventional AD: the ACH2.0, Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing and elicitation of the ISR; usually it can occur only if the extent of the T0 threshold is smaller than that of the T1. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the healthy individual who would develop AD if untreated. AβPP-derived iAβ has been accumulating but its levels are below the T1 threshold. Panel (B): AβPP-derived iAβ crosses the T1 threshold, PKR and/or HRI are activated, eIF2α is phosphorylated at its Ser51 residue, and the neuronal ISR is elicited. Under the ISR conditions the global cellular protein synthesis, including that of the constituents of the AβPP proteolytic pathway, is suppressed and the rate of accumulation of AβPP-derived iAβ is substantially reduced. Concurrently, the neuronal ISR enables the production of components essential for the operation of the AβPP-independent C99 production pathway, and the pathway is activated. C99 generated independently of AβPP rapidly accumulates and drives the progression of AD pathology. Panel (C): The evolution of the initial state in the individual treated with the ISR-inhibiting drug. AβPP-derived iAβ accumulates and crosses the T1 threshold. The drug precludes the elicitation of the neuronal ISR, and the AβPP-independent C99 production pathway remains inoperative. The accumulation of AβPP-derived iAβ continues at the pre-T1 crossing rate; it does not reach the AD pathology-causing levels for the duration of the treatment. On the other hand, since the ISR is suppressed, if AβPP-derived iAβ crosses the T0 threshold, even if it is above the T1 threshold, AACD would commence and persist for the remaining portion of the lifetime; it would be unaffected by the treatment.
Figure 37.
ISR inhibitors in the prevention of conventional AD: the ACH2.0, Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing and elicitation of the ISR; usually it can occur only if the extent of the T0 threshold is smaller than that of the T1. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): The initial state of the levels of AβPP-derived iAβ in the neurons of the healthy individual who would develop AD if untreated. AβPP-derived iAβ has been accumulating but its levels are below the T1 threshold. Panel (B): AβPP-derived iAβ crosses the T1 threshold, PKR and/or HRI are activated, eIF2α is phosphorylated at its Ser51 residue, and the neuronal ISR is elicited. Under the ISR conditions the global cellular protein synthesis, including that of the constituents of the AβPP proteolytic pathway, is suppressed and the rate of accumulation of AβPP-derived iAβ is substantially reduced. Concurrently, the neuronal ISR enables the production of components essential for the operation of the AβPP-independent C99 production pathway, and the pathway is activated. C99 generated independently of AβPP rapidly accumulates and drives the progression of AD pathology. Panel (C): The evolution of the initial state in the individual treated with the ISR-inhibiting drug. AβPP-derived iAβ accumulates and crosses the T1 threshold. The drug precludes the elicitation of the neuronal ISR, and the AβPP-independent C99 production pathway remains inoperative. The accumulation of AβPP-derived iAβ continues at the pre-T1 crossing rate; it does not reach the AD pathology-causing levels for the duration of the treatment. On the other hand, since the ISR is suppressed, if AβPP-derived iAβ crosses the T0 threshold, even if it is above the T1 threshold, AACD would commence and persist for the remaining portion of the lifetime; it would be unaffected by the treatment.
![Ijms 26 04252 g037]()
Figure 38.
ISR inhibitors in the treatment of conventional AD: the ACH2.0, Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): The initial state of the levels of AβPP-derived iAβ and C99 in the neurons of the AD patient. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons, and the neuronal ISR has been elicited. The AβPP proteolytic pathway has been suppressed and the rate of accumulation of AβPP-derived iAβ substantially reduced. Concurrently, the AβPP-independent C99 generation pathway has been activated. C99 accumulated and reached the T2 threshold in a fraction of the neurons, and AD symptoms manifested. Panel (B): The evolution of the initial state in the untreated patient. C99 produced independently of AβPP continues to accumulate and reaches the T2 threshold in a sufficient fraction of the neurons, and the disease enters its end stage. Panel (C): The evolution of the initial state in the AD patient treated with the ISR-inhibiting drug. The ISR state is reversed. The activity of the AβPP proteolytic pathway is restored, and the accumulation of AβPP-derived iAβ resumes at the pre-T1 crossing rate. It would not, however, reach the AD pathology-causing levels. On the other hand, the operation of the AβPP-independent C99 generation pathway ceases. Levels of C99, processed by now available gamma-secretase decline, and the progression of AD is arrested for the duration of the treatment.
Figure 38.
ISR inhibitors in the treatment of conventional AD: the ACH2.0, Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): The initial state of the levels of AβPP-derived iAβ and C99 in the neurons of the AD patient. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons, and the neuronal ISR has been elicited. The AβPP proteolytic pathway has been suppressed and the rate of accumulation of AβPP-derived iAβ substantially reduced. Concurrently, the AβPP-independent C99 generation pathway has been activated. C99 accumulated and reached the T2 threshold in a fraction of the neurons, and AD symptoms manifested. Panel (B): The evolution of the initial state in the untreated patient. C99 produced independently of AβPP continues to accumulate and reaches the T2 threshold in a sufficient fraction of the neurons, and the disease enters its end stage. Panel (C): The evolution of the initial state in the AD patient treated with the ISR-inhibiting drug. The ISR state is reversed. The activity of the AβPP proteolytic pathway is restored, and the accumulation of AβPP-derived iAβ resumes at the pre-T1 crossing rate. It would not, however, reach the AD pathology-causing levels. On the other hand, the operation of the AβPP-independent C99 generation pathway ceases. Levels of C99, processed by now available gamma-secretase decline, and the progression of AD is arrested for the duration of the treatment.
![Ijms 26 04252 g038]()
Figure 39.
ISR inhibitors in the prevention and treatment of unconventional AD: the ACH2.0, Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): ISR inhibitors in the prevention of unconventional AD. Following the occurrence of the unconventional stressor, the neuronal ISR has been unconventionally elicited and the AβPP-independent C99 production pathway unconventionally activated. The neuronal ISR suppresses the AβPP proteolytic pathway and reduces the rate of accumulation of AβPP-derived iAβ. Concurrently, C99 produced independently of AβPP rapidly accumulates but is still below the T1 threshold. The drug reverses the ISR state, restores the activity of the AβPP proteolytic pathway, and the accumulation of the AβPP-derived iAβ resumes at its pre-ISR rate. It also disables the production of C99 in the AβPP-independent pathway, and its levels decline for the duration of the treatment. Panel (B): ISR inhibitors in the treatment of unconventional AD. At the time of the drug’s administration, C99 produced independently of AβPP has crossed the T2 threshold in a fraction of the neurons, and AD symptoms have manifested. The drug reverses the neuronal ISR state, enables the activity of the AβPP proteolytic pathway, and the accumulation of AβPP-derived iAβ resumes at the pre-ISR rate. It also disables the AβPP-independent C99 production pathway. The levels of C99 generated independently of AβPP are declining and the progression of the disease is stopped for the duration of the treatment.
Figure 39.
ISR inhibitors in the prevention and treatment of unconventional AD: the ACH2.0, Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Green box: Duration of the treatment with the ISR-inhibiting drug. Panel (A): ISR inhibitors in the prevention of unconventional AD. Following the occurrence of the unconventional stressor, the neuronal ISR has been unconventionally elicited and the AβPP-independent C99 production pathway unconventionally activated. The neuronal ISR suppresses the AβPP proteolytic pathway and reduces the rate of accumulation of AβPP-derived iAβ. Concurrently, C99 produced independently of AβPP rapidly accumulates but is still below the T1 threshold. The drug reverses the ISR state, restores the activity of the AβPP proteolytic pathway, and the accumulation of the AβPP-derived iAβ resumes at its pre-ISR rate. It also disables the production of C99 in the AβPP-independent pathway, and its levels decline for the duration of the treatment. Panel (B): ISR inhibitors in the treatment of unconventional AD. At the time of the drug’s administration, C99 produced independently of AβPP has crossed the T2 threshold in a fraction of the neurons, and AD symptoms have manifested. The drug reverses the neuronal ISR state, enables the activity of the AβPP proteolytic pathway, and the accumulation of AβPP-derived iAβ resumes at the pre-ISR rate. It also disables the AβPP-independent C99 production pathway. The levels of C99 generated independently of AβPP are declining and the progression of the disease is stopped for the duration of the treatment.
![Ijms 26 04252 g039]()
Figure 40.
BACE1 and/or BACE2 activators in the prevention of conventional AD and in the prevention and treatment of AACD: the ACH2.0, Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing and elicitation of the ISR; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Orange box: Duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The initial state of the levels of AβPP-derived iAβ and C99 in the neurons of an individual. At the time of the drug’s administration these levels are below the T0 threshold. Panel (B): The evolution of the initial state in the untreated individual. AβPP-derived iAβ continues to accumulate. When it crosses the T0 threshold, AACD commences and morphs into AD upon the T1 crossing. At this point PKR and/or HRI kinases are activated, eIF2α is phosphorylated at its Ser51 residue, and the neuronal integrated stress response is elicited. Under the ISR conditions the AβPP proteolytic pathway is suppressed and the rate of accumulation of AβPP-derived iAβ is substantially reduced. Concurrently, the neuronal ISR enables the operation of the AβPP-independent C99 generation pathway. C99 produced independently of AβPP rapidly accumulates and drives the progression of AD pathology. When the T2 threshold is crossed in a sufficient fraction of the neurons, the disease reaches its end stage. Panel (C): The evolution of the initial state following the transient administration of the BACE1 and/or BACE2-activating drug. The treatment substantially depletes AβPP-derived iAβ and its accumulation resumes from low baseline and proceeds at the pre-treatment rate. Neither the T0 or T1 thresholds are reached nor AACD or conventional AD occurs within the remaining lifetime of the treated individual. Panel (D): The outcome of the transient treatment with the BACE1 and/or BACE2 activating drug administered to the AACD patient. At the time of the implementation of the treatment AβPP-derived iAβ has already crossed the T0 threshold and triggered the commencement of AACD. The transient administration of BACE1 and/or BACE2 activators depletes AβPP-derived iAβ well below the T0 threshold. When the T1 is reverse-crossed by AβPP-derived iAβ, AACD is cured. The de novo accumulation of AβPP-derived iAβ commences from a low baseline. It reaches neither T0 nor T1 thresholds within the remaining lifetime of the treated individual. Thus, a single transient iAβ depletion treatment via the activation of BACE1 and/or BACE2 prevents both the occurrence of conventional AD and recurrence of AACD.
Figure 40.
BACE1 and/or BACE2 activators in the prevention of conventional AD and in the prevention and treatment of AACD: the ACH2.0, Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T0: Threshold of cellular concentration of AβPP-derived iAβ triggering the neuronal damage, which manifests as AACD. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink box: The range of cellular concentrations of AβPP-derived iAβ concentrations between the T0 and the highest extent reached by AβPP-derived iAβ or the T1 threshold that support the progression of AACD. The condition commences with the crossing of the T0 threshold and morphs into AD with the T1 crossing and elicitation of the ISR; it can occur only if the extent of the T0 threshold is smaller than that of the T1. Orange box: Duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The initial state of the levels of AβPP-derived iAβ and C99 in the neurons of an individual. At the time of the drug’s administration these levels are below the T0 threshold. Panel (B): The evolution of the initial state in the untreated individual. AβPP-derived iAβ continues to accumulate. When it crosses the T0 threshold, AACD commences and morphs into AD upon the T1 crossing. At this point PKR and/or HRI kinases are activated, eIF2α is phosphorylated at its Ser51 residue, and the neuronal integrated stress response is elicited. Under the ISR conditions the AβPP proteolytic pathway is suppressed and the rate of accumulation of AβPP-derived iAβ is substantially reduced. Concurrently, the neuronal ISR enables the operation of the AβPP-independent C99 generation pathway. C99 produced independently of AβPP rapidly accumulates and drives the progression of AD pathology. When the T2 threshold is crossed in a sufficient fraction of the neurons, the disease reaches its end stage. Panel (C): The evolution of the initial state following the transient administration of the BACE1 and/or BACE2-activating drug. The treatment substantially depletes AβPP-derived iAβ and its accumulation resumes from low baseline and proceeds at the pre-treatment rate. Neither the T0 or T1 thresholds are reached nor AACD or conventional AD occurs within the remaining lifetime of the treated individual. Panel (D): The outcome of the transient treatment with the BACE1 and/or BACE2 activating drug administered to the AACD patient. At the time of the implementation of the treatment AβPP-derived iAβ has already crossed the T0 threshold and triggered the commencement of AACD. The transient administration of BACE1 and/or BACE2 activators depletes AβPP-derived iAβ well below the T0 threshold. When the T1 is reverse-crossed by AβPP-derived iAβ, AACD is cured. The de novo accumulation of AβPP-derived iAβ commences from a low baseline. It reaches neither T0 nor T1 thresholds within the remaining lifetime of the treated individual. Thus, a single transient iAβ depletion treatment via the activation of BACE1 and/or BACE2 prevents both the occurrence of conventional AD and recurrence of AACD.
![Ijms 26 04252 g040]()
Figure 41.
A single transient composite ISR inhibition/BACE activation therapy could prevent for life the recurrence of conventional AD: the ACH2.0 Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Orange box: Duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The initial state of the levels of AβPP-derived iAβ and C99, produced mainly in the AβPP-independent pathway, at the time of the administration of the composite BACE activation/ISR inhibition therapy. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons. PKR and/or HRI have been activated, eIF2α phosphorylated at its Ser51 residue, and the neuronal ISR elicited. As a result, the AβPP proteolytic pathway has been suppressed and the rate of accumulation of AβPP-derived iAβ substantially reduced. Concurrently, the production of C99 in the AβPP-independent pathway has been activated, and AD commenced. It rapidly accumulated, crossed the T2 threshold in a fraction of the neurons, and AD symptoms manifested. Panel (B): The evolution of the initial state in the untreated patient. The accumulation of C99 generated independently of AβPP continues, more neurons cross the T2 threshold, and the disease enters its end stage. Panel (C): The evolution of the initial state following the composite BACE activation/ISR inhibition therapy. The reversal of the neuronal ISR disables the AβPP-independent C99 generation pathway and ceases the influx of its C99 product. It also restores the production of BACE enzymes and ensures their availability. Activated BACE1 and/or BACE2 efficiently and substantially deplete both C99 and iAβ. When drugs are withdrawn, the progression of AD is arrested; the accumulation of AβPP-derived iAβ resumes from low baseline and proceeds at a slow pre-T1 crossing rate. It would not reach the T1 threshold and conventional AD would not recur in the treated individual. Thus, a single composite BACE activation/ISR inhibition treatment can prevent for life the recurrence of conventional AD.
Figure 41.
A single transient composite ISR inhibition/BACE activation therapy could prevent for life the recurrence of conventional AD: the ACH2.0 Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Orange box: Duration of the treatment with the BACE1 and/or BACE2-activating drug. Panel (A): The initial state of the levels of AβPP-derived iAβ and C99, produced mainly in the AβPP-independent pathway, at the time of the administration of the composite BACE activation/ISR inhibition therapy. AβPP-derived iAβ has crossed the T1 threshold in all affected neurons. PKR and/or HRI have been activated, eIF2α phosphorylated at its Ser51 residue, and the neuronal ISR elicited. As a result, the AβPP proteolytic pathway has been suppressed and the rate of accumulation of AβPP-derived iAβ substantially reduced. Concurrently, the production of C99 in the AβPP-independent pathway has been activated, and AD commenced. It rapidly accumulated, crossed the T2 threshold in a fraction of the neurons, and AD symptoms manifested. Panel (B): The evolution of the initial state in the untreated patient. The accumulation of C99 generated independently of AβPP continues, more neurons cross the T2 threshold, and the disease enters its end stage. Panel (C): The evolution of the initial state following the composite BACE activation/ISR inhibition therapy. The reversal of the neuronal ISR disables the AβPP-independent C99 generation pathway and ceases the influx of its C99 product. It also restores the production of BACE enzymes and ensures their availability. Activated BACE1 and/or BACE2 efficiently and substantially deplete both C99 and iAβ. When drugs are withdrawn, the progression of AD is arrested; the accumulation of AβPP-derived iAβ resumes from low baseline and proceeds at a slow pre-T1 crossing rate. It would not reach the T1 threshold and conventional AD would not recur in the treated individual. Thus, a single composite BACE activation/ISR inhibition treatment can prevent for life the recurrence of conventional AD.
![Ijms 26 04252 g041]()
Figure 42.
Transient composite ISR inhibition/BACE activation therapy in the prevention and treatment of unconventional AD: The ACH2.0 Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Orange box: Duration of the treatment with the BACE1 and/or BACE2-activating drug. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Panel (A): The anticipated outcome of the transient composite BACE activation/ISR inhibition therapy in the prevention of unconventional AD. At the time of the administration of the therapy the neuronal ISR has been unconventionally elicited at the levels of AβPP-derived iAβ below the T1 thresholds. Consequently, the AβPP proteolytic pathway and the accumulation of AβPP-derived iAβ have been suppressed, and the AβPP-independent C99 generation pathway unconventionally activated. C99 produced independently of AβPP has rapidly accumulated but is still below the T1 threshold; the disease does not commence. The ISR inhibition enables the production of BACE enzymes, disables the production of C99 in the AβPP-independent C99 pathway, and stops its influx. In this setting, activated BACE1 and/or BACE2 efficiently deplete C99 and iAβ. When drugs are withdrawn, the neuronal ISR is unconventionally re-elicited, and the AβPP-independent C99 generation pathway unconventionally reactivated. C99 produced independently of AβPP rapidly accumulates. When it crosses the T1 threshold the disease commences. Panel (B): Anticipated effects of the transient composite BACE activation/ISR inhibition therapy in the treatment of unconventional AD. At the time of the treatment the neuronal ISR has been unconventionally elicited at levels of AβPP-derived iAβ below the T1 threshold. The AβPP proteolytic pathway has been suppressed and the AβPP-independent C99 production pathway unconventionally activated. C99 generated independently of AβPP has rapidly accumulated and crossed the T1 threshold. The AβPP-independent C99 production pathway was rendered self-sustainable, and AD commenced. C99 has continued to accumulate, crossed the T2 threshold in a fraction of the neurons, and AD symptoms manifested. The composite treatment resulted in a substantial depletion of C99 and iAβ. However, upon the withdrawal of the drugs, the neuronal ISR is unconventionally re-elicited, the AβPP-independent C99 production pathway unconventionally reactivated; C99 rapidly accumulates, re-crosses the T1 threshold, and the disease recurs.
Figure 42.
Transient composite ISR inhibition/BACE activation therapy in the prevention and treatment of unconventional AD: The ACH2.0 Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Orange box: Duration of the treatment with the BACE1 and/or BACE2-activating drug. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Panel (A): The anticipated outcome of the transient composite BACE activation/ISR inhibition therapy in the prevention of unconventional AD. At the time of the administration of the therapy the neuronal ISR has been unconventionally elicited at the levels of AβPP-derived iAβ below the T1 thresholds. Consequently, the AβPP proteolytic pathway and the accumulation of AβPP-derived iAβ have been suppressed, and the AβPP-independent C99 generation pathway unconventionally activated. C99 produced independently of AβPP has rapidly accumulated but is still below the T1 threshold; the disease does not commence. The ISR inhibition enables the production of BACE enzymes, disables the production of C99 in the AβPP-independent C99 pathway, and stops its influx. In this setting, activated BACE1 and/or BACE2 efficiently deplete C99 and iAβ. When drugs are withdrawn, the neuronal ISR is unconventionally re-elicited, and the AβPP-independent C99 generation pathway unconventionally reactivated. C99 produced independently of AβPP rapidly accumulates. When it crosses the T1 threshold the disease commences. Panel (B): Anticipated effects of the transient composite BACE activation/ISR inhibition therapy in the treatment of unconventional AD. At the time of the treatment the neuronal ISR has been unconventionally elicited at levels of AβPP-derived iAβ below the T1 threshold. The AβPP proteolytic pathway has been suppressed and the AβPP-independent C99 production pathway unconventionally activated. C99 generated independently of AβPP has rapidly accumulated and crossed the T1 threshold. The AβPP-independent C99 production pathway was rendered self-sustainable, and AD commenced. C99 has continued to accumulate, crossed the T2 threshold in a fraction of the neurons, and AD symptoms manifested. The composite treatment resulted in a substantial depletion of C99 and iAβ. However, upon the withdrawal of the drugs, the neuronal ISR is unconventionally re-elicited, the AβPP-independent C99 production pathway unconventionally reactivated; C99 rapidly accumulates, re-crosses the T1 threshold, and the disease recurs.
![Ijms 26 04252 g042]()
Figure 43.
Recurrent transient composite BACE1 and/or BACE2 activation/ISR inhibition therapy in the prevention and treatment of unconventional AD: The ACH2.0 Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Brown boxes: The duration of the composite treatment with concurrently administered ISR inhibitors and BACE activators. Panel (A): Effects of the recurrent transient composite BACE activation/ISR inhibition therapy in the prevention of unconventional AD. At the time of the initial treatment the neuronal ISR has been unconventionally elicited. Consequently, the AβPP proteolytic pathway has been suppressed and the rate of accumulation of AβPP-derived iAβ substantially reduced. Concurrently, the production of C99 in the AβPP-independent pathway has been activated. It has rapidly accumulated but has not yet reached the T1 threshold. The inhibition of the neuronal ISR enables the production of BACE enzymes, disables the AβPP-independent C99 generation pathway, stops the influx of its C99 product, and empowers the efficient depletion of the latter by activated BACE1 and/or BACE2. When drugs are withdrawn, the neuronal ISR is unconventionally re-elicited, the AβPP-independent C99 generation pathway unconventionally reactivated, and C99 resumes its accumulation from a low baseline. Before it reaches the T1 threshold, at the time points determined by appropriate biomarkers, the treatment is repeated recurrently for the remaining portion of the lifetime; neither the T1 threshold would be crossed, nor AD would occur in the treated individual. Panel (B): Effects of the recurrent transient composite BACE activation/ISR inhibition therapy in the treatment of unconventional AD. At the time of the initial treatment, the neuronal ISR has been unconventionally elicited, the AβPP proteolytic pathway suppressed, AβPP-independent C99 generation pathway unconventionally activated, the C99 product of the latter has crossed the T1 threshold, the pathway was rendered self-sustainable, and the disease commenced. In a fraction of the neurons C99 has crossed the T2 threshold, and AD symptoms have manifested. The concurrent administration of ISR-inhibiting and BACE-activating drugs enable the production of BACE enzymes, cease the operation of the AβPP-independent C99 generation pathway, and block the influx of its C99 product. In this setting, activated BACE1 and/or BACE2 efficiently deplete C99 and iAβ. Upon the completion of the treatment, the neuronal ISR is unconventionally re-elicited, AβPP-independent C99 production pathway unconventionally reactivated, and the accumulation of its C99 product resumes from a low baseline. It crosses the T1 threshold, but before it reaches AD-pathology-causing levels, at time points defined by suitable biomarkers, the transient composite therapy is re-implemented recurrently for the remaining lifetime. The AD-pathology-causing range of C99 concentrations would not be reached, and the disease would not recur in the treated patient.
Figure 43.
Recurrent transient composite BACE1 and/or BACE2 activation/ISR inhibition therapy in the prevention and treatment of unconventional AD: The ACH2.0 Version Three perspective. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Brown boxes: The duration of the composite treatment with concurrently administered ISR inhibitors and BACE activators. Panel (A): Effects of the recurrent transient composite BACE activation/ISR inhibition therapy in the prevention of unconventional AD. At the time of the initial treatment the neuronal ISR has been unconventionally elicited. Consequently, the AβPP proteolytic pathway has been suppressed and the rate of accumulation of AβPP-derived iAβ substantially reduced. Concurrently, the production of C99 in the AβPP-independent pathway has been activated. It has rapidly accumulated but has not yet reached the T1 threshold. The inhibition of the neuronal ISR enables the production of BACE enzymes, disables the AβPP-independent C99 generation pathway, stops the influx of its C99 product, and empowers the efficient depletion of the latter by activated BACE1 and/or BACE2. When drugs are withdrawn, the neuronal ISR is unconventionally re-elicited, the AβPP-independent C99 generation pathway unconventionally reactivated, and C99 resumes its accumulation from a low baseline. Before it reaches the T1 threshold, at the time points determined by appropriate biomarkers, the treatment is repeated recurrently for the remaining portion of the lifetime; neither the T1 threshold would be crossed, nor AD would occur in the treated individual. Panel (B): Effects of the recurrent transient composite BACE activation/ISR inhibition therapy in the treatment of unconventional AD. At the time of the initial treatment, the neuronal ISR has been unconventionally elicited, the AβPP proteolytic pathway suppressed, AβPP-independent C99 generation pathway unconventionally activated, the C99 product of the latter has crossed the T1 threshold, the pathway was rendered self-sustainable, and the disease commenced. In a fraction of the neurons C99 has crossed the T2 threshold, and AD symptoms have manifested. The concurrent administration of ISR-inhibiting and BACE-activating drugs enable the production of BACE enzymes, cease the operation of the AβPP-independent C99 generation pathway, and block the influx of its C99 product. In this setting, activated BACE1 and/or BACE2 efficiently deplete C99 and iAβ. Upon the completion of the treatment, the neuronal ISR is unconventionally re-elicited, AβPP-independent C99 production pathway unconventionally reactivated, and the accumulation of its C99 product resumes from a low baseline. It crosses the T1 threshold, but before it reaches AD-pathology-causing levels, at time points defined by suitable biomarkers, the transient composite therapy is re-implemented recurrently for the remaining lifetime. The AD-pathology-causing range of C99 concentrations would not be reached, and the disease would not recur in the treated patient.
![Ijms 26 04252 g043]()
Figure 44.
Sustained removal of unconventional stressors converts unconventional AD into conventional one, preventable and treatable by a single transient administration of the composite BACE activation/ISR inhibition therapy. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Orange box: Duration of the treatment with the BACE1 and/or BACE2-activating drug. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Panel (A): The anticipated outcome of the transient composite BACE activation/ISR inhibition therapy administered in concert with the sustained removal of unconventional stressors in the prevention of unconventional AD. At the time of the treatment the neuronal ISR has been unconventionally elicited, the AβPP proteolytic pathway has been suppressed and the rate of accumulation of AβPP-derived iAβ reduced. Concurrently, the AβPP-independent C99 production pathway has been unconventionally activated; its C99 product has accumulated but is still below the T1 threshold. The transient administration of the composite BACE activation/ISR inhibition therapy is carried out in concert with the sustained removal of unconventional stressors (or their depletion below levels below the neuronal ISR-eliciting levels). When the transient BACE activation/ISR inhibition treatment is completed, both C99 and AβPP-derived iAβ are substantially depleted and so are unconventional stressors. The neuronal ISR is not re-elicited, and the AβPP-independent C99 production pathway remains inoperative. The accumulation of AβPP-derived iAβ resumes from a low baseline and proceed at a slow rate. It does not reach the T1 threshold and AD does not occur within the lifetime of the treated individual. Panel (B): The anticipated outcome of the transient composite BACE activation/ISR inhibition therapy administered in concert with the sustained removal of unconventional stressors in the treatment of unconventional AD. At the time of the treatment C99 produced in the unconventionally activated AβPP-independent pathway has crossed the T1 threshold, the pathway became self-sustainable, and AD commenced. C99 has continued to accumulate, crossed the T2 threshold in a fraction of the neurons, and AD symptoms manifested. The transient administration of the composite BACE activation/ISR inhibition therapy is carried out in concert with the sustained removal of unconventional stressors. When BACE-activating and ISR-inhibiting drugs are withdrawn, C99 and AβPP-derived iAβ are substantially depleted. The progression of the disease ceases, and the AβPP-independent C99 generation pathway remains inoperative. The de novo accumulation of AβPP-derived iAβ commences from low baseline and proceeds at a slow rate. It would not reach the T1 threshold, and the disease would not recur within the lifetime of the treated patient. Thus, the sustained removal/depletion of unconventional stressors capable of eliciting the neuronal ISR effectively converts unconventional AD into conventional one, preventable and treatable by a single transient composite ISR inhibition/BACE activation treatment.
Figure 44.
Sustained removal of unconventional stressors converts unconventional AD into conventional one, preventable and treatable by a single transient administration of the composite BACE activation/ISR inhibition therapy. Blue lines: Intraneuronal Aβ, iAβ. Red lines: The C99 fragment of AβPP. T1: Threshold of cellular concentration of iAβ triggering the activation of PKR and/or HRI kinases, phosphorylation of eIF2α, the elicitation of the neuronal ISR, the initiation of the AβPP-independent C99 production pathway, and the commencement of AD; its extent is the only variable in the present figure. T2: Cellular concentration of iAβ triggering neuronal death via apoptosis or necroptosis. Red box: Apoptotic zone, defined as a range of cellular concentrations of iAβ within which cells neuronal cells committed apoptosis or necroptosis or are dead. Green box: Duration of the treatment with the ISR-inhibiting drug. Orange box: Duration of the treatment with the BACE1 and/or BACE2-activating drug. Pink boxes: The duration of the presence of the unconventional stressor capable of activating one or more of eIF2α kinases and triggering the elicitation of the neuronal ISR. Panel (A): The anticipated outcome of the transient composite BACE activation/ISR inhibition therapy administered in concert with the sustained removal of unconventional stressors in the prevention of unconventional AD. At the time of the treatment the neuronal ISR has been unconventionally elicited, the AβPP proteolytic pathway has been suppressed and the rate of accumulation of AβPP-derived iAβ reduced. Concurrently, the AβPP-independent C99 production pathway has been unconventionally activated; its C99 product has accumulated but is still below the T1 threshold. The transient administration of the composite BACE activation/ISR inhibition therapy is carried out in concert with the sustained removal of unconventional stressors (or their depletion below levels below the neuronal ISR-eliciting levels). When the transient BACE activation/ISR inhibition treatment is completed, both C99 and AβPP-derived iAβ are substantially depleted and so are unconventional stressors. The neuronal ISR is not re-elicited, and the AβPP-independent C99 production pathway remains inoperative. The accumulation of AβPP-derived iAβ resumes from a low baseline and proceed at a slow rate. It does not reach the T1 threshold and AD does not occur within the lifetime of the treated individual. Panel (B): The anticipated outcome of the transient composite BACE activation/ISR inhibition therapy administered in concert with the sustained removal of unconventional stressors in the treatment of unconventional AD. At the time of the treatment C99 produced in the unconventionally activated AβPP-independent pathway has crossed the T1 threshold, the pathway became self-sustainable, and AD commenced. C99 has continued to accumulate, crossed the T2 threshold in a fraction of the neurons, and AD symptoms manifested. The transient administration of the composite BACE activation/ISR inhibition therapy is carried out in concert with the sustained removal of unconventional stressors. When BACE-activating and ISR-inhibiting drugs are withdrawn, C99 and AβPP-derived iAβ are substantially depleted. The progression of the disease ceases, and the AβPP-independent C99 generation pathway remains inoperative. The de novo accumulation of AβPP-derived iAβ commences from low baseline and proceeds at a slow rate. It would not reach the T1 threshold, and the disease would not recur within the lifetime of the treated patient. Thus, the sustained removal/depletion of unconventional stressors capable of eliciting the neuronal ISR effectively converts unconventional AD into conventional one, preventable and treatable by a single transient composite ISR inhibition/BACE activation treatment.
![Ijms 26 04252 g044]()
Figure 45.
Mammalian RNA-dependent mRNA amplification occurs in two phases: Principal stages. Single lines: Antisense RNA. Boxed lines: Sense RNA. Blue boxed lines: Single-stranded RNA separated from its complementary RNA strand by helicase. Yellow circles: Helicase complex containing helicase strand-separating activity, nucleotide modifying activity and RNA cleaving activity. Red arrows: positions of the cleavage of the intermediate generating RNA end products of the amplification process. RdRp: RNA-dependent RNA polymerase. AUG: Translation initiation codon. TCE: 3′ Terminal Complementary Element of the antisense RNA strands. ICE: Internal Complementary Element of the antisense RNA strands. Top panel: Conventionally transcribed mRNA molecule referred to as progenitor mRNA. Bottom panel: Stages of the chimeric pathway of the RNA-dependent mRNA amplification; the ICE is located within a portion of antisense RNA corresponding to the 5′ UTR of the progenitor mRNA molecule. (1): The progenitor mRNA is transcribed by RdRp into antisense RNA. (2): Complementary RNA strands are separated by the helicase activity. Helicase complex mounts the 3′ poly(A) segment and moves along the sense RNA strand modifying, on average, every fifth nucleotide. (3): Folding of the antisense RNA into self-priming configuration is guided by the interaction of its TCE and ICE elements. (4): RdRp extends the 3′ terminus of self-primed antisense RNA. This produces a hairpin-like molecule containing both sense and antisense RNA components, referred to as the chimeric RNA intermediate. (5): Complementary strands of the chimeric RNA intermediate are separated by the helicase activity. Nucleotide modifications introduced during the separation prevent the re-annealing of the sense and antisense RNA strands. (6): When the helicase reaches the single-stranded portion of the chimeric intermediate (either the 5′ end of the TCE or a TCE/ICE mismatch) it cleaves the RNA molecule; this cleavage is coupled with polyadenylation of the 3′ terminus of the truncated antisense RNA molecule. (7): End products of RNA-dependent mRNA amplification. Antisense RNA is truncated and polyadenylated at its 3′ end and sense portion of the chimeric RNA is truncated at the 5′ end and acquires the cleaved-off antisense RNA fragment. The polyadenylation of the antisense RNA strand denotes the commencement of the second phase of mRNA amplification. (8): 3′poly(A)-containing antisense RNA is the initial template (progenitor) in the intracellular PCR (iPCR) amplification process; it is transcribed into the sense RNA strand, initiating at the 3′poly (A), by RdRp. (9): The helicase complex separates RNA strands. (10): End products of the first iPCR cycle. Each contains 3′poly (A) and 5′poly(U) and each can serve as a PCR template in the RdRp-driven process.
Figure 45.
Mammalian RNA-dependent mRNA amplification occurs in two phases: Principal stages. Single lines: Antisense RNA. Boxed lines: Sense RNA. Blue boxed lines: Single-stranded RNA separated from its complementary RNA strand by helicase. Yellow circles: Helicase complex containing helicase strand-separating activity, nucleotide modifying activity and RNA cleaving activity. Red arrows: positions of the cleavage of the intermediate generating RNA end products of the amplification process. RdRp: RNA-dependent RNA polymerase. AUG: Translation initiation codon. TCE: 3′ Terminal Complementary Element of the antisense RNA strands. ICE: Internal Complementary Element of the antisense RNA strands. Top panel: Conventionally transcribed mRNA molecule referred to as progenitor mRNA. Bottom panel: Stages of the chimeric pathway of the RNA-dependent mRNA amplification; the ICE is located within a portion of antisense RNA corresponding to the 5′ UTR of the progenitor mRNA molecule. (1): The progenitor mRNA is transcribed by RdRp into antisense RNA. (2): Complementary RNA strands are separated by the helicase activity. Helicase complex mounts the 3′ poly(A) segment and moves along the sense RNA strand modifying, on average, every fifth nucleotide. (3): Folding of the antisense RNA into self-priming configuration is guided by the interaction of its TCE and ICE elements. (4): RdRp extends the 3′ terminus of self-primed antisense RNA. This produces a hairpin-like molecule containing both sense and antisense RNA components, referred to as the chimeric RNA intermediate. (5): Complementary strands of the chimeric RNA intermediate are separated by the helicase activity. Nucleotide modifications introduced during the separation prevent the re-annealing of the sense and antisense RNA strands. (6): When the helicase reaches the single-stranded portion of the chimeric intermediate (either the 5′ end of the TCE or a TCE/ICE mismatch) it cleaves the RNA molecule; this cleavage is coupled with polyadenylation of the 3′ terminus of the truncated antisense RNA molecule. (7): End products of RNA-dependent mRNA amplification. Antisense RNA is truncated and polyadenylated at its 3′ end and sense portion of the chimeric RNA is truncated at the 5′ end and acquires the cleaved-off antisense RNA fragment. The polyadenylation of the antisense RNA strand denotes the commencement of the second phase of mRNA amplification. (8): 3′poly(A)-containing antisense RNA is the initial template (progenitor) in the intracellular PCR (iPCR) amplification process; it is transcribed into the sense RNA strand, initiating at the 3′poly (A), by RdRp. (9): The helicase complex separates RNA strands. (10): End products of the first iPCR cycle. Each contains 3′poly (A) and 5′poly(U) and each can serve as a PCR template in the RdRp-driven process.
![Ijms 26 04252 g045]()
Figure 46.
Folding of the antisense RNA molecules corresponding to human AβPP mRNAs initiated at the known transcription start sites. TSS: Transcription start site. (-)150, (-)149, (-)146, (-)144, (-)143: Known transcription start sites of human AβPP mRNA. Distances are given in numbers of nucleotides upstream from the translation-initiating AUG codon. Asterisk: The nucleotide of the antisense RNA molecule corresponding to the transcription-initiating nucleotide of human AβPP mRNA. Highlighted in blue: “C”, which is transcribed from the 5′-terminal cap “G” of AβPP mRNA. Only the antisense RNA strand originating from human AβPP mRNA transcribed from the TSS (-)149 is capable of forming self-primed structure without the 3′-overhang.
Figure 46.
Folding of the antisense RNA molecules corresponding to human AβPP mRNAs initiated at the known transcription start sites. TSS: Transcription start site. (-)150, (-)149, (-)146, (-)144, (-)143: Known transcription start sites of human AβPP mRNA. Distances are given in numbers of nucleotides upstream from the translation-initiating AUG codon. Asterisk: The nucleotide of the antisense RNA molecule corresponding to the transcription-initiating nucleotide of human AβPP mRNA. Highlighted in blue: “C”, which is transcribed from the 5′-terminal cap “G” of AβPP mRNA. Only the antisense RNA strand originating from human AβPP mRNA transcribed from the TSS (-)149 is capable of forming self-primed structure without the 3′-overhang.
Figure 47.
Current understanding of AD: The neuronal ISR suppresses the AβPP proteolytic pathway but enables the AβPP-independent generation of C99, which drives the disease; AβPP is produced in the ISR-compatible process but not processed; Tau is translated from IRES in the 5′UTR of its mRNA. eIF2α: eukaryotic translation initiation factor 2α. PKR and HRI: kinases that phosphorylate eIF2α at its Ser51 residue. PACT: PKR activator. TNFα: tumor necrosis factor α. OMA1: mitochondrial protease activated during mitochondrial dysfunction. DELE1: substrate of OMA1 protease. Cleavage of DELE1 leads to the activation of HRI. Phosphorylation of eIF2α elicits the integrated stress response, which, in turn, enables the generation of components essential for the activity of the AβPP-independent pathway of C99 and iAβ production. iAβ: intraneuronal Aβ. AβPP-derived iAβ accumulates physiologically via the importation of extracellular Aβ and the retention of iAβ produced by gamma-cleavages on intracellular membranes in an exceedingly slow process. In most individuals it does not reach the PKR- and/or HRI-activating, the neuronal ISR-eliciting threshold within their lifetimes and no conventional AD occurs. When this threshold is crossed, the neuronal ISR is elicited and the disease ensues. In unconventional AD, stressors that are distinct from AβPP-derived iAβ and capable of activating one or more of eIF2α kinases trigger the elicitation of the neuronal ISR. Under the ISR conditions the AβPP proteolytic pathway is suppressed; this happens within the framework of the ISR-mediated inhibition of the global cellular protein synthesis and includes AβPP, BACE enzymes, components of the gamma-secretase complex and, consequently, Aβ and iAβ. Concurrently, the neuronal ISR provides the components essential for, and thus enables, the operation of the AβPP-independent C99 generation pathway, and it is this C99, which drives AD pathology. Simultaneously, in the second phase of the same mechanism that underlies AβPP-independent generation of C99, AβPP is produced in the ISR-compatible process but is not processed further. In general, in the AD-affected neurons, any new protein synthesis has to occur in an ISR-compatible manner. In cases of C99 and AβPP this is achieved, apparently, via the nucleotide modifications of their mRNAs. Translation of tau protein, on the other hand, persists under the ISR conditions because its mRNA contains an internal ribosomal entry site in the 5′UTR.
Figure 47.
Current understanding of AD: The neuronal ISR suppresses the AβPP proteolytic pathway but enables the AβPP-independent generation of C99, which drives the disease; AβPP is produced in the ISR-compatible process but not processed; Tau is translated from IRES in the 5′UTR of its mRNA. eIF2α: eukaryotic translation initiation factor 2α. PKR and HRI: kinases that phosphorylate eIF2α at its Ser51 residue. PACT: PKR activator. TNFα: tumor necrosis factor α. OMA1: mitochondrial protease activated during mitochondrial dysfunction. DELE1: substrate of OMA1 protease. Cleavage of DELE1 leads to the activation of HRI. Phosphorylation of eIF2α elicits the integrated stress response, which, in turn, enables the generation of components essential for the activity of the AβPP-independent pathway of C99 and iAβ production. iAβ: intraneuronal Aβ. AβPP-derived iAβ accumulates physiologically via the importation of extracellular Aβ and the retention of iAβ produced by gamma-cleavages on intracellular membranes in an exceedingly slow process. In most individuals it does not reach the PKR- and/or HRI-activating, the neuronal ISR-eliciting threshold within their lifetimes and no conventional AD occurs. When this threshold is crossed, the neuronal ISR is elicited and the disease ensues. In unconventional AD, stressors that are distinct from AβPP-derived iAβ and capable of activating one or more of eIF2α kinases trigger the elicitation of the neuronal ISR. Under the ISR conditions the AβPP proteolytic pathway is suppressed; this happens within the framework of the ISR-mediated inhibition of the global cellular protein synthesis and includes AβPP, BACE enzymes, components of the gamma-secretase complex and, consequently, Aβ and iAβ. Concurrently, the neuronal ISR provides the components essential for, and thus enables, the operation of the AβPP-independent C99 generation pathway, and it is this C99, which drives AD pathology. Simultaneously, in the second phase of the same mechanism that underlies AβPP-independent generation of C99, AβPP is produced in the ISR-compatible process but is not processed further. In general, in the AD-affected neurons, any new protein synthesis has to occur in an ISR-compatible manner. In cases of C99 and AβPP this is achieved, apparently, via the nucleotide modifications of their mRNAs. Translation of tau protein, on the other hand, persists under the ISR conditions because its mRNA contains an internal ribosomal entry site in the 5′UTR.
![Ijms 26 04252 g047]()


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}