

PPARα Genetic Deletion Reveals Global Transcriptional Changes in the Brain and Exacerbates Cerebral Infarction in a Mouse Model of Stroke

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
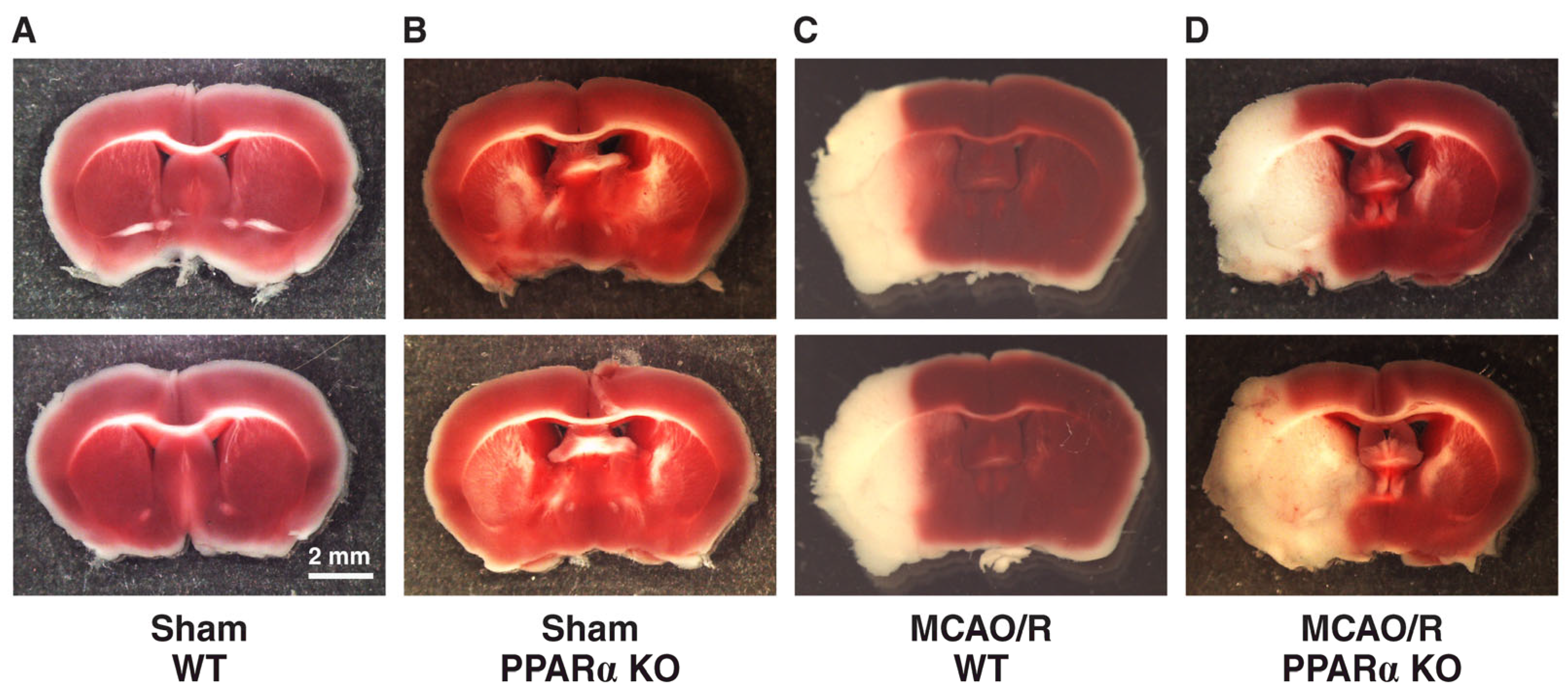
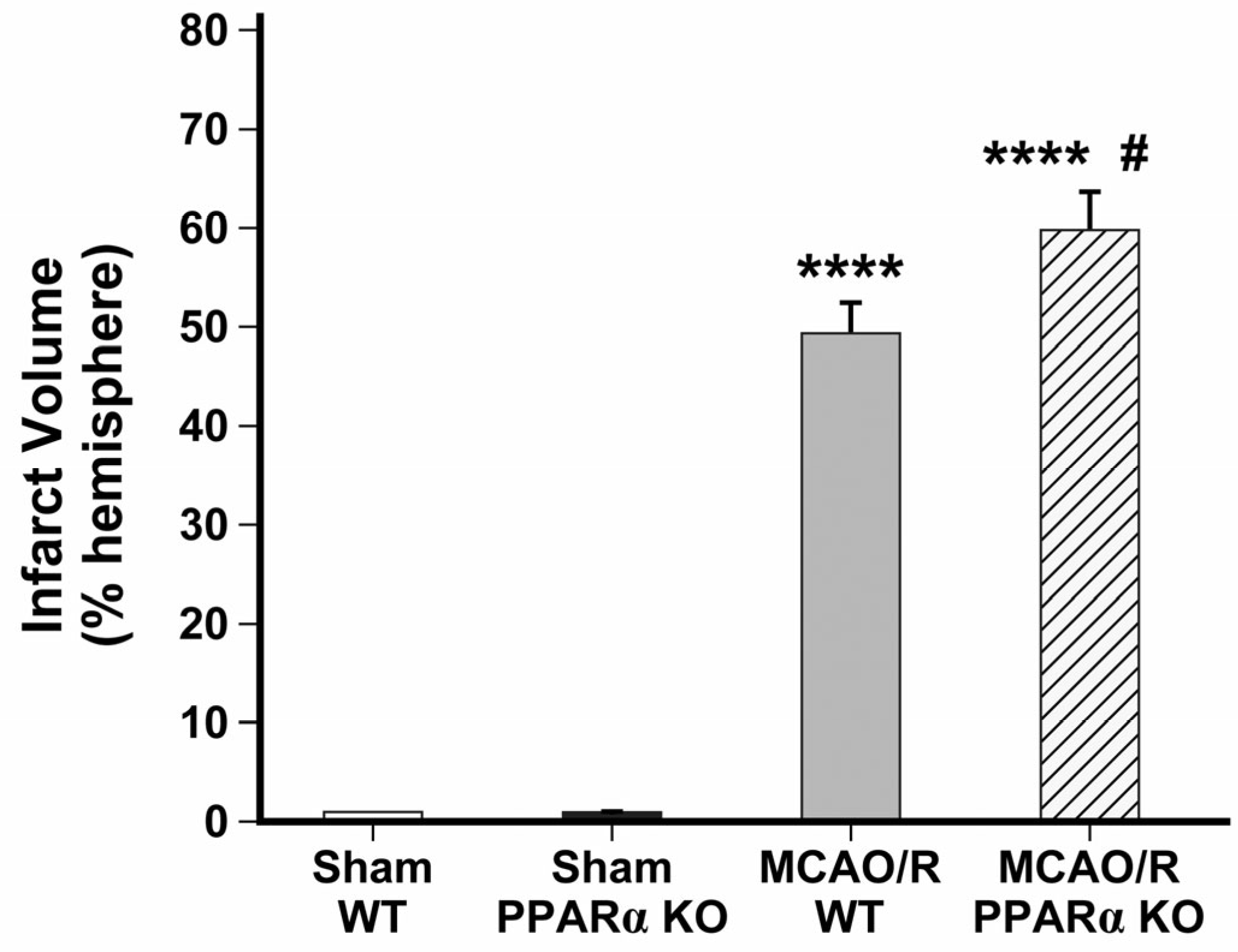
2.1. PPARα KO Increases Infarct Volume in Mouse Stroke Brains
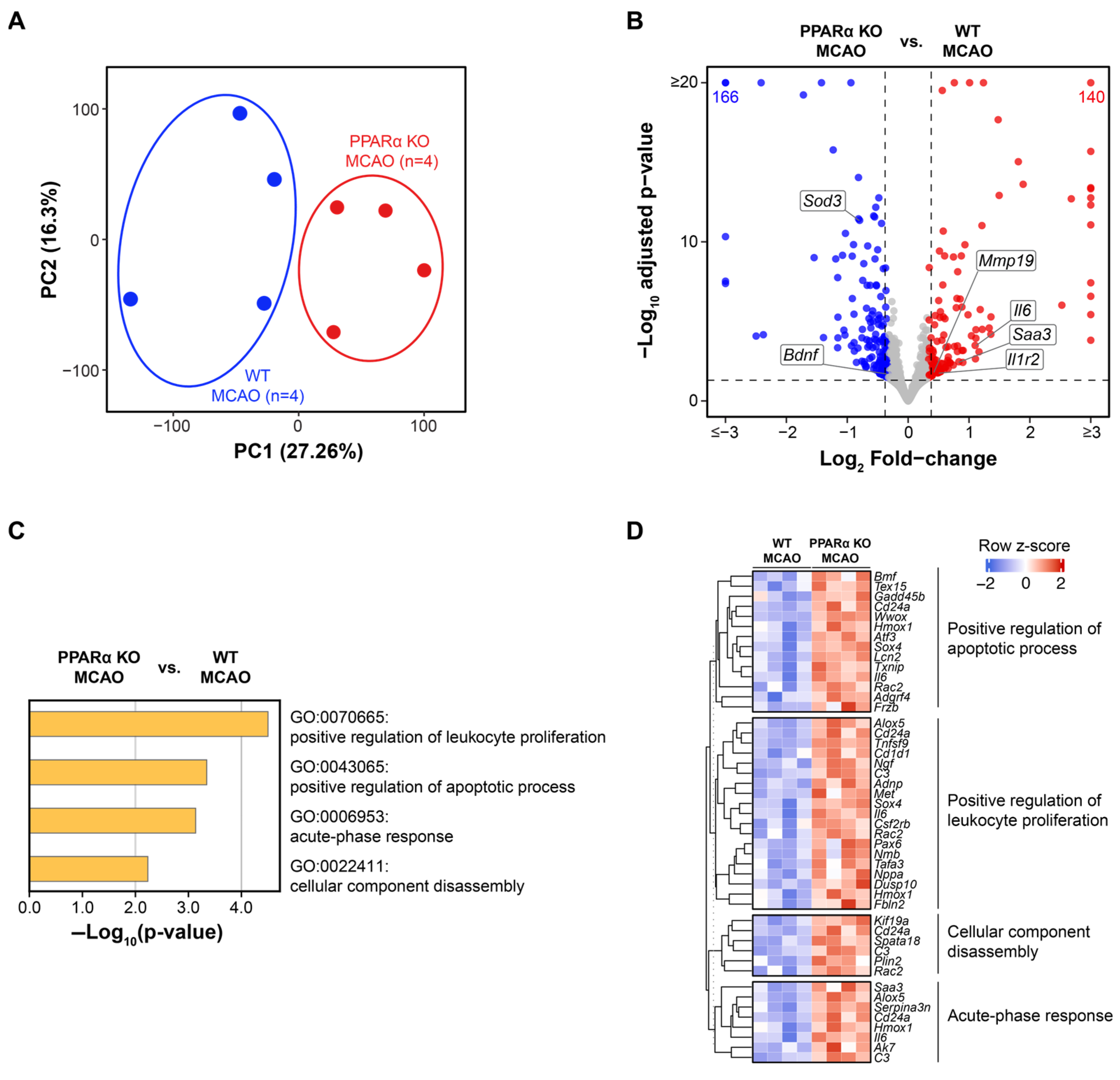
2.2. Genetic Deletion of PPARα Elicits Global Transcriptional Changes in Mouse Brains After Stroke
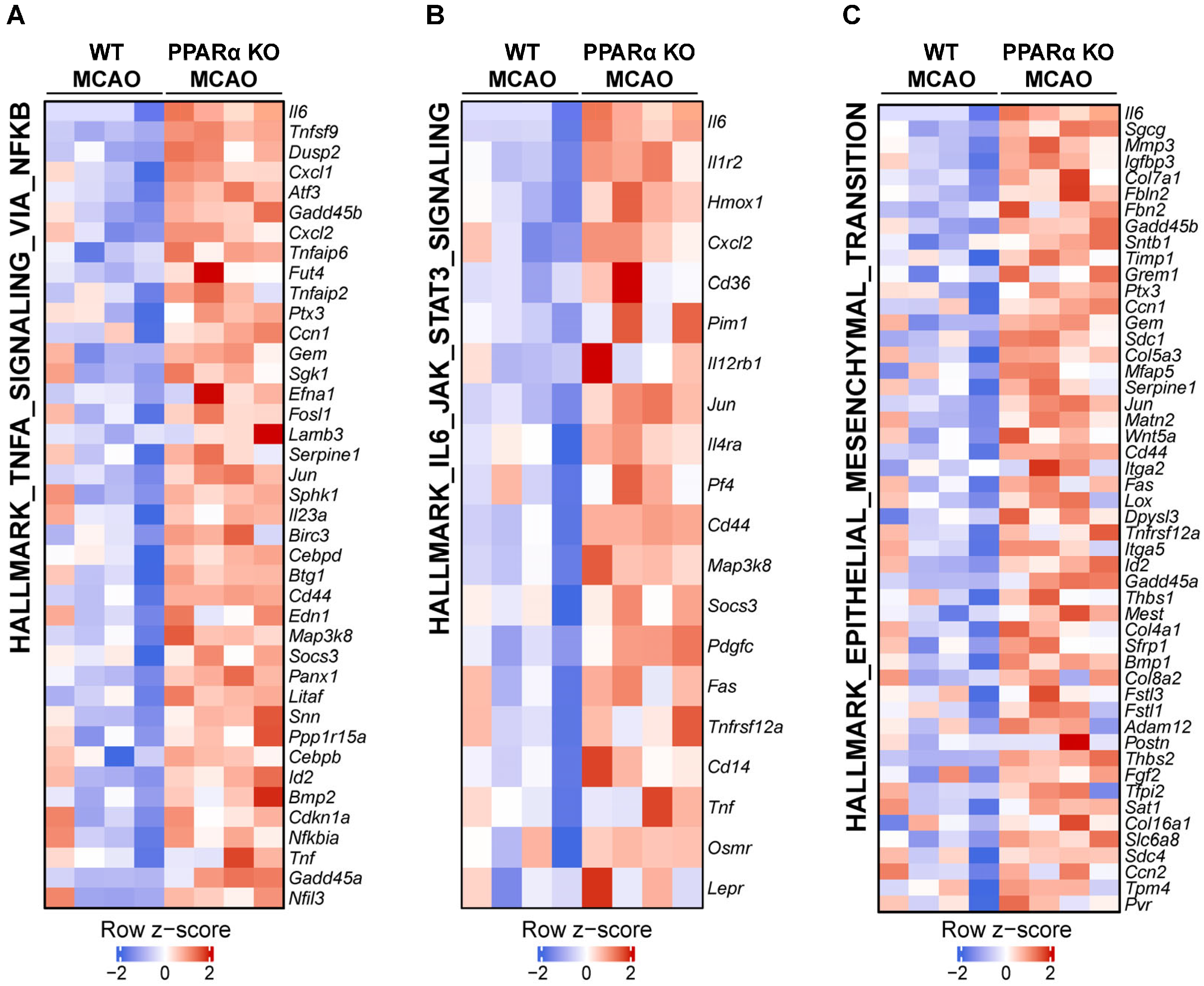
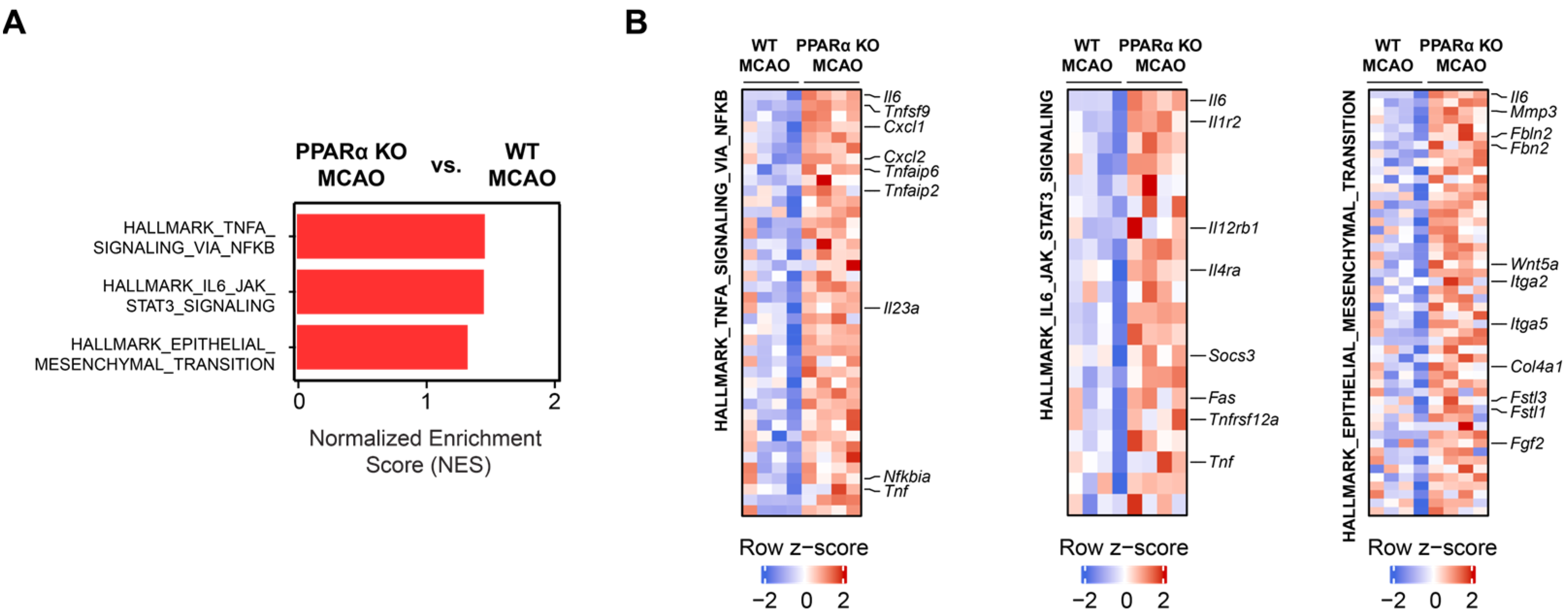
2.3. PPARα Deletion Alters Gene Expression Signatures for TNFα Signaling, IL6 Signaling, and Epithelial–Mesenchymal Transition (EMT) in Stroke Brains
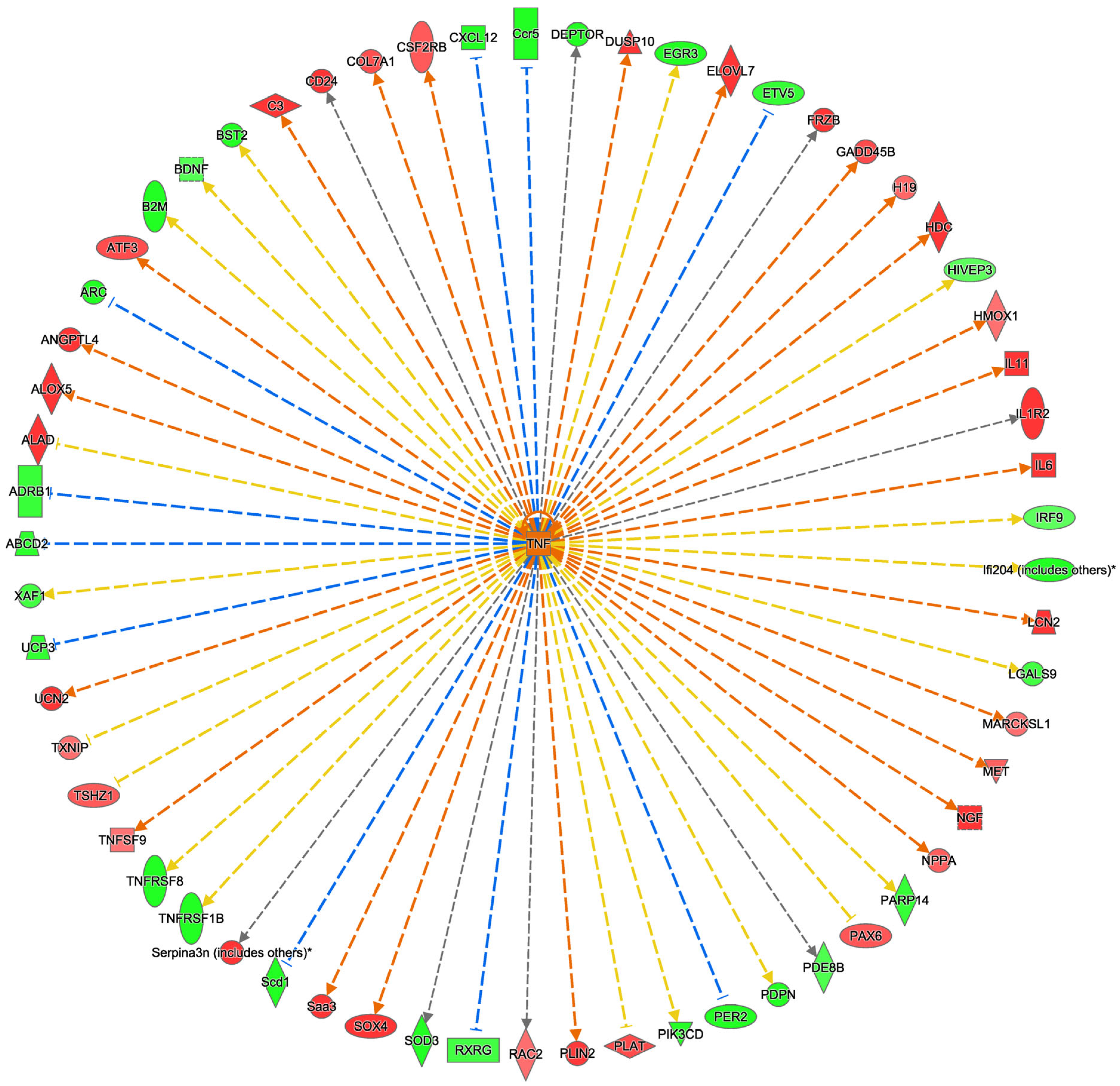
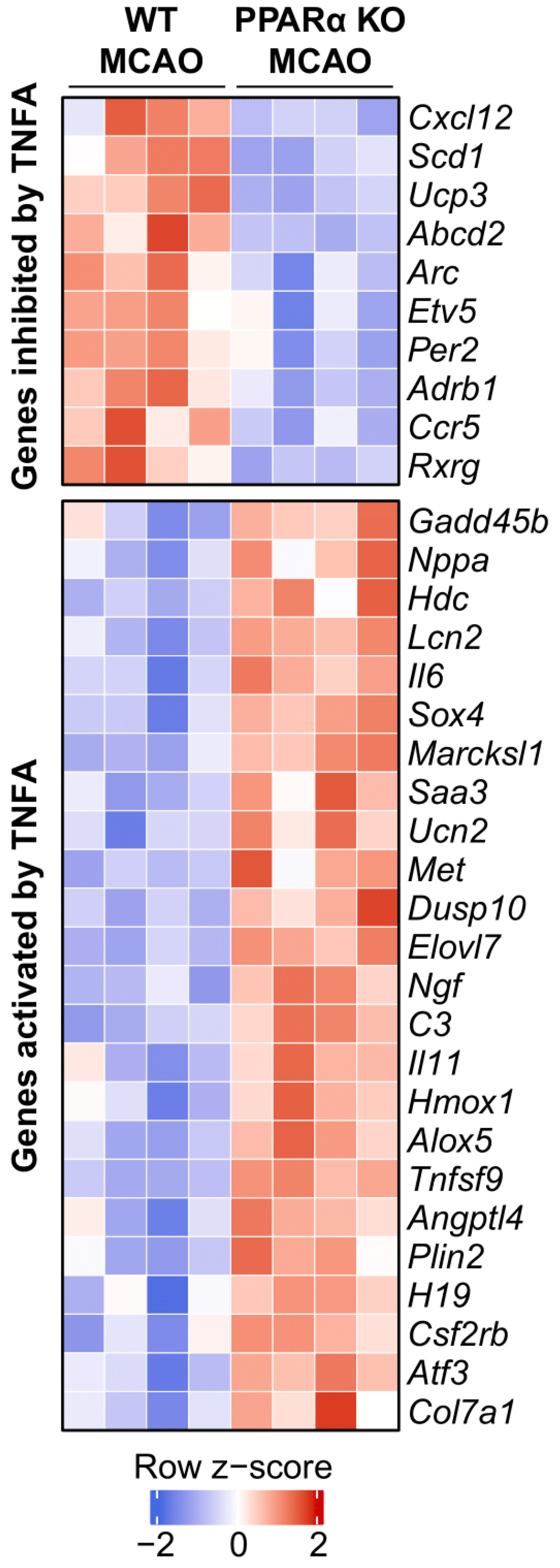
2.4. Ingenuity Pathway Analysis Reveals TNFα as an Upstream Regulator Affected by PPARα KO in Mouse Stroke Brains
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Mouse Model of Stroke
4.3. Infarct Volume Quantification
4.4. RNA Sequencing (RNA-Seq)
4.4.1. RNA-Seq Data Processing
4.4.2. Quality Control and Assurance (QC/QA) of RNA-Seq Data
4.4.3. Differential Gene Expression and Gene Set Enrichment Analysis (GSEA)
4.4.4. Pathway Analysis with Metascape and IPA
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Martin, S.S.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Barone Gibbs, B.; Beaton, A.Z.; Boehme, A.K.; et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data from the American Heart Association. Circulation 2024, 149, e347–e913. [Google Scholar] [CrossRef]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef]
- Wang, J.; Fields, J.; Zhao, C.; Langer, J.; Thimmulappa, R.K.; Kensler, T.W.; Yamamoto, M.; Biswal, S.; Doré, S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic. Biol. Med. 2007, 43, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.I.; Ali, Z.; Suri, M.F.; Shuaib, A.; Baker, G.; Todd, K.; Guterman, L.R.; Hopkins, L.N. Extracellular glutamate and other amino acids in experimental intracerebral hemorrhage: An in vivo microdialysis study. Crit. Care Med. 2003, 31, 1482–1489. [Google Scholar] [CrossRef]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 2010, 1802, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARα: Energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal. 2010, 8, e002. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.S.; McCaw, S.E.; Marcus, S.L.; Rachubinski, R.A.; Capone, J.P. The peroxisome proliferator-activated receptor interacts with the retinoid X receptor in vivo. Gene 1994, 148, 327–330. [Google Scholar] [CrossRef]
- Hamblin, M.; Chang, L.; Fan, Y.; Zhang, J.; Chen, Y.E. PPARs and the cardiovascular system. Antioxid. Redox Signal. 2009, 11, 1415–1452. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef]
- Khuchua, Z.; Glukhov, A.I.; Strauss, A.W.; Javadov, S. Elucidating the Beneficial Role of PPAR Agonists in Cardiac Diseases. Int. J. Mol. Sci. 2018, 19, 3464. [Google Scholar] [CrossRef]
- Deplanque, D.; Gelé, P.; Pétrault, O.; Six, I.; Furman, C.; Bouly, M.; Nion, S.; Dupuis, B.; Leys, D.; Fruchart, J.C.; et al. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J. Neurosci. 2003, 23, 6264–6271. [Google Scholar] [CrossRef] [PubMed]
- Ouk, T.; Laprais, M.; Bastide, M.; Mostafa, K.; Gautier, S.; Bordet, R. Withdrawal of fenofibrate treatment partially abrogates preventive neuroprotection in stroke via loss of vascular protection. Vascul. Pharmacol. 2009, 51, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, X.; Guo, Q.; Namura, S. Chronic treatment with fibrates elevates superoxide dismutase in adult mouse brain microvessels. Brain Res. 2010, 1359, 247–255. [Google Scholar] [CrossRef]
- Matzen, J.S.; Krogh, C.L.; Forman, J.L.; Garred, P.; Møller, K.; Bache, S. Lectin complement pathway initiators after subarachnoid hemorrhage—An observational study. J. Neuroinflamm. 2020, 17, 338. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.G.; Chen, Y.; Long, D.H.; Zhang, M.; Ji, W.D.; Zhang, W.J.; Liu, J.H.; Hong, L.P.; He, X.S.; Chen, W.L. PPARα Agonist Fenofibrate Ameliorates Learning and Memory Deficits in Rats Following Global Cerebral Ischemia. Mol. Neurobiol. 2015, 52, 601–609. [Google Scholar] [CrossRef]
- Collino, M.; Aragno, M.; Mastrocola, R.; Benetti, E.; Gallicchio, M.; Dianzani, C.; Danni, O.; Thiemermann, C.; Fantozzi, R. Oxidative stress and inflammatory response evoked by transient cerebral ischemia/reperfusion: Effects of the PPAR-α agonist WY14643. Free Radic. Biol. Med. 2006, 41, 579–589. [Google Scholar] [CrossRef]
- Ouk, T.; Gautier, S.; Pétrault, M.; Montaigne, D.; Maréchal, X.; Masse, I.; Devedjian, J.C.; Deplanque, D.; Bastide, M.; Nevière, R.; et al. Effects of the PPAR-α agonist fenofibrate on acute and short-term consequences of brain ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 542–551. [Google Scholar] [CrossRef]
- Mysiorek, C.; Culot, M.; Dehouck, L.; Derudas, B.; Staels, B.; Bordet, R.; Cecchelli, R.; Fenart, L.; Berezowski, V. Peroxisome-proliferator-activated receptor-alpha activation protects brain capillary endothelial cells from oxygen-glucose deprivation-induced hyperpermeability in the blood-brain barrier. Curr. Neurovasc. Res. 2009, 6, 181–193. [Google Scholar] [CrossRef]
- Jenny, N.S.; Callas, P.W.; Judd, S.E.; McClure, L.A.; Kissela, B.; Zakai, N.A.; Cushman, M. Inflammatory cytokines and ischemic stroke risk: The REGARDS cohort. Neurology 2019, 92, e2375–e2384. [Google Scholar] [CrossRef]
- Yao, H.; Zhang, Y.; Shu, H.; Xie, B.; Tao, Y.; Yuan, Y.; Shang, Y.; Yuan, S.; Zhang, J. Hyperforin Promotes Post-stroke Neuroangiogenesis via Astrocytic IL-6-Mediated Negative Immune Regulation in the Ischemic Brain. Front. Cell. Neurosci. 2019, 13, 201. [Google Scholar] [CrossRef]
- Băcilă, C.-I.; Vlădoiu, M.-G.; Văleanu, M.; Moga, D.-F.-C.; Pumnea, P.-M. The Role of IL-6 and TNF-Alpha Biomarkers in Predicting Disability Outcomes in Acute Ischemic Stroke Patients. Life 2025, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Watters, O.; O’Connor, J.J. A role for tumor necrosis factor-α in ischemia and ischemic preconditioning. J. Neuroinflamm. 2011, 8, 87. [Google Scholar] [CrossRef]
- Clausen, B.H.; Wirenfeldt, M.; Høgedal, S.S.; Frich, L.H.; Nielsen, H.H.; Schrøder, H.D.; Østergaard, K.; Finsen, B.; Kristensen, B.W.; Lambertsen, K.L. Characterization of the TNF and IL-1 systems in human brain and blood after ischemic stroke. Acta Neuropathol. Commun. 2020, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Portier, I.; Rustad, J.L.; Cody, M.J.; de Araujo, C.V.; Hoki, C.; Alexander, M.D.; Grandhi, R.; Dyer, M.R.; Neal, M.D.; et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J. Clin. Investig. 2022, 132, e154225. [Google Scholar] [CrossRef]
- Hansen, C.E.; Hollaus, D.; Kamermans, A.; de Vries, H.E. Tension at the gate: Sensing mechanical forces at the blood–brain barrier in health and disease. J. Neuroinflamm. 2024, 21, 325. [Google Scholar] [CrossRef]
- Chen, D.; Li, L.; Wang, Y.; Xu, R.; Peng, S.; Zhou, L.; Deng, Z. Ischemia-reperfusion injury of brain induces endothelial-mesenchymal transition and vascular fibrosis via activating let-7i/TGF-βR1 double-negative feedback loop. FASEB J 2020, 34, 7178–7191. [Google Scholar] [CrossRef]
- Qin, C.; Yang, S.; Chu, Y.-H.; Zhang, H.; Pang, X.-W.; Chen, L.; Zhou, L.-Q.; Chen, M.; Tian, D.-S.; Wang, W. Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 215. [Google Scholar] [CrossRef]
- Inoue, I.; Shino, K.; Noji, S.; Awata, T.; Katayama, S. Expression of peroxisome proliferator-activated receptor α (PPAR α) in primary cultures of human vascular endothelial cells. Biochem. Biophys. Res. Commun. 1998, 246, 370–374. [Google Scholar] [CrossRef]
- Staels, B.; Koenig, W.; Habib, A.; Merval, R.; Lebret, M.; Torra, I.P.; Delerive, P.; Fadel, A.; Chinetti, G.; Fruchart, J.C.; et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature 1998, 393, 790–793. [Google Scholar] [CrossRef]
- Chinetti, G.; Griglio, S.; Antonucci, M.; Torra, I.P.; Delerive, P.; Majd, Z.; Fruchart, J.C.; Chapman, J.; Najib, J.; Staels, B. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 1998, 273, 25573–25580. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar] [CrossRef]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.M.; Chan, P.H. Superoxide Dismutases in Stroke. In Handbook of Neurochemistry and Molecular Neurobiology: Acute Ischemic Injury and Repair in the Nervous System; Lajtha, A., Chan, P.H., Eds.; Springer US: New York, NY, USA, 2007; pp. 121–144. [Google Scholar]
- Mai, N.; Miller-Rhodes, K.; Prifti, V.; Kim, M.; O’Reilly, M.A.; Halterman, M.W. Lung-Derived SOD3 Attenuates Neurovascular Injury After Transient Global Cerebral Ischemia. J. Am. Heart Assoc. 2019, 8, e011801. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X.; O’Connor, M.; Wang, G.; Han, F. Brain-Derived Neurotrophic Factor and Its Potential Therapeutic Role in Stroke Comorbidities. Neural Plast. 2020, 2020, 1969482. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, A.; Wu, J.; Mo, L.; Lu, F.; Wan, G. Il1r2 and Tnfrsf12a in transcranial magnetic stimulation effect of ischemic stroke via bioinformatics analysis. Medicine 2024, 103, e36109. [Google Scholar] [CrossRef]
- Oh, S.H.; Kim, O.J.; Shin, D.A.; Song, J.; Yoo, H.; Kim, Y.K.; Kim, J.K. Alteration of immunologic responses on peripheral blood in the acute phase of ischemic stroke: Blood genomic profiling study. J. Neuroimmunol. 2012, 249, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhu, Z.; Zang, Y.; Bu, X.; Xu, T.; Zhong, C.; Wang, A.; Peng, H.; Guo, D.; Zheng, X.; et al. Increased Serum Complement C3 Levels Are Associated With Adverse Clinical Outcomes After Ischemic Stroke. Stroke 2021, 52, 868–877. [Google Scholar] [CrossRef]
- Eriksson, U.; Kurrer, M.O.; Sebald, W.; Brombacher, F.; Kopf, M. Dual Role of the IL-12/IFN-γ Axis in the Development of Autoimmune Myocarditis: Induction by IL-12 and Protection by IFN-γ1. J. Immunol. 2001, 167, 5464–5469. [Google Scholar] [CrossRef]
- Ji, X.C.; Shi, Y.J.; Zhang, Y.; Chang, M.Z.; Zhao, G. Reducing Suppressors of Cytokine Signaling-3 (SOCS3) Expression Promotes M2 Macrophage Polarization and Functional Recovery After Intracerebral Hemorrhage. Front. Neurol. 2020, 11, 586905. [Google Scholar] [CrossRef]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wu, Y.; Zhang, R.; Xu, L.; Xie, F. TNFSF9 Silence Impedes Cerebral Ischemia–Reperfusion Injury via Modulating SLC3A2 Expression in Brain Microvascular Endothelial Cells. J. Mol. Neurosci. 2025, 75, 12. [Google Scholar] [CrossRef]
- Chen, A.-Q.; Fang, Z.; Chen, X.-L.; Yang, S.; Zhou, Y.-F.; Mao, L.; Xia, Y.-P.; Jin, H.-J.; Li, Y.-N.; You, M.-F.; et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain–barrier disruption after ischemic stroke. Cell Death Dis. 2019, 10, 487. [Google Scholar] [CrossRef]
- Xue, S.; Zhou, X.; Yang, Z.-H.; Si, X.-K.; Sun, X. Stroke-induced damage on the blood–brain barrier. Front. Neurol. 2023, 14, 1248970. [Google Scholar] [CrossRef] [PubMed]
- Broughton, B.R.S.; Reutens, D.C.; Sobey, C.G. Apoptotic Mechanisms After Cerebral Ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: The PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478. [Google Scholar] [CrossRef]
- Engel, O.; Kolodziej, S.; Dirnagl, U.; Prinz, V. Modeling Stroke in Mice—Middle Cerebral Artery Occlusion with the Filament Model. J. Vis. Exp. 2011, 47, e2423. [Google Scholar] [CrossRef]
- Huang, L.; Wong, S.; Snyder, E.Y.; Hamblin, M.H.; Lee, J.P. Human neural stem cells rapidly ameliorate symptomatic inflammation in early-stage ischemic-reperfusion cerebral injury. Stem Cell Res. Ther. 2014, 5, 129. [Google Scholar] [CrossRef]
- Eckert, A.; Huang, L.; Gonzalez, R.; Kim, H.S.; Hamblin, M.H.; Lee, J.P. Bystander Effect Fuels Human Induced Pluripotent Stem Cell-Derived Neural Stem Cells to Quickly Attenuate Early Stage Neurological Deficits After Stroke. Stem Cells Transl. Med. 2015, 4, 841–851. [Google Scholar] [CrossRef]
- Boese, A.C.; Eckert, A.; Hamblin, M.H.; Lee, J.P. Human neural stem cells improve early stage stroke outcome in delayed tissue plasminogen activator-treated aged stroke brains. Exp. Neurol. 2020, 329, 113275. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.H.; Boese, A.C.; Murad, R.; Lee, J.P. MMP-3 Knockout Induces Global Transcriptional Changes and Reduces Cerebral Infarction in Both Male and Female Models of Ischemic Stroke. Int. J. Mol. Sci. 2024, 25, 7383. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A.; Morton, M.T.; Tsao-Wu, G.; Savalos, R.A.; Davidson, C.; Sharp, F.R. A semiautomated method for measuring brain infarct volume. J. Cereb. Blood Flow Metab. 1990, 10, 290–293. [Google Scholar] [CrossRef]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef]
- Mudge, J.M.; Carbonell-Sala, S.; Diekhans, M.; Martinez, J.G.; Hunt, T.; Jungreis, I.; Loveland, J.E.; Arnan, C.; Barnes, I.; Bennett, R.; et al. GENCODE 2025: Reference gene annotation for human and mouse. Nucleic Acids Res. 2025, 53, D966–D975. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamblin, M.H.; Boese, A.C.; Murad, R.; Lee, J.-P. PPARα Genetic Deletion Reveals Global Transcriptional Changes in the Brain and Exacerbates Cerebral Infarction in a Mouse Model of Stroke. Int. J. Mol. Sci. 2025, 26, 4082. https://doi.org/10.3390/ijms26094082
Hamblin MH, Boese AC, Murad R, Lee J-P. PPARα Genetic Deletion Reveals Global Transcriptional Changes in the Brain and Exacerbates Cerebral Infarction in a Mouse Model of Stroke. International Journal of Molecular Sciences. 2025; 26(9):4082. https://doi.org/10.3390/ijms26094082
Chicago/Turabian StyleHamblin, Milton H., Austin C. Boese, Rabi Murad, and Jean-Pyo Lee. 2025. "PPARα Genetic Deletion Reveals Global Transcriptional Changes in the Brain and Exacerbates Cerebral Infarction in a Mouse Model of Stroke" International Journal of Molecular Sciences 26, no. 9: 4082. https://doi.org/10.3390/ijms26094082
APA StyleHamblin, M. H., Boese, A. C., Murad, R., & Lee, J.-P. (2025). PPARα Genetic Deletion Reveals Global Transcriptional Changes in the Brain and Exacerbates Cerebral Infarction in a Mouse Model of Stroke. International Journal of Molecular Sciences, 26(9), 4082. https://doi.org/10.3390/ijms26094082