
Dysregulation of Labile Iron Predisposes Chemotherapy Resistant Cancer Cells to Ferroptosis

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
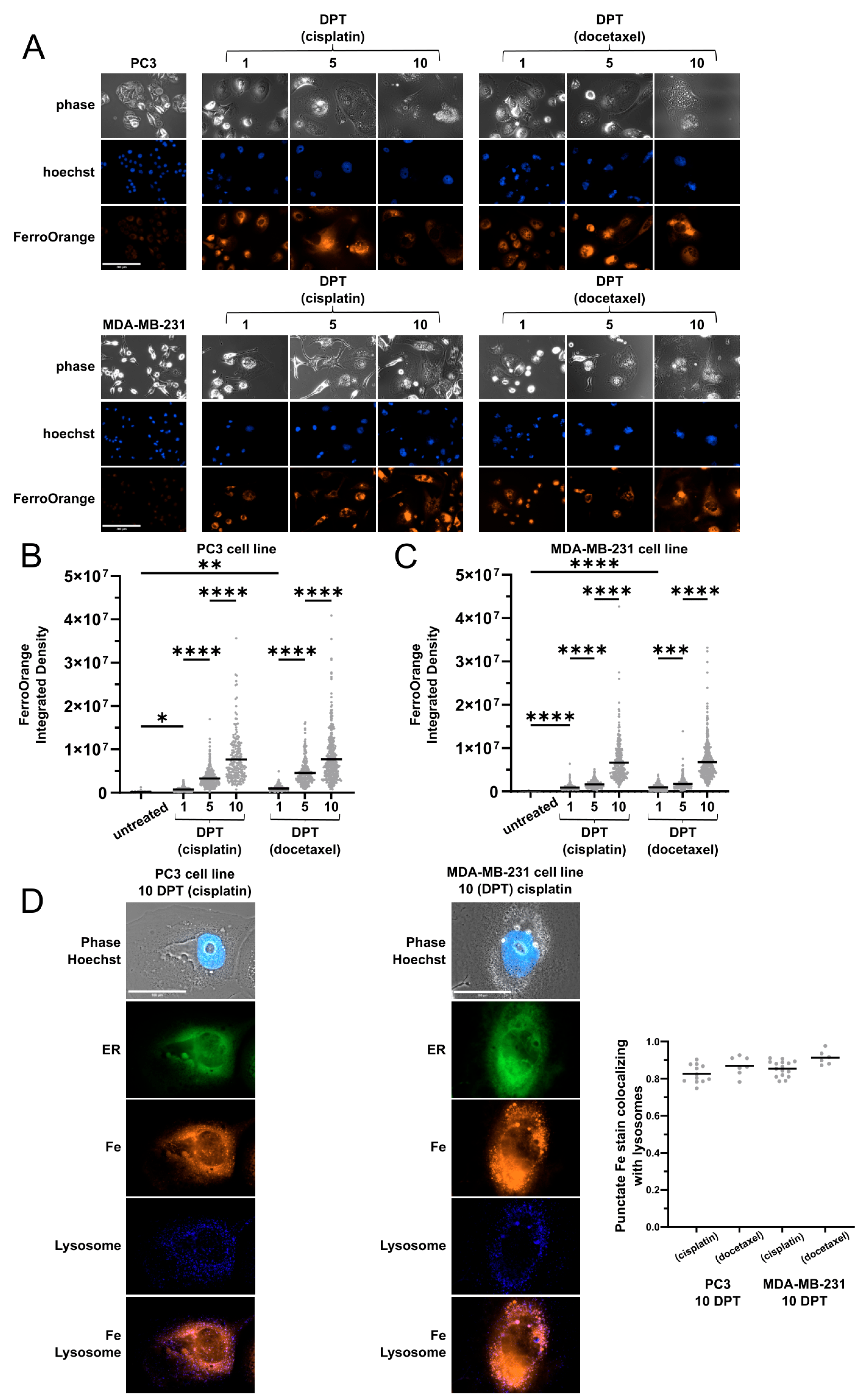
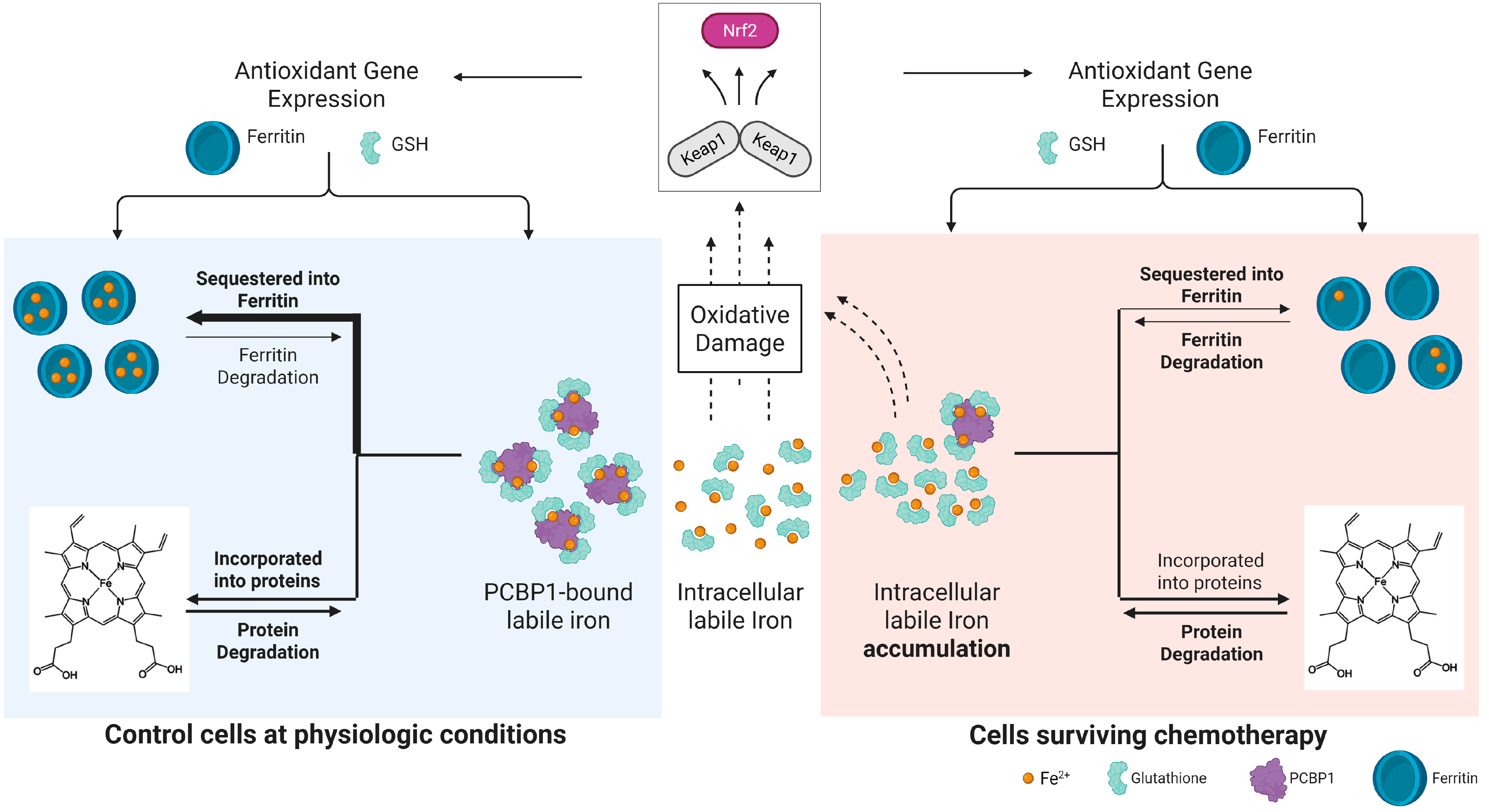
2.1. Cells Surviving Chemotherapy Have High Labile Iron
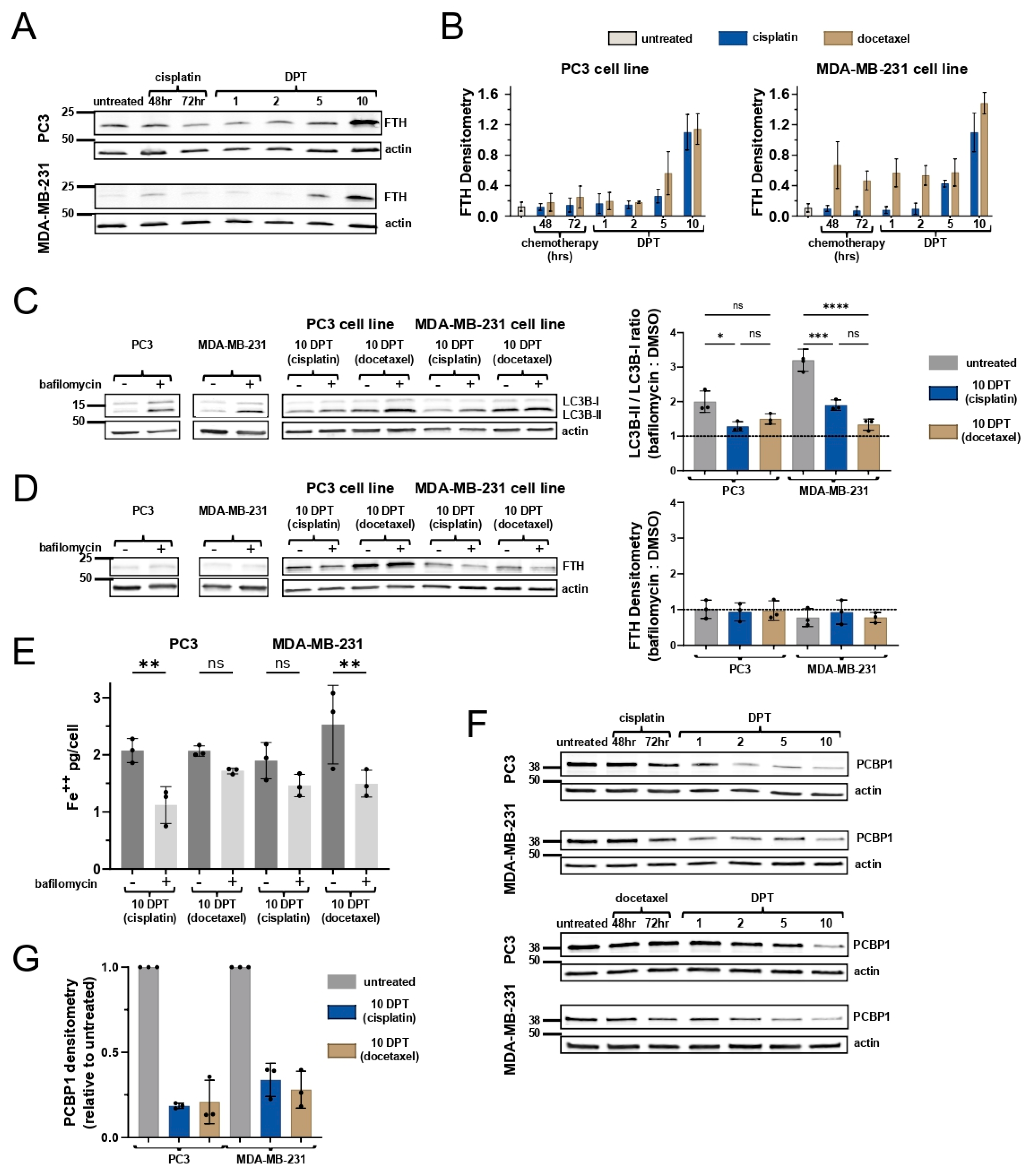
2.2. Ferritin Expression Does Not Attenuate Labile Iron Levels
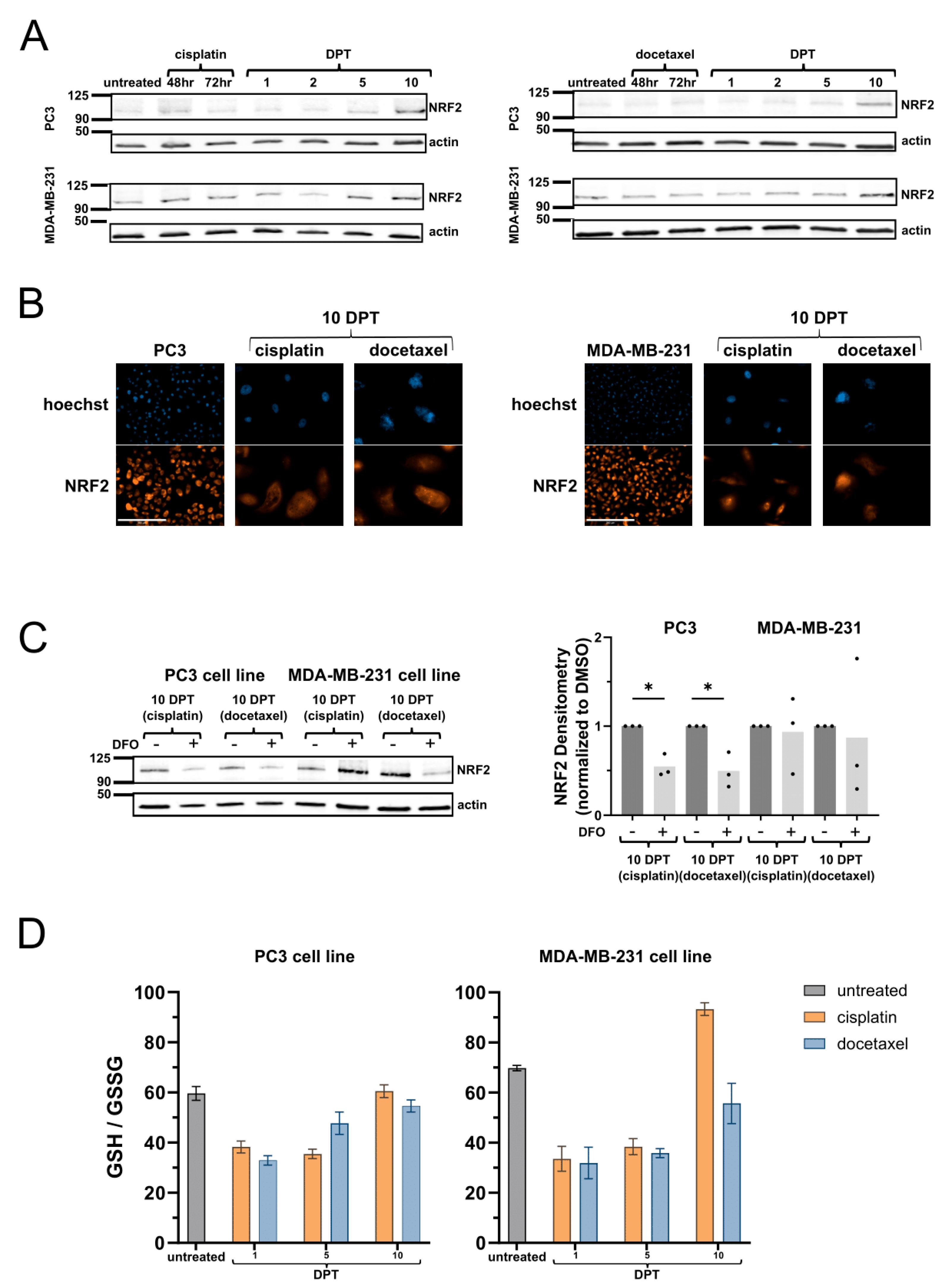
2.3. Surviving Cells Have Increased NRF2-Mediated Antioxidant Response
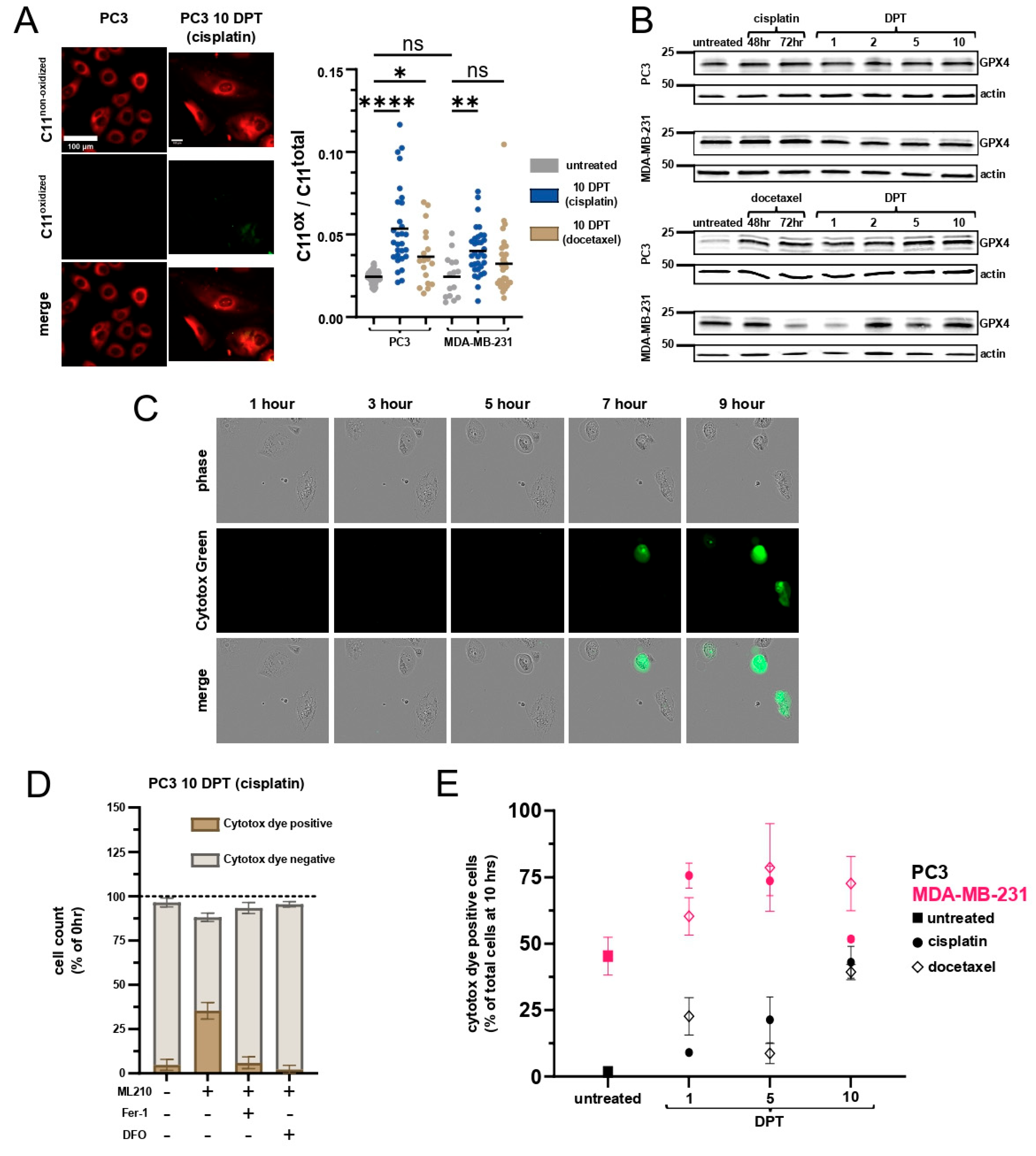
2.4. Cells Surviving Chemotherapy Are Vulnerable to Ferroptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Generation of Surviving Cells
4.2. Drug Treatments
4.3. Labile Iron Live Cell Imaging
4.4. Labile Iron Assay from Lysates
4.5. Western Blot
4.6. GSH/GSSG Assay
4.7. C11 BODIPY Analysis
4.8. Ferroptosis Cell Death Assay
4.9. Release of Immunomodulatory Markers During Ferroptosis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loftus, L.V.; Amend, S.R.; Pienta, K.J. Interplay between Cell Death and Cell Proliferation Reveals New Strategies for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 4723. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Olzmann, J.A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell Biol. 2024, 25, 424–442. [Google Scholar] [CrossRef]
- Minotti, G.; Aust, S.D. The role of iron in the initiation of lipid peroxidation. Chem. Phys. Lipids 1987, 44, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.-C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Dixon, S.J.; Pratt, D.A. Ferroptosis: A flexible constellation of related biochemical mechanisms. Mol. Cell 2023, 83, 1030–1042. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; A Valenzuela, C.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Camprubi, E.; Jordan, S.F.; Vasiliadou, R.; Lane, N. Iron catalysis at the origin of life. IUBMB Life 2017, 69, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Synthesis and breakdown of universal metabolic precursors promoted by iron. Nature 2019, 569, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.C.; Preiner, M.; Xavier, J.C.; Zimorski, V.; Martin, W.F. The last universal common ancestor between ancient Earth chemistry and the onset of genetics. PLoS Genet. 2018, 14, e1007518. [Google Scholar] [CrossRef]
- Wade, J.; Byrne, D.J.; Ballentine, C.J.; Drakesmith, H. Temporal variation of planetary iron as a driver of evolution. Proc. Natl. Acad. Sci. USA 2021, 118, e2109865118. [Google Scholar] [CrossRef]
- Posey, J.E.; Gherardini, F.C. Lack of a role for iron in the Lyme disease pathogen. Science 2000, 288, 1651–1653. [Google Scholar] [CrossRef]
- Andreini, C.; Putignano, V.; Rosato, A.; Banci, L. The human iron-proteome. Metallomics 2018, 10, 1223–1231. [Google Scholar] [CrossRef]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2023, 25, 133–155. [Google Scholar] [CrossRef]
- Zhang, C. Essential functions of iron-requiring proteins in DNA replication, repair and cell cycle control. Protein Cell 2014, 5, 750–760. [Google Scholar] [CrossRef]
- Eid, R.; Arab, N.T.; Greenwood, M.T. Iron mediated toxicity and programmed cell death: A review and a re-examination of existing paradigms. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 399–430. [Google Scholar] [CrossRef]
- Eaton, J.W.; Qian, M. Molecular bases of cellular iron toxicity. Free. Radic. Biol. Med. 2002, 32, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.; Aviles, M.V.; Chen, Y.L.; Latunde-Dada, G.O. The Role of GSH in Intracellular Iron Trafficking. Int. J. Mol. Sci. 2021, 22, 1278. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Protchenko, O.; Wang, Y.; Novoa-Aponte, L.; Leon-Torres, A.; Grounds, S.; Tietgens, A.J. Iron-tracking strategies: Chaperones capture iron in the cytosolic labile iron pool. Front. Mol. Biosci. 2023, 10, 1127690. [Google Scholar] [CrossRef]
- Andrews, S.C. The Ferritin-like superfamily: Evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim. Biophys. Acta 2010, 1800, 691–705. [Google Scholar] [CrossRef]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef]
- Patel, S.J.; Frey, A.G.; Palenchar, D.J.; Achar, S.; Bullough, K.Z.; Vashisht, A.; Wohlschlegel, J.A.; Philpott, C.C. A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat. Chem. Biol. 2019, 15, 872–881. [Google Scholar] [CrossRef]
- Kim, C.-J.; Gonye, A.L.; Truskowski, K.; Lee, C.-F.; Cho, Y.-K.; Austin, R.H.; Pienta, K.J.; Amend, S.R. Nuclear morphology predicts cell survival to cisplatin chemotherapy. Neoplasia 2023, 42, 100906. [Google Scholar] [CrossRef]
- Schmidt, M.J.; Naghdloo, A.; Prabakar, R.K.; Kamal, M.; Cadaneanu, R.; Garraway, I.P.; Lewis, M.; Aparicio, A.; Zurita-Saavedra, A.; Corn, P.; et al. Polyploid cancer cells reveal signatures of chemotherapy resistance. Oncogene 2024, 44, 439–449. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef]
- Patel, S.J.; Protchenko, O.; Shakoury-Elizeh, M.; Baratz, E.; Jadhav, S.; Philpott, C.C. The iron chaperone and nucleic acid-binding activities of poly(rC)-binding protein 1 are separable and independently essential. Proc. Natl. Acad. Sci. USA 2021, 118, e2104666118. [Google Scholar] [CrossRef]
- Protchenko, O.; Baratz, E.; Jadhav, S.; Li, F.; Shakoury-Elizeh, M.; Gavrilova, O.; Ghosh, M.C.; Cox, J.E.; Maschek, J.A.; Tyurin, V.A.; et al. Iron Chaperone Poly rC Binding Protein 1 Protects Mouse Liver From Lipid Peroxidation and Steatosis. Hepatology 2021, 73, 1176–1193. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Daiber, A. (Eds.) Environmental Stressors and OxInflammatory Tissues Responses; CRC Press: Boca Raton, FL, USA, 2023; ISBN 1032357002/978-1032357003. [Google Scholar]
- Buha, A.; Baralić, K.; Djukic-Cosic, D.; Bulat, Z.; Tinkov, A.; Panieri, E.; Saso, L. The Role of Toxic Metals and Metalloids in Nrf2 Signaling. Antioxidants 2021, 10, 630. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Kumar, H.; Aldridge, K.; Rieger, S.; Han, E.; Jiang, E.; Chan, E.R.; Soliman, A.; Mahdi, H.; Letterio, J.J. A Core NRF2 Gene Set Defined Through Comprehensive Transcriptomic Analysis Predicts Selective Drug Resistance and Poor Multicancer Prognosis. Antioxidants Redox Signal. 2024, 41, 1031–1050. [Google Scholar] [CrossRef]
- Hamed, M.Y.; Silver, J. Studies on the Reactions of Ferric Iron with Glutathione and Some Related Thiols. 2. Complex-Formation in the Ph Range 3 to 7. Inorg. Chim. A-Bioinor. 1983, 80, 115–122. [Google Scholar] [CrossRef]
- Hider, R.C.; Kong, X.L. Glutathione: A key component of the cytoplasmic labile iron pool. Biometals 2011, 24, 1179–1187. [Google Scholar] [CrossRef]
- Kumar, C.; Igbaria, A.; D’Autreaux, B.; Planson, A.-G.; Junot, C.; Godat, E.; Bachhawat, A.K.; Delaunay-Moisan, A.; Toledano, M.B. Glutathione revisited: A vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011, 30, 2044–2056. [Google Scholar] [CrossRef]
- Sánchez-Alcázar, J.; Villalón-García, I.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Talaverón-Rey, M.; Suárez-Rivero, J.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration. Neural Regen. Res. 2023, 18, 1196–1202. [Google Scholar] [CrossRef]
- Weiwer, M.; Bittker, J.A.; Lewis, T.A.; Shimada, K.; Yang, W.S.; MacPherson, L.; Dandapani, S.; Palmer, M.; Stockwell, B.R.; Schreiber, S.L.; et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 2012, 22, 1822–1826. [Google Scholar] [CrossRef]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Travain, V.B.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Hollestelle, A.; Elstrodt, F.; Nagel, J.H.; Kallemeijn, W.W.; Schutte, M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol. Cancer Res. 2007, 5, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Rao, Z.; Zhang, J.; Gollowitzer, A.; Siebenkäs, K.; Bindel, N.; D’avanzo, E.; van Roey, R.; Hajjaj, Y.; Özel, E.; et al. Zeb1 mediates EMT/plasticity-associated ferroptosis sensitivity in cancer cells by regulating lipogenic enzyme expression and phospholipid composition. Nat. Cell Biol. 2024, 26, 1470–1481. [Google Scholar] [CrossRef]
- Catanzaro, E.; Demuynck, R.; Naessens, F.; Galluzzi, L.; Krysko, D.V. Immunogenicity of ferroptosis in cancer: A matter of context? Trends Cancer 2024, 10, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Howley, B.V.; Howe, P.H. TGF-beta signaling in cancer: Post-transcriptional regulation of EMT via hnRNP E1. Cytokine 2019, 118, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.Y.; Lei, C.Q.; Shu, H.B. PCBP1 modulates the innate immune response by facilitating the binding of cGAS to DNA. Cell. Mol. Immunol. 2021, 18, 2334–2343. [Google Scholar] [CrossRef]
- Taylor, K.E.; Miller, L.G.; Contreras, L.M. RNA-binding proteins that preferentially interact with 8-oxoG-modified RNAs: Our current understanding. Biochem. Soc. Trans. 2024, 52, 111–122. [Google Scholar] [CrossRef]
- Saraev, D.D.; Pratt, D.A. Reactions of lipid hydroperoxides and how they may contribute to ferroptosis sensitivity. Curr. Opin. Chem. Biol. 2024, 81, 102478. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Mason, A.B.; Ponka, P. The long history of iron in the Universe and in health and disease. Biochim. Biophys. Acta 2012, 1820, 161–187. [Google Scholar] [CrossRef]
- Levi, S.; Ripamonti, M.; Moro, A.S.; Cozzi, A. Iron imbalance in neurodegeneration. Mol. Psychiatry 2024, 29, 1139–1152. [Google Scholar] [CrossRef]
- Rodriguez, R.; Cañeque, T.; Baron, L.; Müller, S.; Carmona, A.; Colombeau, L.; Versini, A.; Sabatier, M.; Sampaio, J.; Mishima, E.; et al. Activation of lysosomal iron triggers ferroptosis in cancer. Res. Sq. 2024. [Google Scholar] [CrossRef]
- Antoszczak, M.; Müller, S.; Cañeque, T.; Colombeau, L.; Dusetti, N.; Santofimia-Castaño, P.; Gaillet, C.; Puisieux, A.; Iovanna, J.L.; Rodriguez, R. Iron-Sensitive Prodrugs That Trigger Active Ferroptosis in Drug-Tolerant Pancreatic Cancer Cells. J. Am. Chem. Soc. 2022, 144, 11536–11545. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Allevato, M.; Trinh, S.; Koshizuka, K.; Nachmanson, D.; Nguyen, T.-T.C.; Yokoyama, Y.; Wu, X.; Andres, A.; Wang, Z.; Watrous, J.; et al. A genome-wide CRISPR screen reveals that antagonism of glutamine metabolism sensitizes head and neck squamous cell carcinoma to ferroptotic cell death. Cancer Lett. 2024, 598, 217089. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loftus, L.V.; Rolle, L.T.A.; Wang, B.; Pienta, K.J.; Amend, S.R. Dysregulation of Labile Iron Predisposes Chemotherapy Resistant Cancer Cells to Ferroptosis. Int. J. Mol. Sci. 2025, 26, 4193. https://doi.org/10.3390/ijms26094193
Loftus LV, Rolle LTA, Wang B, Pienta KJ, Amend SR. Dysregulation of Labile Iron Predisposes Chemotherapy Resistant Cancer Cells to Ferroptosis. International Journal of Molecular Sciences. 2025; 26(9):4193. https://doi.org/10.3390/ijms26094193
Chicago/Turabian StyleLoftus, Luke V., Louis T. A. Rolle, Bowen Wang, Kenneth J. Pienta, and Sarah R. Amend. 2025. "Dysregulation of Labile Iron Predisposes Chemotherapy Resistant Cancer Cells to Ferroptosis" International Journal of Molecular Sciences 26, no. 9: 4193. https://doi.org/10.3390/ijms26094193
APA StyleLoftus, L. V., Rolle, L. T. A., Wang, B., Pienta, K. J., & Amend, S. R. (2025). Dysregulation of Labile Iron Predisposes Chemotherapy Resistant Cancer Cells to Ferroptosis. International Journal of Molecular Sciences, 26(9), 4193. https://doi.org/10.3390/ijms26094193