
GMP-like and MLP-like Subpopulations of Hematopoietic Stem and Progenitor Cells Harboring Mutated EZH2 and TP53 at Diagnosis Promote Acute Myeloid Leukemia Relapse: Data of Combined Molecular, Functional, and Genomic Single-Stem-Cell Analyses

 , , , and
, , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
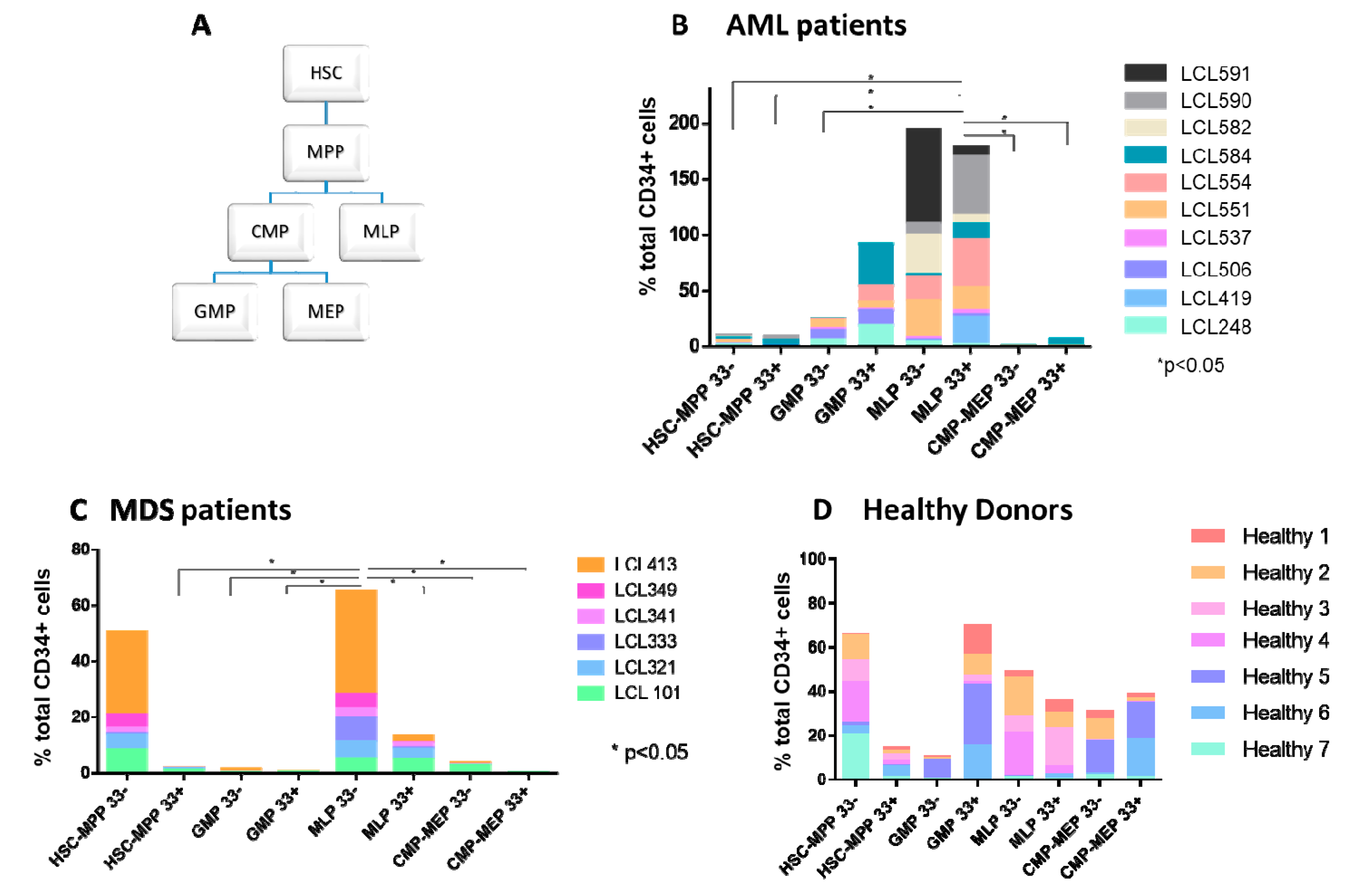
2.1. Abnormal Distribution of HSPC Subpopulations in Samples Derived from MDS and AML Patients
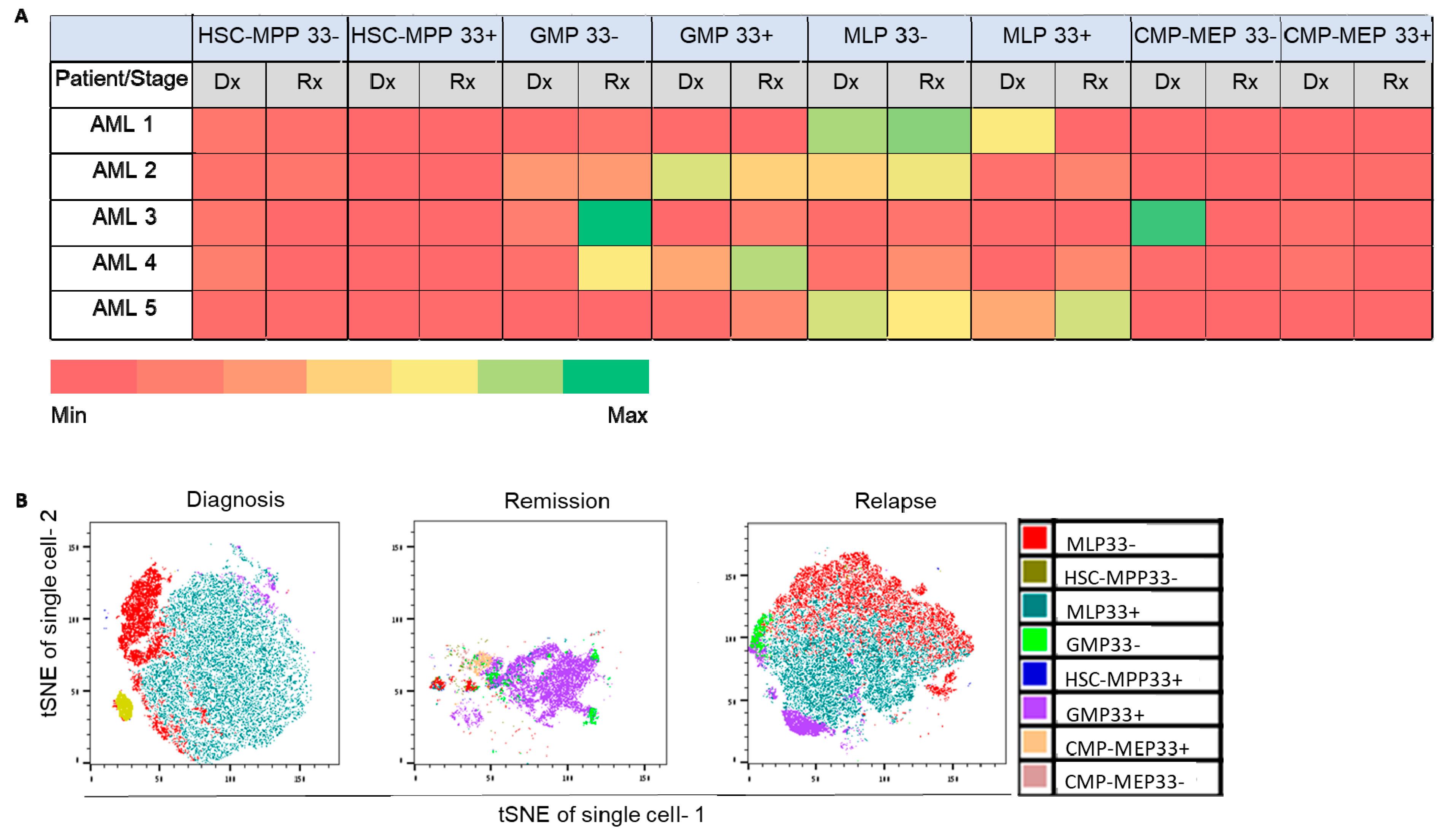
2.2. Involvement of Specific HSPC Subpopulations in Ex Vivo Chemo-Resistance and AML Recurrence
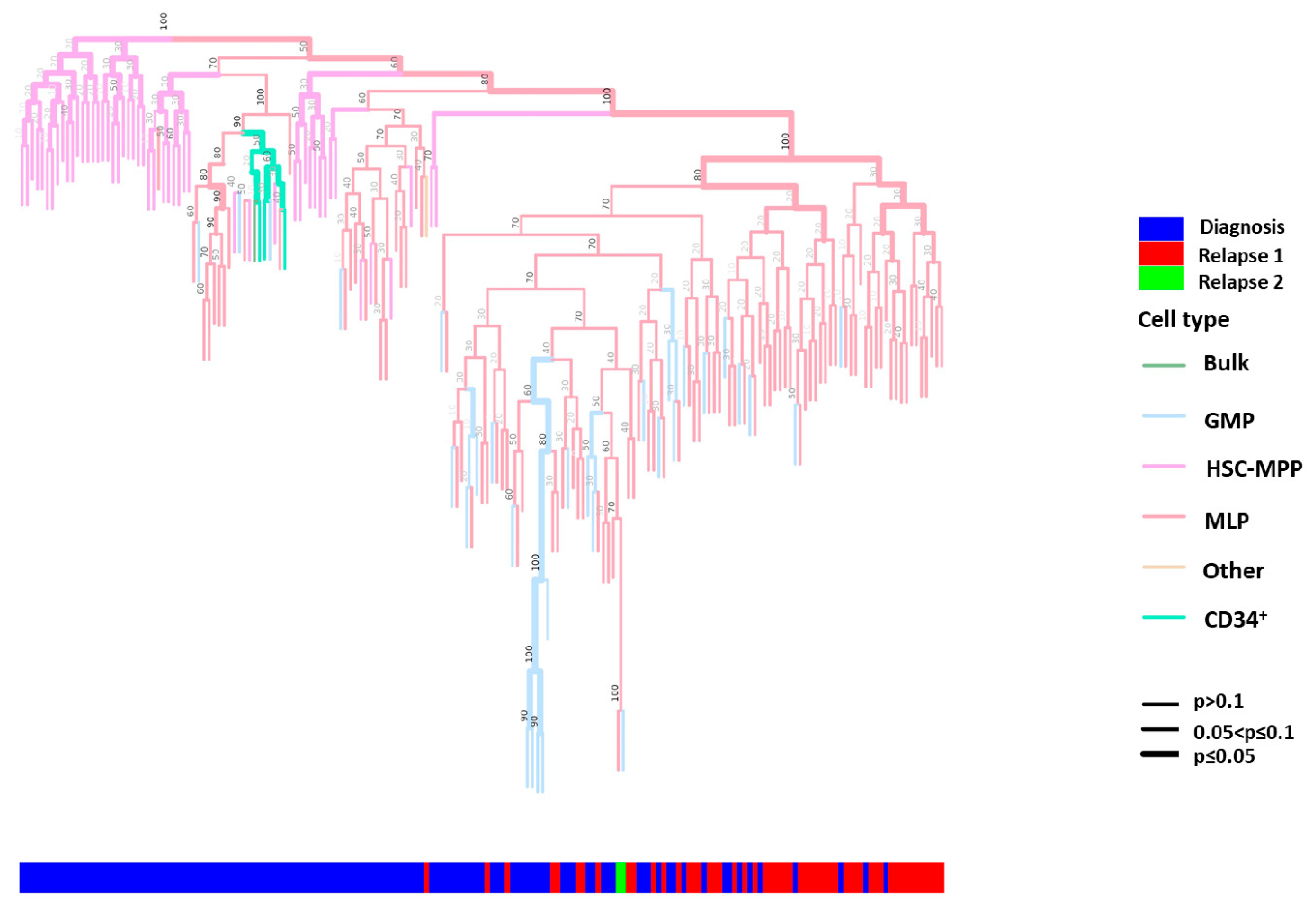
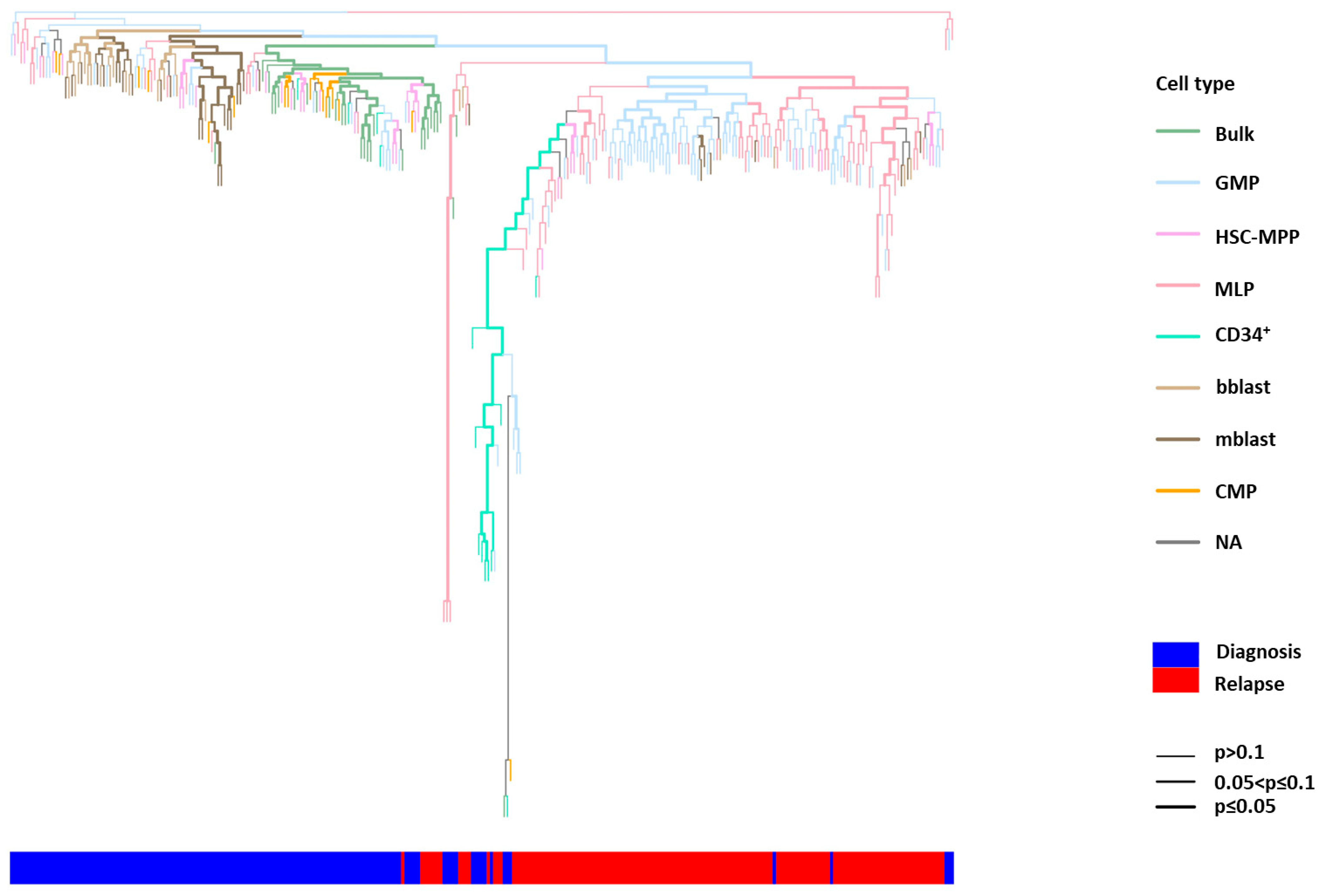
2.3. Phylogenetic Analysis of Single HSPCs and HSPC-Derived Subclones
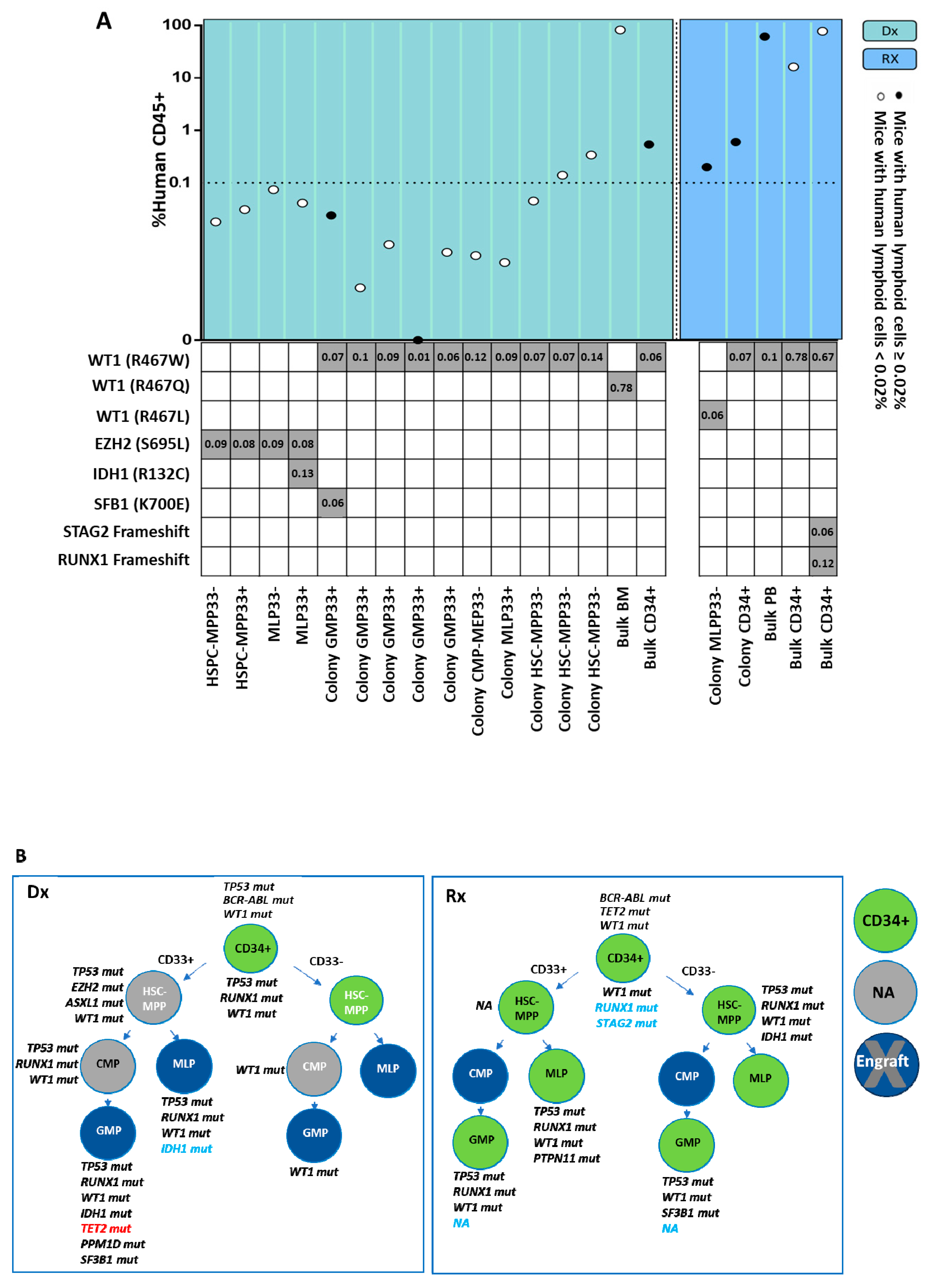
2.4. Leukemogenic Potential of Various HSPC Subpopulations In Vivo
2.5. Mutational Profile Analysis of Single-HSPC-Derived Subclones Demonstrates the Presence of a Relapse-Associated Clone at Diagnosis
3. Discussion
4. Materials and Methods
4.1. Human Specimens
4.2. HSPC Isolation and Culture
4.3. Generation of Single-Cell-Derived Subclones from Sorted HSPCs
4.4. In Vivo Leukemogenic Potential Assays
4.5. Lineage Tree Reconstruction
4.6. MIP Analysis for Mutational Profile Determination
4.7. Targeted MIP Panel Sequencing Variant Calling
4.8. Chimerism Analysis
4.9. Ex Vivo Chemo-Resistance Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Dohner, K.; Thiede, C.; Jahn, N.; Panina, E.; Gambietz, A.; Larson, R.A.; Prior, T.W.; Marcucci, G.; Jones, D.; Krauter, J.; et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood 2020, 135, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Fennell, K.A.; Vassiliadis, D.; Lam, E.Y.N.; Martelotto, L.G.; Balic, J.J.; Hollizeck, S.; Weber, T.S.; Semple, T.; Wang, Q.; Miles, D.C.; et al. Non-genetic determinants of malignant clonal fitness at single-cell resolution. Nature 2022, 601, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef]
- Schwede, M.; Jahn, K.; Kuipers, J.; Miles, L.A.; Bowman, R.L.; Robinson, T.; Furudate, K.; Uryu, H.; Tanaka, T.; Sasaki, Y.; et al. Mutation order in acute myeloid leukemia identifies uncommon patterns of evolution and illuminates phenotypic heterogeneity. Leukemia 2024, 38, 1501–1510. [Google Scholar] [CrossRef]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef]
- Karantanos, T.; Jones, R.J. Acute Myeloid Leukemia Stem Cell Heterogeneity and Its Clinical Relevance. Adv. Exp. Med. Biol. 2019, 1139, 153–169. [Google Scholar] [CrossRef]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Chopra, M.; Bohlander, S.K. The cell of origin and the leukemia stem cell in acute myeloid leukemia. Genes Chromosomes Cancer 2019, 58, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, R.; Jha, D.K.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef]
- Notta, F.; Doulatov, S.; Laurenti, E.; Poeppl, A.; Jurisica, I.; Dick, J.E. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 2011, 333, 218–221. [Google Scholar] [CrossRef]
- Vosberg, S.; Greif, P.A. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer 2019, 58, 839–849. [Google Scholar] [CrossRef]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, I.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussig, D.; Greaves, M. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia 2019, 33, 1113–1123. [Google Scholar] [CrossRef]
- Hughes, A.E.; Magrini, V.; Demeter, R.; Miller, C.A.; Fulton, R.; Fulton, L.L.; Eades, W.C.; Elliott, K.; Heath, S.; Westervelt, P.; et al. Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing. PLoS Genet. 2014, 10, e1004462. [Google Scholar] [CrossRef]
- Paguirigan, A.L.; Smith, J.; Meshinchi, S.; Carroll, M.; Maley, C.; Radich, J.P. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci. Transl. Med. 2015, 7, 281re2. [Google Scholar] [CrossRef]
- Shouval, R.; Shlush, L.I.; Yehudai-Resheff, S.; Ali, S.; Pery, N.; Shapiro, E.; Tzukerman, M.; Rowe, J.M.; Zuckerman, T. Single cell analysis exposes intratumor heterogeneity and suggests that FLT3-ITD is a late event in leukemogenesis. Exp. Hematol. 2014, 42, 457–463. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Dimitriou, M.; Mortera-Blanco, T.; Tobiasson, M.; Mazzi, S.; Lehander, M.; Hogstrand, K.; Karimi, M.; Walldin, G.; Jansson, M.; Vonlanthen, S.; et al. Identification and surveillance of rare relapse-initiating stem cells during complete remission after transplantation. Blood 2024, 143, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, A.; Klein, T.; Born, G.; Hilpert, M.; Scherberich, A.; Lengerke, C.; Skoda, R.C.; Bourgine, P.E.; Martin, I. Culturing patient-derived malignant hematopoietic stem cells in engineered and fully humanized 3D niches. Proc. Natl. Acad. Sci. USA 2021, 118, e2114227118. [Google Scholar] [CrossRef]
- Woo, J.; Howard, N.P.; Storer, B.E.; Fang, M.; Yeung, C.C.; Scott, B.L.; Deeg, H.J. Mutational analysis in serial marrow samples during azacitidine treatment in patients with post-transplant relapse of acute myeloid leukemia or myelodysplastic syndromes. Haematologica 2017, 102, e216–e218. [Google Scholar] [CrossRef]
- van Spronsen, M.F.; Hanekamp, D.; Westers, T.M.; van Gils, N.; Vermue, E.; Rutten, A.; Jansen, J.H.; Lissenberg-Witte, B.I.; Smit, L.; Schuurhuis, G.J.; et al. Immunophenotypic aberrant hematopoietic stem cells in myelodysplastic syndromes: A biomarker for leukemic progression. Leukemia 2023, 37, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Lahnemann, D.; Koster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Ebinger, S.; Ozdemir, E.Z.; Ziegenhain, C.; Tiedt, S.; Castro Alves, C.; Grunert, M.; Dworzak, M.; Lutz, C.; Turati, V.A.; Enver, T.; et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 849–862. [Google Scholar] [CrossRef]
- Behbehani, G.K.; Samusik, N.; Bjornson, Z.B.; Fantl, W.J.; Medeiros, B.C.; Nolan, G.P. Mass Cytometric Functional Profiling of Acute Myeloid Leukemia Defines Cell-Cycle and Immunophenotypic Properties That Correlate with Known Responses to Therapy. Cancer Discov. 2015, 5, 988–1003. [Google Scholar] [CrossRef]
- Malani, D.; Kumar, A.; Bruck, O.; Kontro, M.; Yadav, B.; Hellesoy, M.; Kuusanmaki, H.; Dufva, O.; Kankainen, M.; Eldfors, S.; et al. Implementing a Functional Precision Medicine Tumor Board for Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 388–401. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Chapal-Ilani, N.; Adar, R.; Pery, N.; Maruvka, Y.; Spiro, A.; Shouval, R.; Rowe, J.M.; Tzukerman, M.; Bercovich, D.; et al. Cell lineage analysis of acute leukemia relapse uncovers the role of replication-rate heterogeneity and microsatellite instability. Blood 2012, 120, 603–612. [Google Scholar] [CrossRef]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Rollig, C.; Kramer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brummendorf, T.H.; Naumann, R.; et al. EZH2 mutations and impact on clinical outcome: An analysis in 1,604 patients with newly diagnosed acute myeloid leukemia. Haematologica 2020, 105, e228–e231. [Google Scholar] [CrossRef] [PubMed]
- Gollner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Yan, B.; Claxton, D.; Huang, S.; Qiu, Y. AML chemoresistance: The role of mutant TP53 subclonal expansion and therapy strategy. Exp. Hematol. 2020, 87, 13–19. [Google Scholar] [CrossRef]
- Tao, L.; Raz, O.; Marx, Z.; Ghosh, M.S.; Huber, S.; Greindl-Junghans, J.; Biezuner, T.; Amir, S.; Milo, L.; Adar, R.; et al. Retrospective cell lineage reconstruction in humans by using short tandem repeats. Cell Rep. Methods 2021, 1, 100054. [Google Scholar] [CrossRef] [PubMed]
- Raz, O.; Biezuner, T.; Spiro, A.; Amir, S.; Milo, L.; Titelman, A.; Onn, A.; Chapal-Ilani, N.; Tao, L.; Marx, T.; et al. Short tandem repeat stutter model inferred from direct measurement of in vitro stutter noise. Nucleic Acids Res. 2019, 47, 2436–2445. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Lemoine, F.; Domelevo Entfellner, J.B.; Wilkinson, E.; Correia, D.; Davila Felipe, M.; De Oliveira, T.; Gascuel, O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452–456. [Google Scholar] [CrossRef]
- Neumeier, Y.; Raz, O.; Tao, L.; Marx, Z.; Shapiro, E. Tracking Somatic Mutations for Lineage Reconstruction. Methods Mol. Biol. 2025, 2886, 23–45. [Google Scholar] [CrossRef]
- Biezuner, T.; Brilon, Y.; Arye, A.B.; Oron, B.; Kadam, A.; Danin, A.; Furer, N.; Minden, M.D.; Hwan Kim, D.D.; Shapira, S.; et al. An improved molecular inversion probe based targeted sequencing approach for low variant allele frequency. NAR Genom. Bioinform. 2022, 4, lqab125. [Google Scholar] [CrossRef]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge-Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar]
- Li, W.; Freudenberg, J. Mappability and read length. Front. Genet. 2014, 5, 381. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Consortium, W.G.S.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahar Gabay, T.; Stolero, N.; Rabhun, N.; Sabah, R.; Raz, O.; Neumeier, Y.; Marx, Z.; Tao, L.; Biezuner, T.; Amir, S.; et al. GMP-like and MLP-like Subpopulations of Hematopoietic Stem and Progenitor Cells Harboring Mutated EZH2 and TP53 at Diagnosis Promote Acute Myeloid Leukemia Relapse: Data of Combined Molecular, Functional, and Genomic Single-Stem-Cell Analyses. Int. J. Mol. Sci. 2025, 26, 4224. https://doi.org/10.3390/ijms26094224
Shahar Gabay T, Stolero N, Rabhun N, Sabah R, Raz O, Neumeier Y, Marx Z, Tao L, Biezuner T, Amir S, et al. GMP-like and MLP-like Subpopulations of Hematopoietic Stem and Progenitor Cells Harboring Mutated EZH2 and TP53 at Diagnosis Promote Acute Myeloid Leukemia Relapse: Data of Combined Molecular, Functional, and Genomic Single-Stem-Cell Analyses. International Journal of Molecular Sciences. 2025; 26(9):4224. https://doi.org/10.3390/ijms26094224
Chicago/Turabian StyleShahar Gabay, Tal, Nofar Stolero, Niv Rabhun, Rawan Sabah, Ofir Raz, Yaara Neumeier, Zipora Marx, Liming Tao, Tamir Biezuner, Shiran Amir, and et al. 2025. "GMP-like and MLP-like Subpopulations of Hematopoietic Stem and Progenitor Cells Harboring Mutated EZH2 and TP53 at Diagnosis Promote Acute Myeloid Leukemia Relapse: Data of Combined Molecular, Functional, and Genomic Single-Stem-Cell Analyses" International Journal of Molecular Sciences 26, no. 9: 4224. https://doi.org/10.3390/ijms26094224
APA StyleShahar Gabay, T., Stolero, N., Rabhun, N., Sabah, R., Raz, O., Neumeier, Y., Marx, Z., Tao, L., Biezuner, T., Amir, S., Adar, R., Levy, R., Chapal-Ilani, N., Evtiugina, N., Shlush, L. I., Shapiro, E., Yehudai-Resheff, S., & Zuckerman, T. (2025). GMP-like and MLP-like Subpopulations of Hematopoietic Stem and Progenitor Cells Harboring Mutated EZH2 and TP53 at Diagnosis Promote Acute Myeloid Leukemia Relapse: Data of Combined Molecular, Functional, and Genomic Single-Stem-Cell Analyses. International Journal of Molecular Sciences, 26(9), 4224. https://doi.org/10.3390/ijms26094224