
Transcriptome-Wide Analysis and Experimental Validation from FFPE Tissue Identifies Stage-Specific Gene Expression Profiles Differentiating Adenoma, Carcinoma In-Situ and Adenocarcinoma in Colorectal Cancer Progression

 ,
,  ,
,  ,
,  , , , , ,
, , , , , 
Abstract
1. Introduction
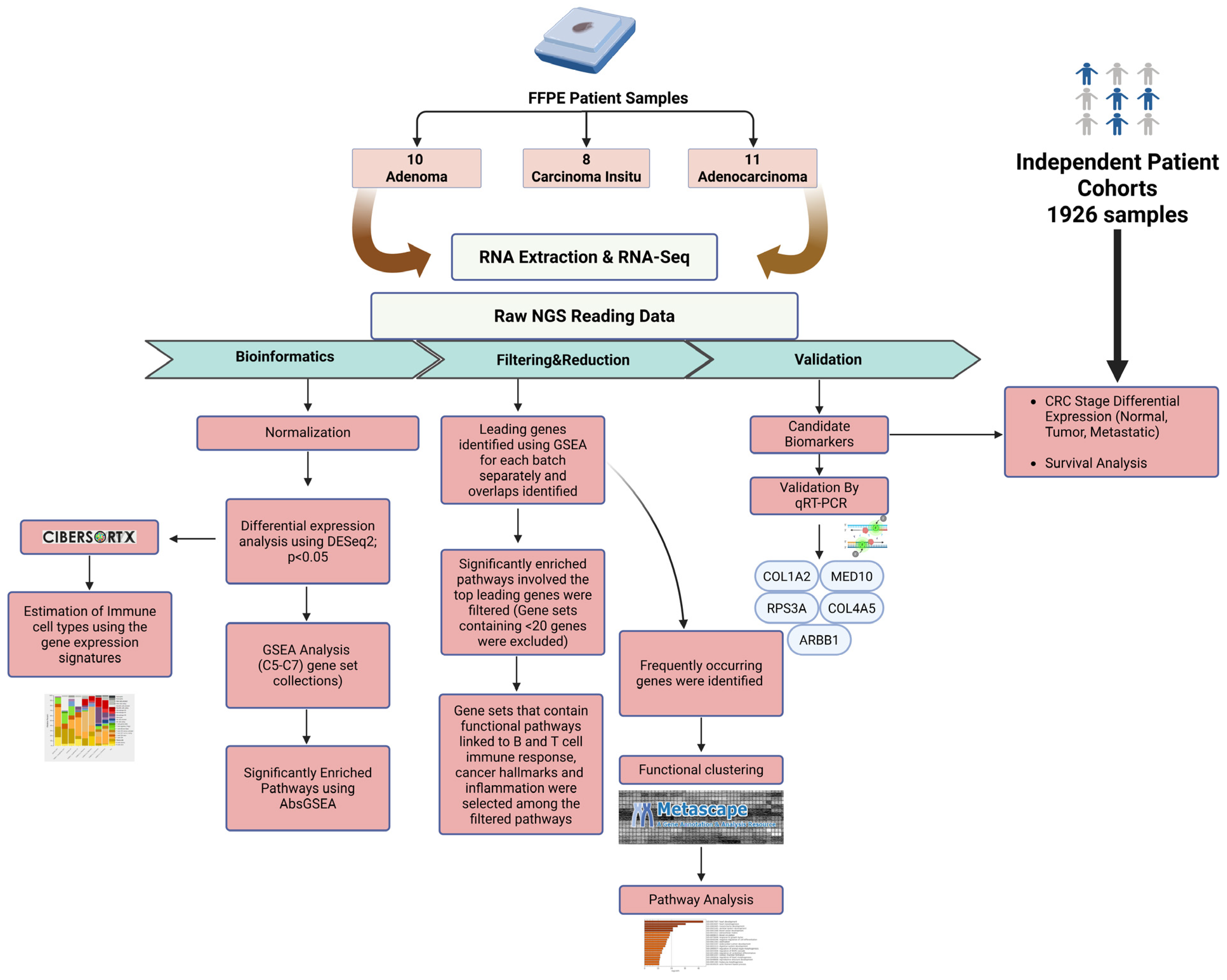
2. Results
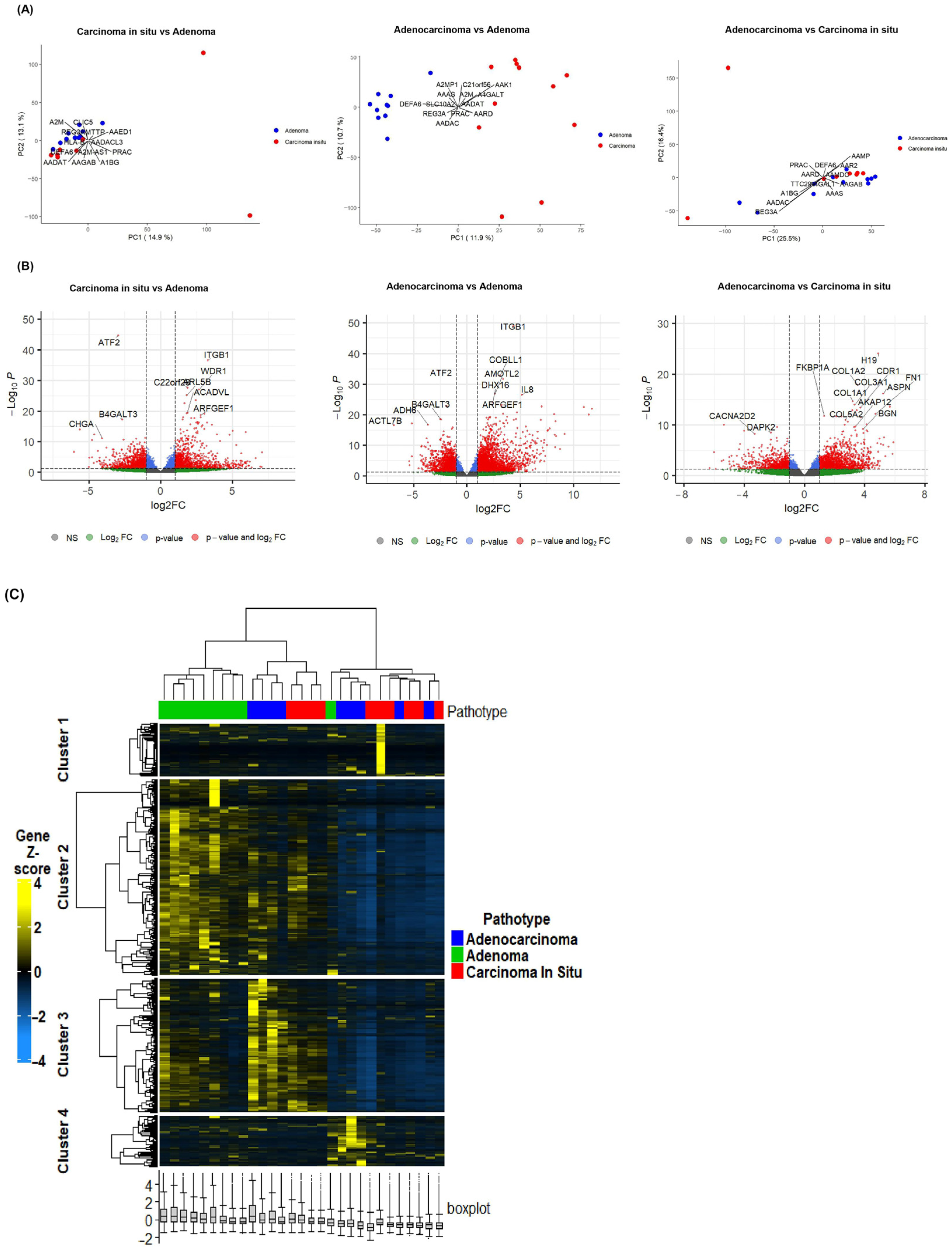
2.1. Differential Gene Expression Signatures Across Adenoma, CIS and Adenocarcinoma Stages in CRC Patients
2.2. Gene-Set Enrichment Analysis of Differentially Expressed Genes Highlights Distinct Molecular Signatures Between Adenoma, CIS and Adenocarcinoma
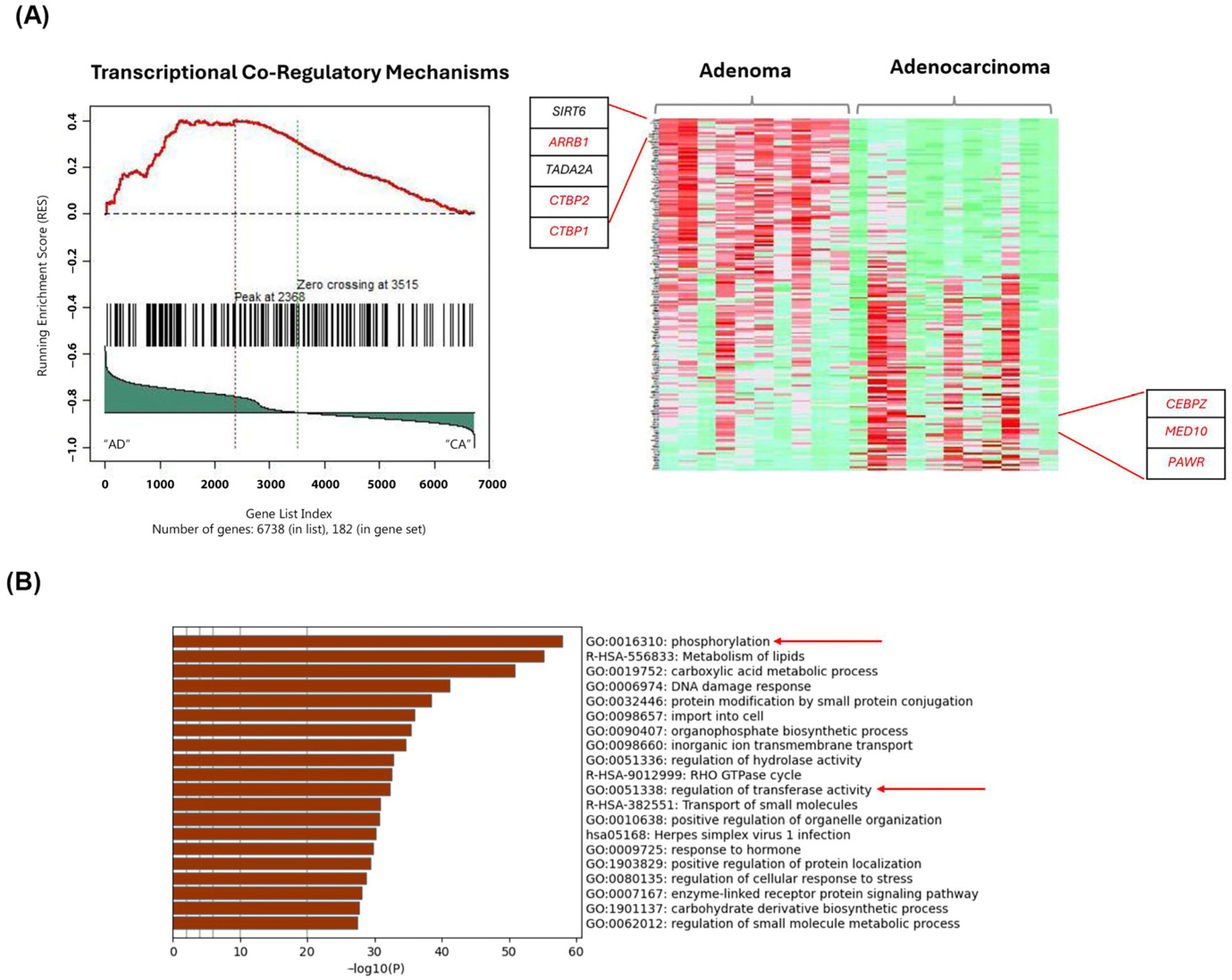
2.3. Gene-Set Enrichment Analysis on Patient Samples Revealed Distinctive Tumor Stage-Mediated Activation of Transcriptional Co-Regulatory Mechanisms, Protein Kinase Functional Pathways and Cellular Metabolic Processes
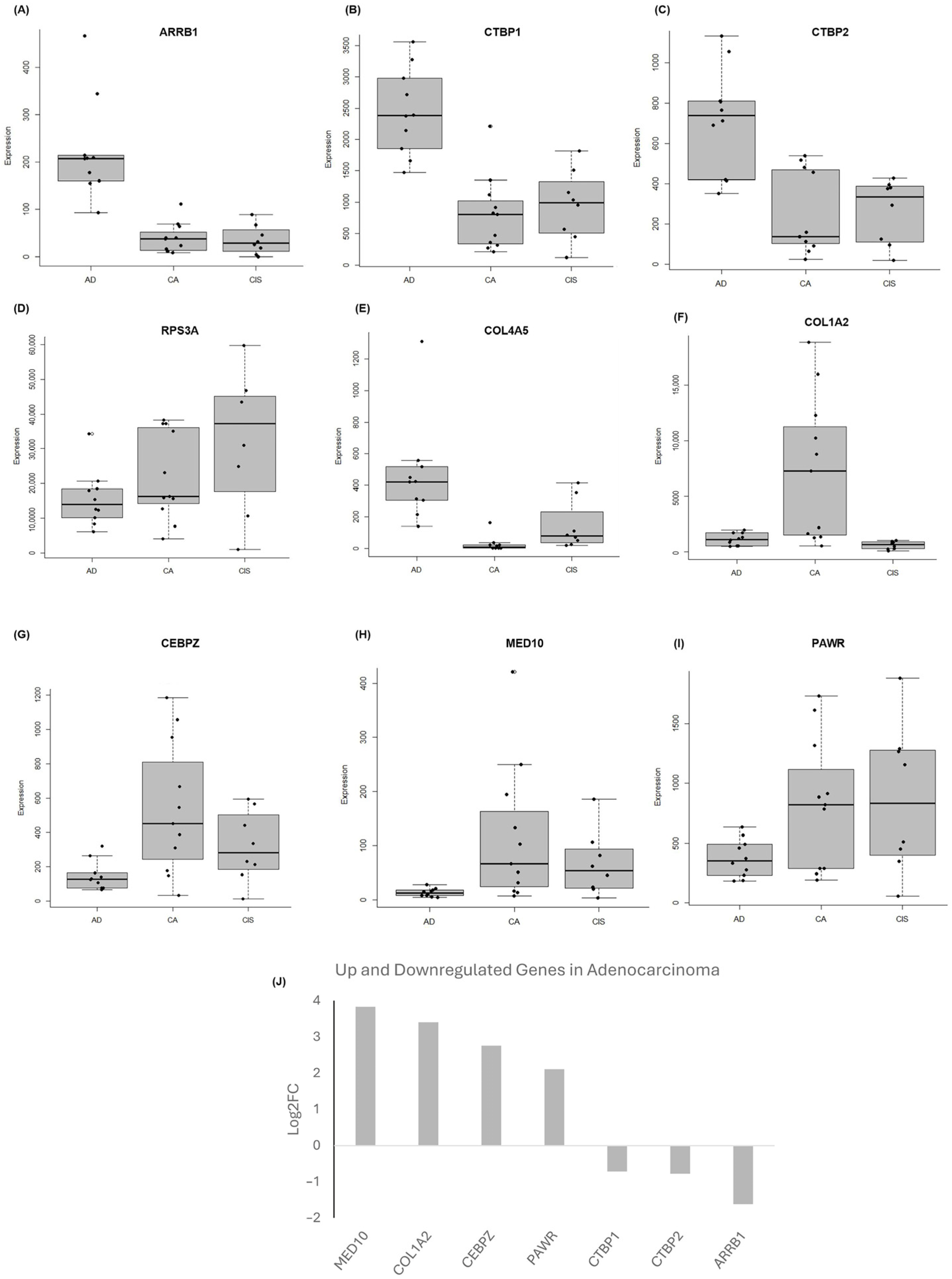
2.4. Validation of the Selected Candidate Biomarkers Using RT-qPCR
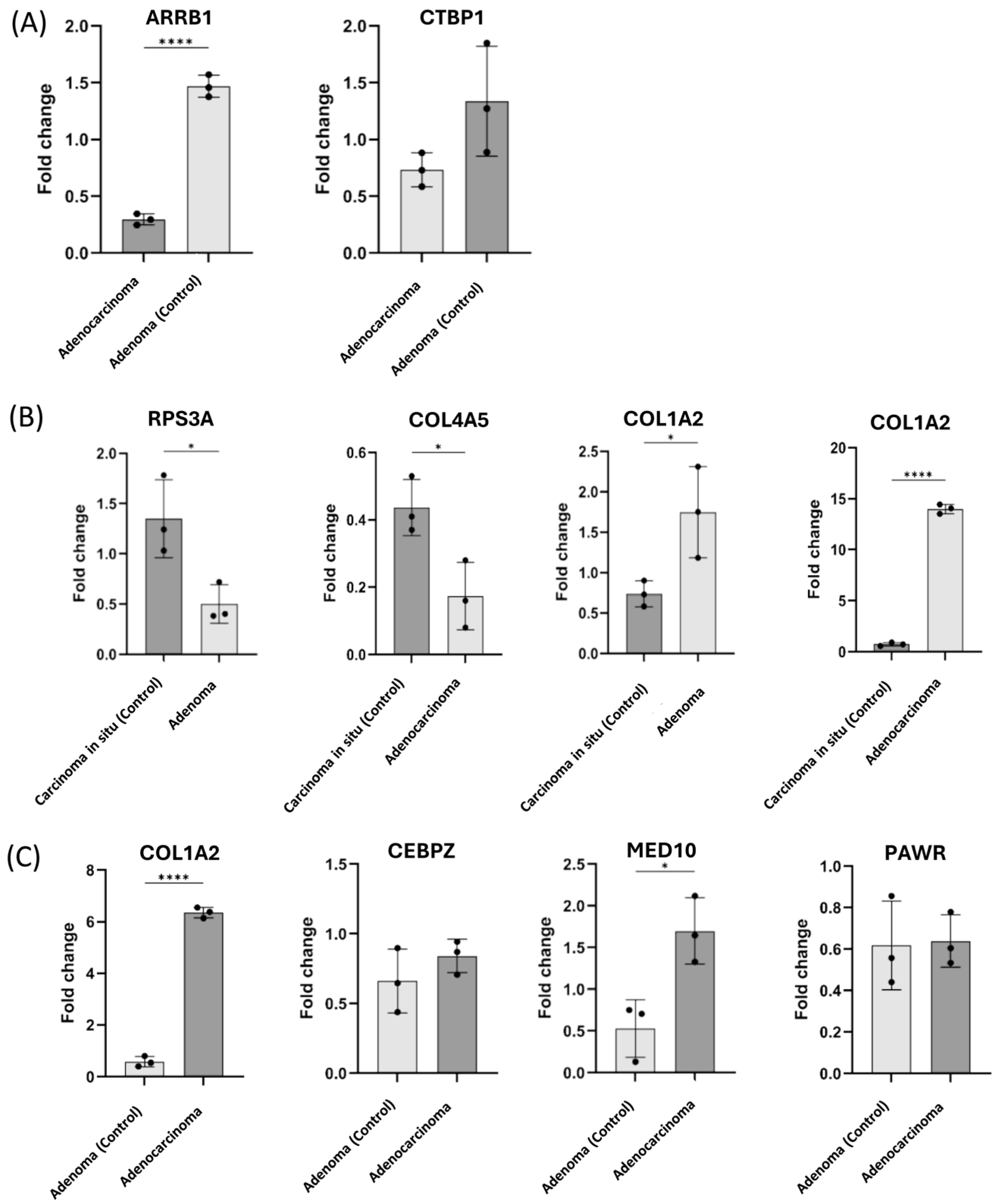
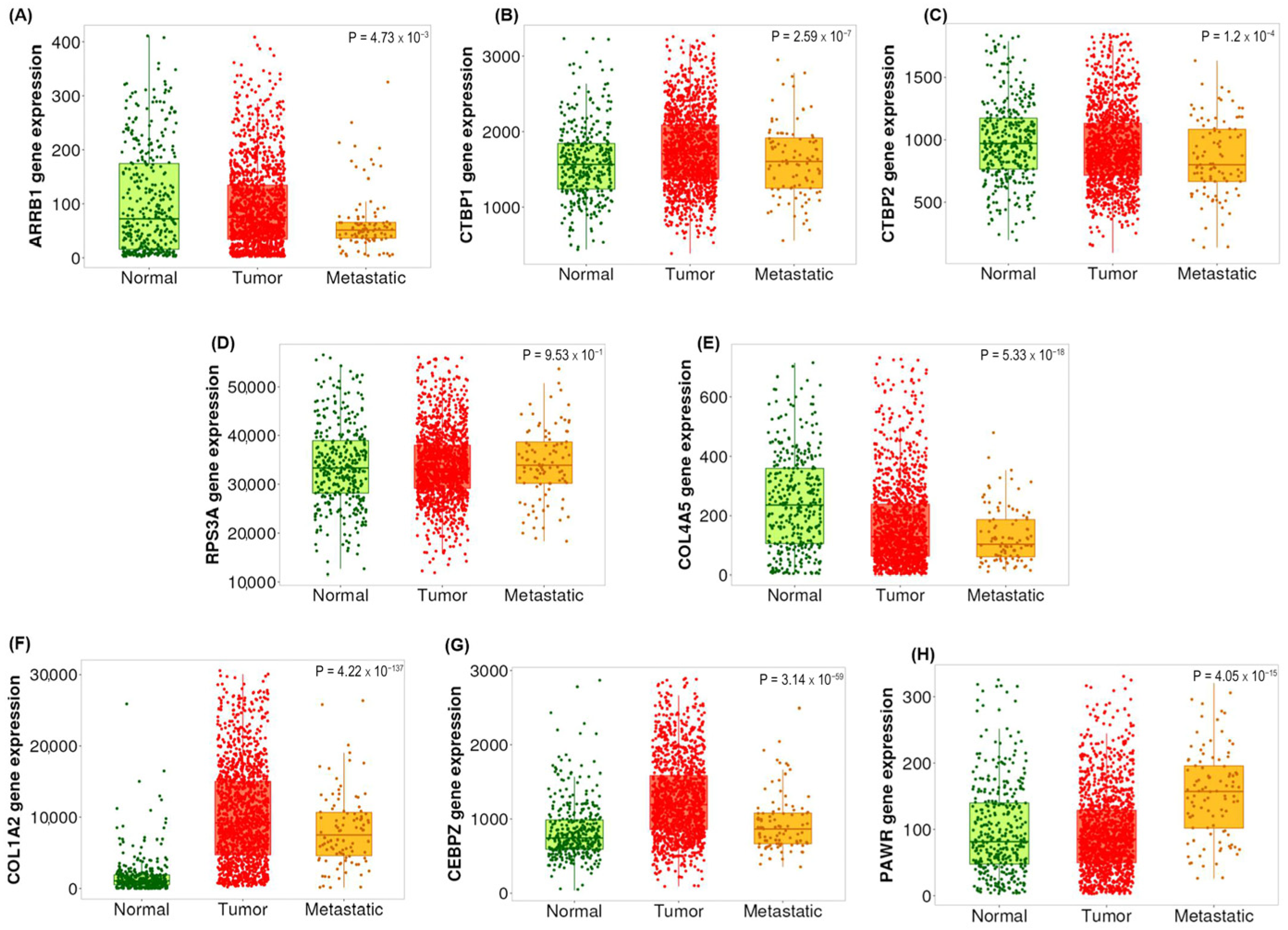
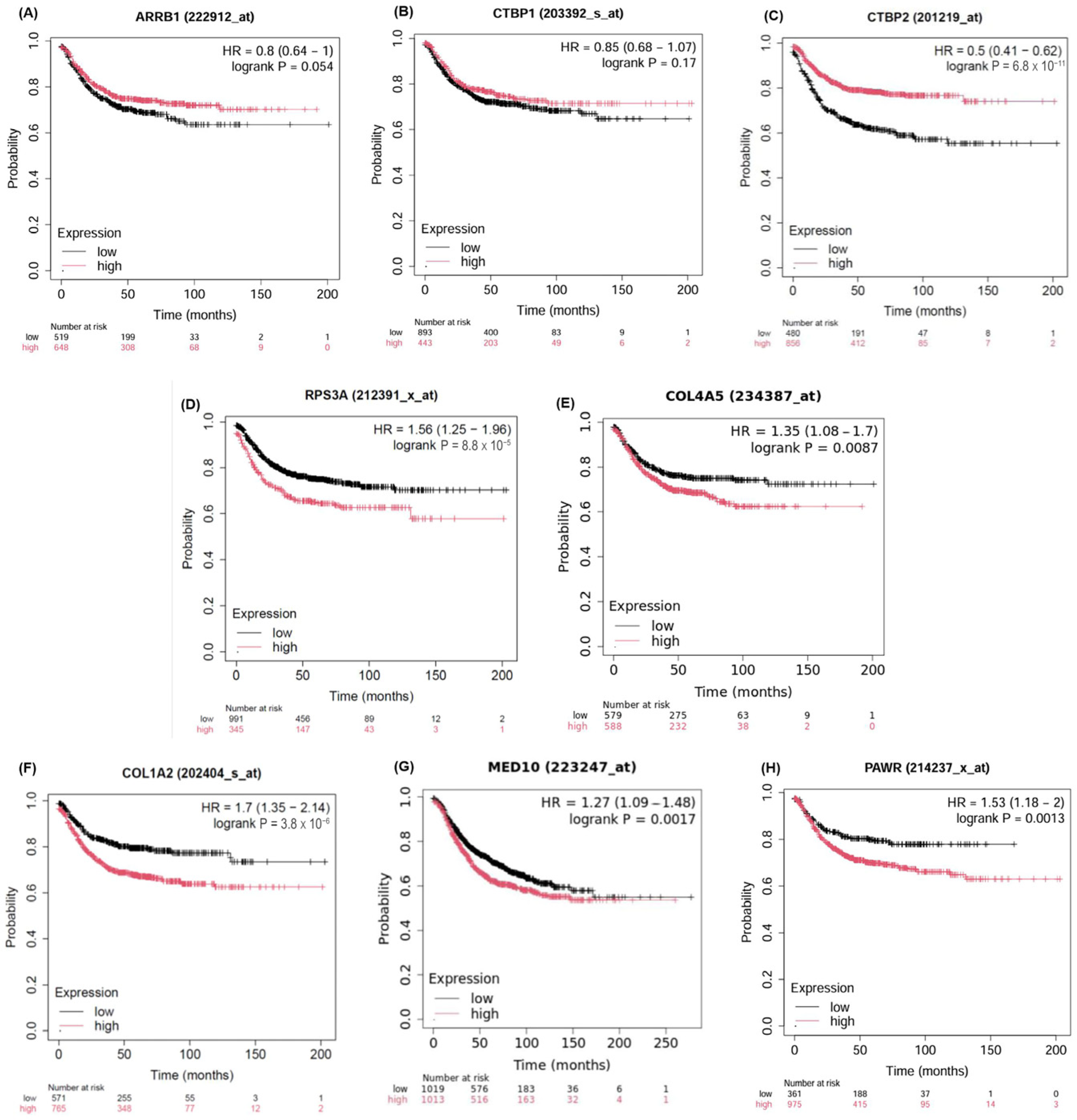
2.5. Validation of the Candidate Biomarkers Differentiating Between Adenoma, CIS and Adenocarcinoma Using Independent Cohort of CRC Patients
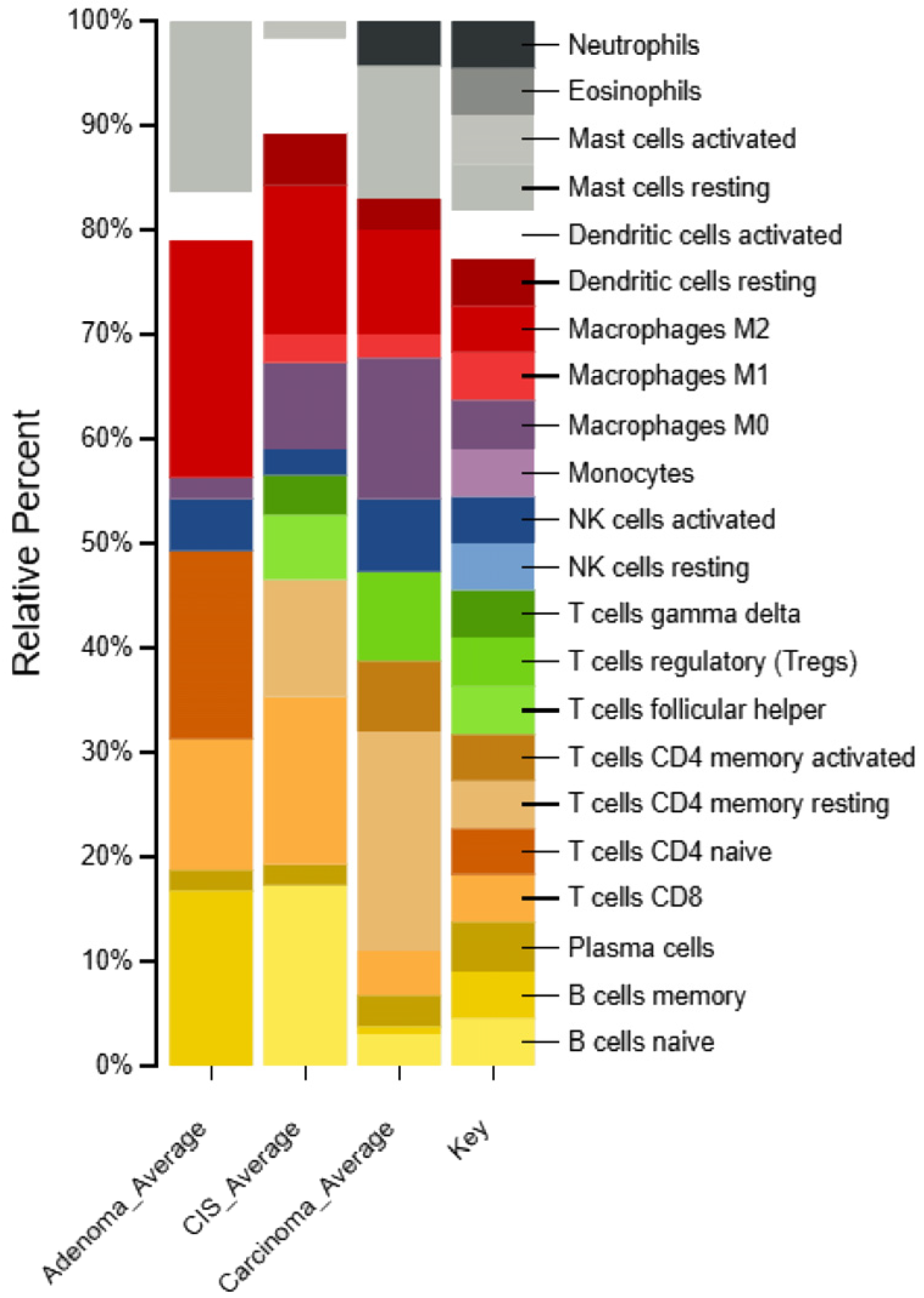
2.6. Identification of Immune Cell Types in Different Types of CRC Patients
3. Discussion
4. Materials and Methods
4.1. FFPE Tissue Specimens from Endoscopic Biopsies of CRC Patients and Independent Validation Cohort
4.2. RNA Extraction
4.3. Whole Transcriptome Sequencing
4.4. RNA-Seq Data Analysis
4.5. Gene Set Enrichment Analysis
4.6. Functional Enrichment Analysis by Metascape
4.7. Validation of Selected Candidate Biomarkers Using Quantitative Reverse Transcriptase-PCR (RT-qPCR)
4.8. Validation of Putative Biomarkers in Independent CRC Patient Cohorts via TNMPlot, KMplot and cBioPortal
4.9. Exploration of Immune Cell Characteristics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- La Vecchia, S.; Sebastián, C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Ru, Z.; Ny, K.T.; Ns, L.R.; Kk, W.W.; Jy, S.J.; Jun, Y.; Wong, S.H. Serrated neoplasia in the colorectum: Gut microbiota and molecular pathways. Gut Microbes 2021, 13, 1863135. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Guo, S.; Du, S.; Cao, Z.; Yang, Y.; Su, X.; Wei, W. Efficacy and safety of berberine in preventing recurrence of colorectal adenomas: A systematic review and meta-analysis. J. Ethnopharmacol. 2022, 282, 114617. [Google Scholar] [CrossRef] [PubMed]
- Aghabozorgi, A.S.; Bahreyni, A.; Soleimani, A.; Bahrami, A.; Khazaei, M.; Ferns, G.A.; Avan, A.; Hassanian, S.M. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie 2019, 157, 64–71. [Google Scholar] [CrossRef]
- Kantor, M.; Sobrado, J.; Patel, S.; Eiseler, S.; Ochner, C. Hereditary Colorectal Tumors: A Literature Review on MUTYH-Associated Polyposis. Gastroenterol. Res. Pract. 2017, 2017, 8693182. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, P.; Li, J.; Wang, B.; Tan, X.; Yin, H.; Han, Y.; Wang, H.; Shi, X.; Li, X.; Xie, C.; et al. Molecular characterization of colorectal adenoma and colorectal cancer via integrated genomic transcriptomic analysis. Front. Oncol. 2023, 13, 1067849. [Google Scholar] [CrossRef]
- Tian, Y.; Kharazmi, E.; Brenner, H.; Xu, X.; Sundquist, K.; Sundquist, J.; Fallah, M. Importance of Family History of Colorectal Carcinoma In Situ Versus Invasive Colorectal Cancer: A Nationwide Cohort Study. J. Natl. Compr. Cancer Netw. 2021, 19, 1252–1257. [Google Scholar] [CrossRef]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedźwiedzka, E.; Arłukowicz, T.; Przybyłowicz, K.E. A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis. Cancers 2021, 13, 2025. [Google Scholar] [CrossRef]
- Vacante, M.; Borzì, A.M.; Basile, F.; Biondi, A. Biomarkers in colorectal cancer: Current clinical utility and future perspectives. World J. Clin. Cases 2018, 6, 869–881. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, J.B.; Li, X.J.; Liu, H.; Liu, Y.J.; Liu, X.P. Association of KRAS, NRAS, BRAF and PIK3CA gene mutations with clinicopathological features, prognosis and ring finger protein 215 expression in patients with colorectal cancer. Biomed. Rep. 2023, 19, 104. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dubansky, B.; Lewis, S.; Telgenhoff, D. Classification and Histological Characteristics of Colorectal Cancer. Am. Soc. Clin. Lab. Sci. 2024, 36, 50–54. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Valenzuela, G.; Canepa, J.; Simonetti, C.; Solo de Zaldívar, L.; Marcelain, K.; González-Montero, J. Consensus molecular subtypes of colorectal cancer in clinical practice: A translational approach. World J. Clin. Oncol. 2021, 12, 1000–1008. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Holtedahl, K.; Borgquist, L.; Donker, G.A.; Buntinx, F.; Weller, D.; Campbell, C.; Månsson, J.; Hammersley, V.; Braaten, T.; Parajuli, R. Symptoms and signs of colorectal cancer, with differences between proximal and distal colon cancer: A prospective cohort study of diagnostic accuracy in primary care. BMC Fam. Pract. 2021, 22, 148. [Google Scholar] [CrossRef]
- Gonzalez-Pons, M.; Cruz-Correa, M. Colorectal Cancer Biomarkers: Where Are We Now? BioMed Res. Int. 2015, 2015, 149014. [Google Scholar] [CrossRef]
- Duan, B.; Zhang, Y.; Bai, J.; Wang, J.; Duan, X.; Luo, X.; Zhang, R.; Pu, Y.; Kou, M.; Lei, J.; et al. Colorectal Cancer: An Overview. In Gastrointestinal Cancers; Morgado-Diaz, J.A., Ed.; Exon Publications: Brisbane, Australia, 2022; Available online: https://exonpublications.com/index.php/exon/article/view/gastrointestinal-cancers-colorectal-cancer (accessed on 9 July 2022).
- Nachar, O.M. The role of positron emission tomography in colorectal carcinoma. Ochsner J. 2002, 4, 146–155. [Google Scholar] [PubMed] [PubMed Central]
- Bompou, E.; Vassiou, A.; Baloyiannis, I.; Perivoliotis, K.; Fezoulidis, I.; Tzovaras, G. Comparative evaluation of CT and MRI in the preoperative staging of colon cancer. Sci. Rep. 2024, 14, 17145. [Google Scholar] [CrossRef]
- Bor, R.; Fábián, A.; Szepes, Z. Role of ultrasound in colorectal diseases. World J. Gastroenterol. 2016, 22, 9477–9487. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hong, S.W.; Byeon, J.S. Endoscopic diagnosis and treatment of early colorectal cancer. Intest. Res. 2022, 20, 281–290. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Committee, A.S.o.P.; Fisher, D.A.; Shergill, A.K.; Early, D.S.; Acosta, R.D.; Chandrasekhara, V.; Chathadi, K.V.; Decker, G.A.; Evans, J.A.; Fanelli, R.D.; et al. Role of endoscopy in the staging and management of colorectal cancer. Gastrointest. Endosc. 2013, 78, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Strul, H.; Arber, N. Fecal occult blood test for colorectal cancer screening. Ann. Oncol. 2002, 13, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Hasana, U.; Nadeem, I.A.; Khatri, S.P.; Nawaz, S.; Makhdoom, Q.U.; Wazir, S.; Patel, K.; Ghaly, M. Advances in colorectal cancer screening and detection: A narrative review on biomarkers, imaging and preventive strategies. J. Egypt. Natl. Cancer Inst. 2025, 37, 20. [Google Scholar] [CrossRef] [PubMed]
- Hančinová, M.; Ondruš, D.; Ondrušová, M.; Mrinakova, B. Colorectal carcinoma—Epidemiology, risk factors, prognostic bio markers. Klin. Onkol. 2025, 38, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pan, J.; Wang, L.; Ji, G.; Dang, Y. Colorectal Cancer: Pathogenesis and Targeted Therapy. MedComm 2025, 6, e70127. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hamoudi, R.A.; Appert, A.; Ye, H.; Ruskone-Fourmestraux, A.; Streubel, B.; Chott, A.; Raderer, M.; Gong, L.; Wlodarska, I.; De Wolf-Peeters, C.; et al. Differential expression of NF-kappaB target genes in MALT lymphoma with and without chromosome translocation: Insights into molecular mechanism. Leukemia 2010, 24, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Meng, L.; Gao, J.; Lu, M.; Guo, C.; Li, Y.; Rong, Z.; Ye, Y. PHB2 promotes colorectal cancer cell proliferation and tumorigenesis through NDUFS1-mediated oxidative phosphorylation. Cell Death Dis. 2023, 14, 44. [Google Scholar] [CrossRef]
- Ahmad, R.; Singh, J.K.; Wunnava, A.; Al-Obeed, O.; Abdulla, M.; Srivastava, S.K. Emerging trends in colorectal cancer: Dysregulated signaling pathways (Review). Int. J. Mol. Med. 2021, 47, 14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, H.; Zheng, H.; Cao, X.; Meng, P.; Liu, J.; Zuo, H.; Zhang, T.; Wang, Z. Association between serum gamma-glutamyl transferase and advanced colorectal adenoma among inpatients: A case-control study. Front. Oncol. 2023, 13, 1188017. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Soncin, I.; Sheng, J.; Chen, Q.; Foo, S.; Duan, K.; Lum, J.; Poidinger, M.; Zolezzi, F.; Karjalainen, K.; Ruedl, C. The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat. Commun. 2018, 9, 582. [Google Scholar] [CrossRef]
- Kuwahara, T.; Hazama, S.; Suzuki, N.; Yoshida, S.; Tomochika, S.; Nakagami, Y.; Matsui, H.; Shindo, Y.; Kanekiyo, S.; Tokumitsu, Y.; et al. Intratumoural-infiltrating CD4 + and FOXP3 + T cells as strong positive predictive markers for the prognosis of resectable colorectal cancer. Br. J. Cancer 2019, 121, 659–665. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Schlößer, H.A.; Gryschok, L.; Malcher, J.; Wennhold, K.; Garcia-Marquez, M.; Herbold, T.; Neuhaus, L.S.; Becker, H.J.; Fiedler, A.; et al. Characterization of tumor-associated B-cell subsets in patients with colorectal cancer. Oncotarget 2014, 5, 4651–4664. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, E.; Ding, C.; Li, S.; Zhou, X.; Aikemu, B.; Fan, X.; Sun, J.; Zheng, M.; Yang, X. Roles and mechanisms of tumour-infiltrating B cells in human cancer: A new force in immunotherapy. Biomark. Res. 2023, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Rezende, R.M.; Lanser, A.J.; Rubino, S.; Kuhn, C.; Skillin, N.; Moreira, T.G.; Liu, S.; Gabriely, G.; David, B.A.; Menezes, G.B.; et al. γδ T cells control humoral immune response by inducing T follicular helper cell differentiation. Nat. Commun. 2018, 9, 3151. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Corsale, A.M.; Di Simone, M.; Lo Presti, E.; Dieli, F.; Meraviglia, S. gammadelta T cells and their clinical application in colon cancer. Front. Immunol. 2023, 14, 1098847. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, Y.; Chen, Z.; Han, J.; Ma, X.; Zheng, X.; Chen, J. Functional and Therapeutic Significance of Tumor-Associated Macrophages in Colorectal Cancer. Front. Oncol. 2022, 12, 781233. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Du, W.; Quan, X.; Wang, C.; Song, Q.; Mou, J.; Pei, D. Regulation of tumor metastasis and CD8+ T cells infiltration by circRNF216/miR-576-5p/ZC3H12C axis in colorectal cancer. Cell. Mol. Biol. Lett. 2024, 29, 19. [Google Scholar] [CrossRef]
- Masle-Farquhar, E.; Jeelall, Y.; White, J.; Bier, J.; Deenick, E.K.; Brink, R.; Horikawa, K.; Goodnow, C.C. CARD11 gain-of-function mutation drives cell-autonomous accumulation of PD-1(+) ICOS(high) activated T cells, T-follicular, T-regulatory and T-follicular regulatory cells. Front. Immunol. 2023, 14, 1095257. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- MacLeod, M.K.; Clambey, E.T.; Kappler, J.W.; Marrack, P. CD4 memory T cells: What are they and what can they do? Semin. Immunol. 2009, 21, 53–61. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Slattery, M.L.; Herrick, J.S.; Mullany, L.E.; Samowitz, W.S.; Sevens, J.R.; Sakoda, L.; Wolff, R.K. The co-regulatory networks of tumor suppressor genes, oncogenes, and miRNAs in colorectal cancer. Genes Chromosomes Cancer 2017, 56, 769–787. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Hunter, T. Protein kinase signaling networks in cancer. Curr. Opin. Genet. Dev. 2011, 21, 4–11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Patel, S.; Alam, A.; Pant, R.; Chattopadhyay, S. Wnt Signaling and Its Significance Within the Tumor Microenvironment: Novel Therapeutic Insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Nyström, H. The extracellular matrix in colorectal cancer and its metastatic settling—Alterations and biological implications. Crit. Rev. Oncol. Hematol. 2022, 175, 103712. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Ha, S.E.; Wu, M.; Zogg, H.; Ronkon, C.F.; Lee, M.Y.; Ro, S. Extracellular Matrix Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 9185. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rosanò, L.; Bagnato, A. β-arrestin1 at the cross-road of endothelin-1 signaling in cancer. J. Exp. Clin. Cancer Res. 2016, 35, 121. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef]
- Song, Q.; Han, Z.; Wu, X.; Wang, Y.; Zhou, L.; Yang, L.; Liu, N.; Sui, H.; Cai, J.; Ji, Q.; et al. β-Arrestin1 Promotes Colorectal Cancer Metastasis Through GSK-3β/β-Catenin Signaling- Mediated Epithelial-to-Mesenchymal Transition. Front. Cell Dev. Biol. 2021, 9, 650067. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grivas, P.D.; Papavassiliou, A.G. Transcriptional corepressors in cancer: Emerging targets for therapeutic intervention. Cancer 2013, 119, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Asangani, I.A.; Chakravarthi, B.V.S.K.; Ateeq, B.; Lonigro, R.J.; Cao, Q.; Ram-Shankar, M.; Camacho, D.F.; McGregor, N.; Schumann, T.E.W.; et al. Role of Transcriptional Corepressor CtBP1 in Prostate Cancer Progression. Neoplasia 2012, 14, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.B.; Anderson, M.; Diaz, M.O.; Zeleznik-Le, N.J. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc. Natl. Acad. Sci. USA 2003, 100, 8342–8347. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Palmer, S.; Brouillet, J.P.; Kilbey, A.; Fulton, R.; Walker, M.; Crossley, M.; Bartholomew, C. Evi-1 transforming and repressor activities are mediated by CtBP co-repressor proteins. J. Biol. Chem. 2001, 276, 25834–25840. [Google Scholar] [CrossRef] [PubMed]
- May, T.; Yang, J.; Shoni, M.; Liu, S.; He, H.; Gali, R.; Ng, S.K.; Crum, C.; Berkowitz, R.S.; Ng, S.W. BRCA1 expression is epigenetically repressed in sporadic ovarian cancer cells by overexpression of C-terminal binding protein 2. Neoplasia 2013, 15, 600–608. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Birts, C.N.; Harding, R.; Soosaipillai, G.; Halder, T.; Azim-Araghi, A.; Darley, M.; Cutress, R.I.; Bateman, A.C.; Blaydes, J.P. Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol. Cell 2010, 103, 1–19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peña, C.; García, J.M.; García, V.; Silva, J.; Domínguez, G.; Rodríguez, R.; Maximiano, C.; García de Herreros, A.; Muñoz, A.; Bonilla, F. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int. J. Cancer 2006, 119, 2098–2104. [Google Scholar] [CrossRef] [PubMed]
- Blevins, M.A.; Huang, M.; Zhao, R. The Role of CtBP1 in Oncogenic Processes and Its Potential as a Therapeutic Target. Mol. Cancer Ther. 2017, 16, 981–990. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hamada, F.; Bienz, M. The APC tumor suppressor binds to C-terminal binding protein to divert nuclear beta-catenin from TCF. Dev. Cell 2004, 7, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.; Yoshida, T.; Joazeiro, C.A.; Jones, K.A. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes. Dev. 2006, 20, 586–600. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yuan, X.; He, Y.; Wang, W. ceRNA network-regulated COL1A2 high expression correlates with poor prognosis and immune infiltration in colon adenocarcinoma. Sci. Rep. 2023, 13, 16932. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Zheng, T.; Xu, X.X.; Zheng, L.; Li, F.Y.; Li, X.X.; Zhou, L. Comprehensive analysis of the differential expression and prognostic value of COL1A2 in colon adenocarcinoma. Aging 2022, 14, 7390–7407. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mortezapour, M.; Tapak, L.; Bahreini, F.; Najafi, R.; Afshar, S. Identification of key genes in colorectal cancer diagnosis by weighted gene co-expression network analysis. Comput. Biol. Med. 2023, 157, 106779. [Google Scholar] [CrossRef]
- Wu, C.-C.; Wang, Y.-H.; Hu, S.-W.; Wu, W.-L.; Yeh, C.-T.; Bamodu, O.A. MED10 Drives the Oncogenicity and Refractory Phenotype of Bladder Urothelial Carcinoma Through the Upregulation of hsa-miR-590. Front. Oncol. 2022, 11, 744937. [Google Scholar] [CrossRef]
- Cheratta, A.R.; Thayyullathil, F.; Pallichankandy, S.; Subburayan, K.; Alakkal, A.; Galadari, S. Prostate apoptosis response-4 and tumor suppression: It’s not just about apoptosis anymore. Cell Death Dis. 2021, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Tao, K.; Zheng, X.; Liu, D.; Ma, D.; Gao, Q. Expression of PAWR predicts prognosis of ovarian cancer. Cancer Cell Int. 2020, 20, 598. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shen, J.; Tan, F.Q.; Zheng, X.Y.; Xie, L.P. Antitumor Activity of Small Activating RNAs Induced PAWR Gene Activation in Human Bladder Cancer Cells. Int. J. Med. Sci. 2021, 18, 3039–3049. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, C.; Weng, J.; Liu, C.; Zhou, Q.; Chen, W.; Hsu, J.L.; Sun, J.; Atyah, M.; Xu, Y.; Shi, Y.; et al. High RPS3A expression correlates with low tumor immune cell infiltration and unfavorable prognosis in hepatocellular carcinoma patients. Am. J. Cancer Res. 2020, 10, 2768–2784. [Google Scholar] [PubMed] [PubMed Central]
- Zhou, X.; Liao, W.J.; Liao, J.M.; Liao, P.; Lu, H. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeng, X.; Wang, H.Y.; Wang, Y.P.; Bai, S.Y.; Pu, K.; Zheng, Y.; Guo, Q.H.; Guan, Q.L.; Ji, R.; Zhou, Y.N. COL4A family: Potential prognostic biomarkers and therapeutic targets for gastric cancer. Transl. Cancer Res. 2020, 9, 5218–5232. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, H.; Wang, Y.; Wang, S.; Xie, Y.; Sun, K.; Li, S.; Cui, W.; Wang, K. The involvement of collagen family genes in tumor enlargement of gastric cancer. Sci. Rep. 2023, 13, 100. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wallin, E.F.; Jolly, E.C.; Suchánek, O.; Bradley, J.A.; Espéli, M.; Jayne, D.R.W.; Linterman, M.A.; Smith, K.G.C. Human T-follicular helper and T-follicular regulatory cell maintenance is independent of germinal centers. Blood 2014, 124, 2666–2674. [Google Scholar] [CrossRef]
- Jackson, S.; Smriti, M.; Hilal, A. Gamma Delta T Cell: A Unique Member of the T Cell Family with a Significant Role in Cancer Immunotherapy. In Biology of T Cells in Health and Disease; Hilal, A., Ed.; IntechOpen: Rijeka, Croatia, 2025; Chapter 1. [Google Scholar] [CrossRef]
- Battaglin, F.; Naseem, M.; Lenz, H.J.; Salem, M.E. Microsatellite instability in colorectal cancer: Overview of its clinical significance and novel perspectives. Clin. Adv. Hematol. Oncol. 2018, 16, 735–745. [Google Scholar] [PubMed] [PubMed Central]
- Cai, L.; Chen, A.; Tang, D. A new strategy for immunotherapy of microsatellite-stable (MSS)-type advanced colorectal cancer: Multi-pathway combination therapy with PD-1/PD-L1 inhibitors. Immunology 2024, 173, 209–226. [Google Scholar] [CrossRef]
- Ding, X.; Huang, H.; Fang, Z.; Jiang, J. From Subtypes to Solutions: Integrating CMS Classification with Precision Therapeutics in Colorectal Cancer. Curr. Treat. Options Oncol. 2024, 25, 1580–1593. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.; Nicholls, M.E.; Gajjar, A.; Senagore, A.J.; Qiu, S.; Szabo, C.; Hellmich, M.R.; Chao, C. Consensus Molecular Subtypes of Colorectal Cancer and their Clinical Implications. Int. Biol. Biomed. J. 2017, 3, 105–111. [Google Scholar] [PubMed] [PubMed Central]
- Valdeolivas, A.; Amberg, B.; Giroud, N.; Richardson, M.; Gálvez, E.J.C.; Badillo, S.; Julien-Laferrière, A.; Túrós, D.; Voith von Voithenberg, L.; Wells, I.; et al. Profiling the heterogeneity of colorectal cancer consensus molecular subtypes using spatial transcriptomics. NPJ Precis. Oncol. 2024, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.E.; Huang, Z.P.; Wang, X.; Cai, D.; Li, C.H.; Gao, F.; Wu, X.J.; Wang, W. A Transcription Factor Signature Can Identify the CMS4 Subtype and Stratify the Prognostic Risk of Colorectal Cancer. Front. Oncol. 2022, 12, 902974. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Győrffy, B. Integrated analysis of public datasets for the discovery and validation of survival-associated genes in solid tumors. Innovation 2024, 5, 100625. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alhosani, F.; Ilce, B.Y.; Alhamidi, R.S.; Bhamidimarri, P.M.; Hamad, A.M.; Alkhayyal, N.; Künstner, A.; Khandanpour, C.; Busch, H.; Al-Ramadi, B.; et al. Transcriptome Profiling Associated with CARD11 Overexpression in Colorectal Cancer Implicates a Potential Role for Tumor Immune Microenvironment and Cancer Pathways Modulation via NF-κB. Int. J. Mol. Sci. 2024, 25, 10367. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Sets Used | Type |
|---|---|
| C1 | Positional Gene Sets |
| C2 | Curated Gene Sets |
| C3 | Regulatory-Related Gene Sets |
| C4 | Computational Gene Sets |
| C5 | Gene Ontology Gene Sets (Biological Processes, Molecular Functions, and Cellular Components) |
| C6 | Oncogenic Related Gene Sets |
| C7 | Immunology-Related Gene Sets |
| C8 | Cell type Signature Gene Sets |
| No | Gender | Age | Nationality | Subtype |
|---|---|---|---|---|
| 1 | Male | 61 | Italian | Tubular Adenoma |
| 2 | Male | 61 | Qatari | Tubular Adenoma |
| 3 | Male | 54 | Emirati | Tubular Adenoma |
| 4 | Male | 61 | Emirati | Tubular Adenoma |
| 5 | Female | 39 | Italian | Tubular Adenoma |
| 6 | Male | 75 | Indian | Tubular Adenoma |
| 7 | Female | 64 | British | Tubular Adenoma |
| 8 | Male | 48 | Portuguese | Tubular Adenoma |
| 9 | Female | 51 | Emirati | Tubular Adenoma |
| 10 | Male | 50 | South African | Tubular Adenoma |
| 11 | Female | 59 | French | Adenocarcinoma |
| 12 | Female | 43 | Filipino | Adenocarcinoma |
| 13 | Male | 56 | Swiss | Adenocarcinoma |
| 14 | Female | 64 | Emirati | Adenocarcinoma |
| 15 | Female | 38 | Iraqi | Adenocarcinoma |
| 16 | Female | 44 | Sudanese | Adenocarcinoma |
| 17 | Female | 32 | Emirati | Adenocarcinoma |
| 18 | Female | 65 | Egyptian | Adenocarcinoma |
| 19 | Male | 77 | Indian | Adenocarcinoma |
| 20 | Male | 84 | Syrian | Adenocarcinoma |
| 21 | Female | 65 | Egyptian | Adenocarcinoma |
| 22 | Male | 70 | Romanian | Carcinoma in situ |
| 23 | Female | 80 | Emirati | Carcinoma in situ |
| 24 | NA | NA | NA | Carcinoma in situ |
| 25 | Male | NA | Filipino | Carcinoma in situ |
| 26 | Female | NA | Emirati | Carcinoma in situ |
| 27 | Male | NA | Emirati | Carcinoma in situ |
| 28 | Female | NA | Egyptian | Carcinoma in situ |
| 29 | NA | NA | NA | Carcinoma in situ |
| Gene ID | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| COL1A2 | TCAAAGGCATTAGGGGACACA | CATTTTCACCAGGGGCACCA |
| CEBPZ | GAAGAGTTTGGCCATCTATTGG | CATCTAAGCTGTTTGAGACTTG |
| MED10 | AGATCGACACCATGAAGAAAT | TGAGCTGGTTAAGAAGGCGG |
| PAWR | GCGGAAACGAGAAGATGCAA | TTCTAGGTGGCTCCTGTAG |
| ARRB1 | GCGGAGAGTCTATGTGACG | ACAGGTCCTTGCGAAAGGT |
| CTBP1 | GATGTCAGGCGTCCGACC | ACAGTGGCCACGTCCTTCA |
| CTBP2 | AATCCCCCACAGCGGTCCA | GTGACGCCACTATGAACCTG |
| RPS3A | GGACCCAAGGAACCAAAATT | AGTTTTTACCCTGAACATCTTC |
| COL4A5 | TCCCAAAGGATTACCAGGCA | GGAACCCAACTCTCCTTTGT |
| 18S | TGACTCAACACGGGAAACC | TCGCTCCACCAACTAAGAAC |
| GAPDH | TGTCAGTGG TGGACCTGACCT | TCGCTGTTGAAGTCA GACGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhosani, F.; Alhamidi, R.S.; Ilce, B.Y.; Altaie, A.M.; Ali, N.; Hamad, A.M.; Künstner, A.; Khandanpour, C.; Busch, H.; Al-Ramadi, B.; et al. Transcriptome-Wide Analysis and Experimental Validation from FFPE Tissue Identifies Stage-Specific Gene Expression Profiles Differentiating Adenoma, Carcinoma In-Situ and Adenocarcinoma in Colorectal Cancer Progression. Int. J. Mol. Sci. 2025, 26, 4194. https://doi.org/10.3390/ijms26094194
Alhosani F, Alhamidi RS, Ilce BY, Altaie AM, Ali N, Hamad AM, Künstner A, Khandanpour C, Busch H, Al-Ramadi B, et al. Transcriptome-Wide Analysis and Experimental Validation from FFPE Tissue Identifies Stage-Specific Gene Expression Profiles Differentiating Adenoma, Carcinoma In-Situ and Adenocarcinoma in Colorectal Cancer Progression. International Journal of Molecular Sciences. 2025; 26(9):4194. https://doi.org/10.3390/ijms26094194
Chicago/Turabian StyleAlhosani, Faisal, Reem Sami Alhamidi, Burcu Yener Ilce, Alaa Muayad Altaie, Nival Ali, Alaa Mohamed Hamad, Axel Künstner, Cyrus Khandanpour, Hauke Busch, Basel Al-Ramadi, and et al. 2025. "Transcriptome-Wide Analysis and Experimental Validation from FFPE Tissue Identifies Stage-Specific Gene Expression Profiles Differentiating Adenoma, Carcinoma In-Situ and Adenocarcinoma in Colorectal Cancer Progression" International Journal of Molecular Sciences 26, no. 9: 4194. https://doi.org/10.3390/ijms26094194
APA StyleAlhosani, F., Alhamidi, R. S., Ilce, B. Y., Altaie, A. M., Ali, N., Hamad, A. M., Künstner, A., Khandanpour, C., Busch, H., Al-Ramadi, B., Harati, R., Sayed, K., AlFazari, A., Bendardaf, R., & Hamoudi, R. (2025). Transcriptome-Wide Analysis and Experimental Validation from FFPE Tissue Identifies Stage-Specific Gene Expression Profiles Differentiating Adenoma, Carcinoma In-Situ and Adenocarcinoma in Colorectal Cancer Progression. International Journal of Molecular Sciences, 26(9), 4194. https://doi.org/10.3390/ijms26094194