
Genome-Wide Characterization, Comparative Analysis, and Expression Profiling of SWEET Genes Family in Four Cymbidium Species (Orchidaceae)

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
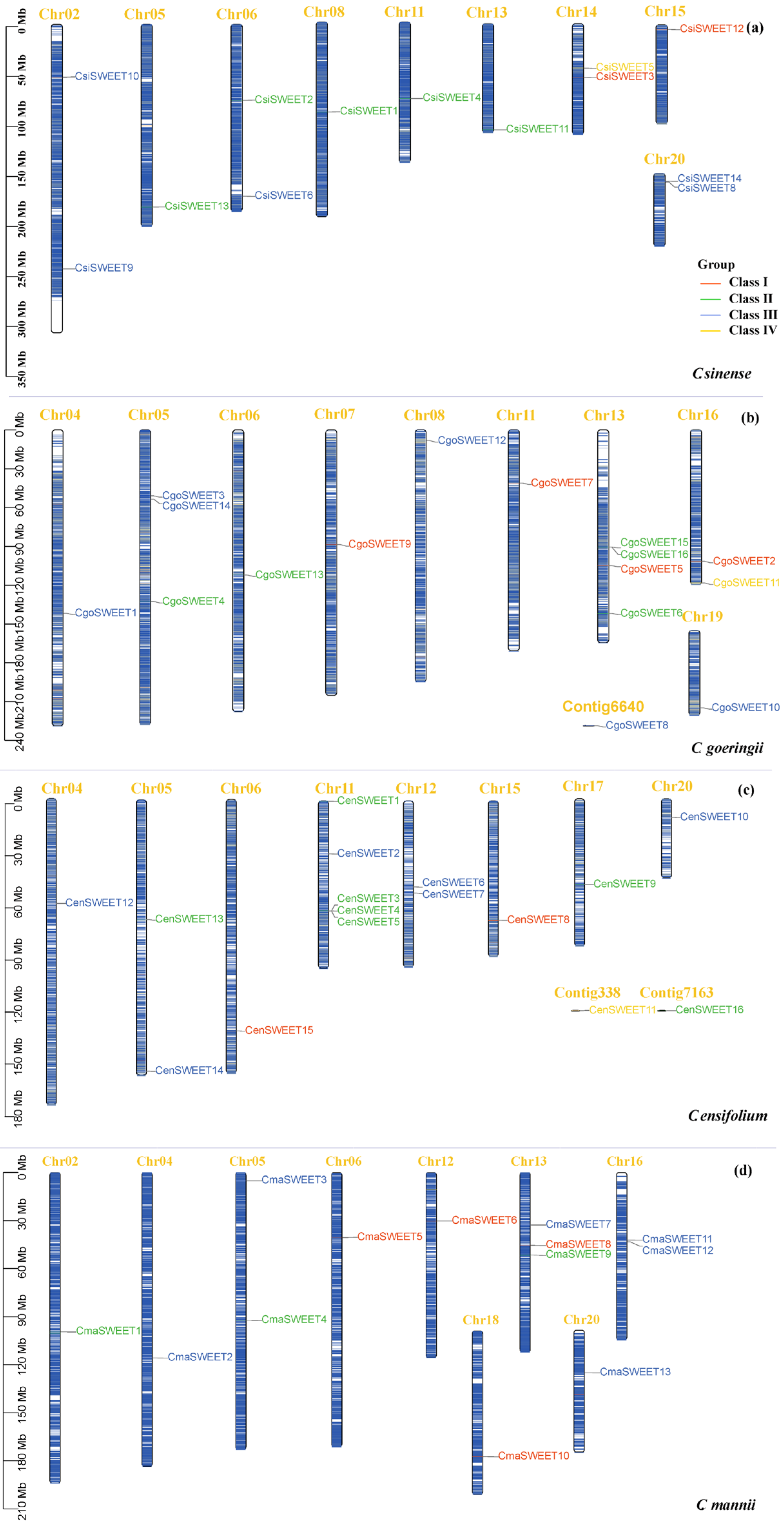
2.1. Identification and Characterization of SWEET Genes in Four Cymbidium Species Genomes
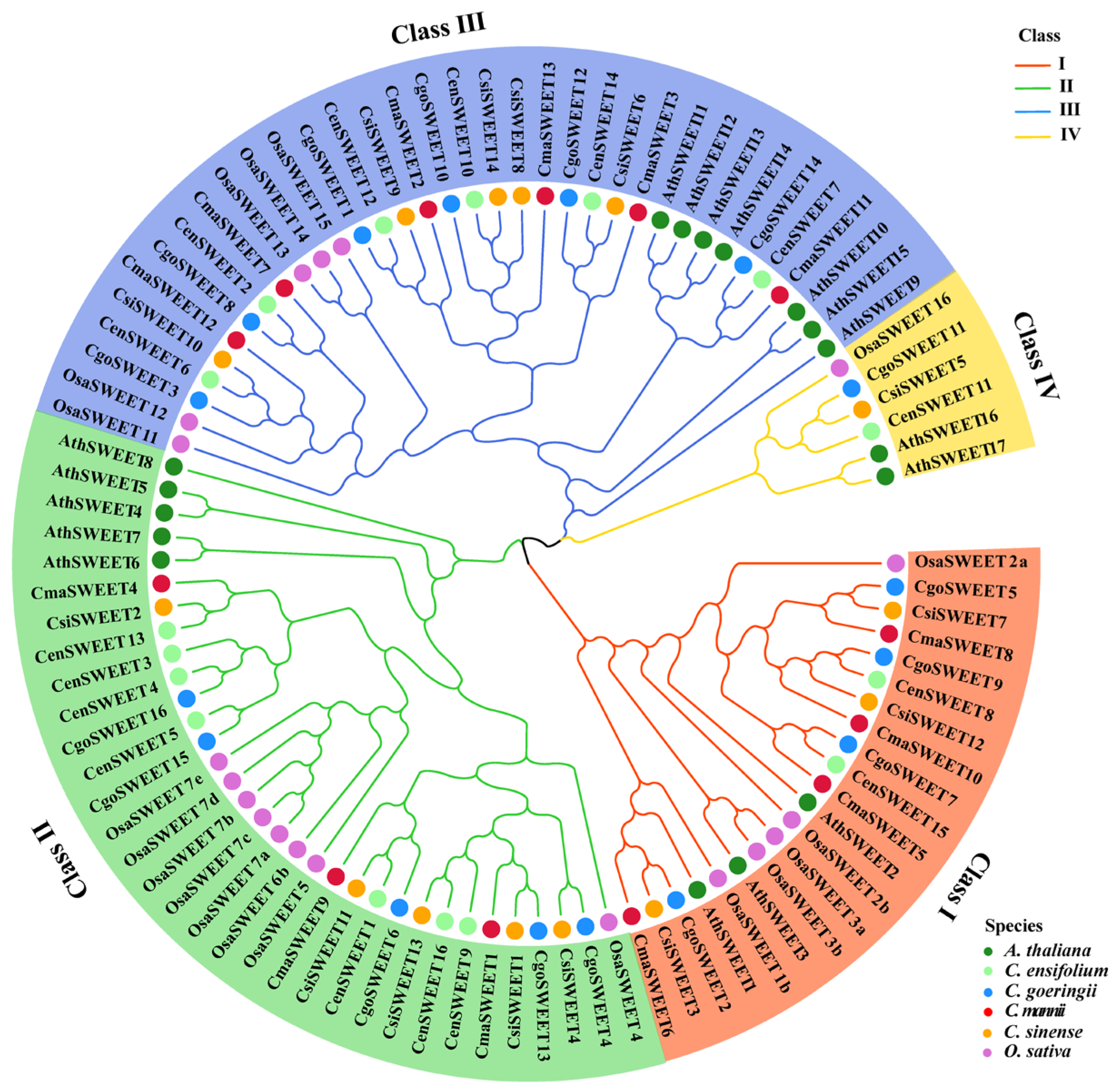
2.2. Phylogenetic Analysis of SWEET Family
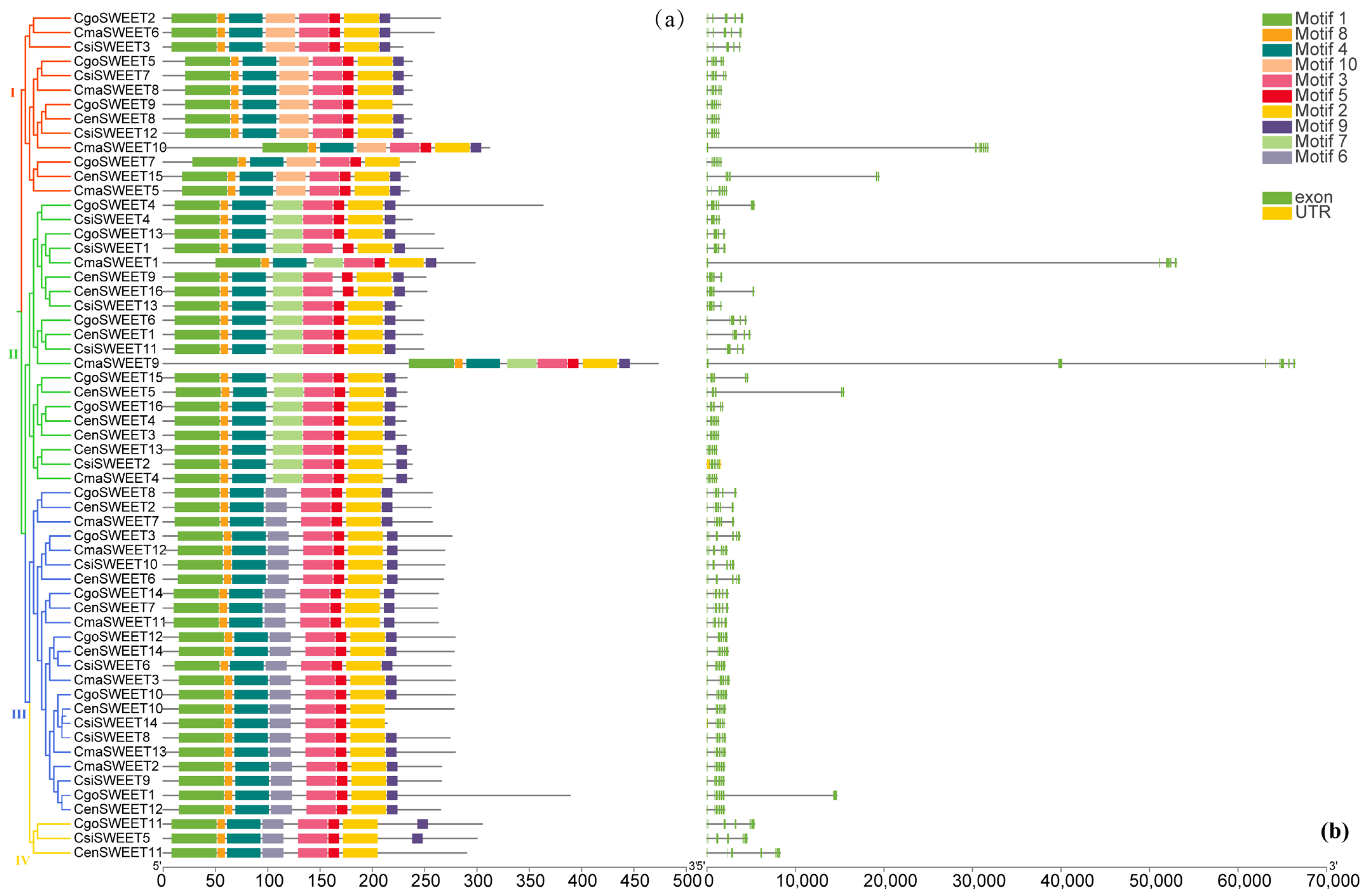
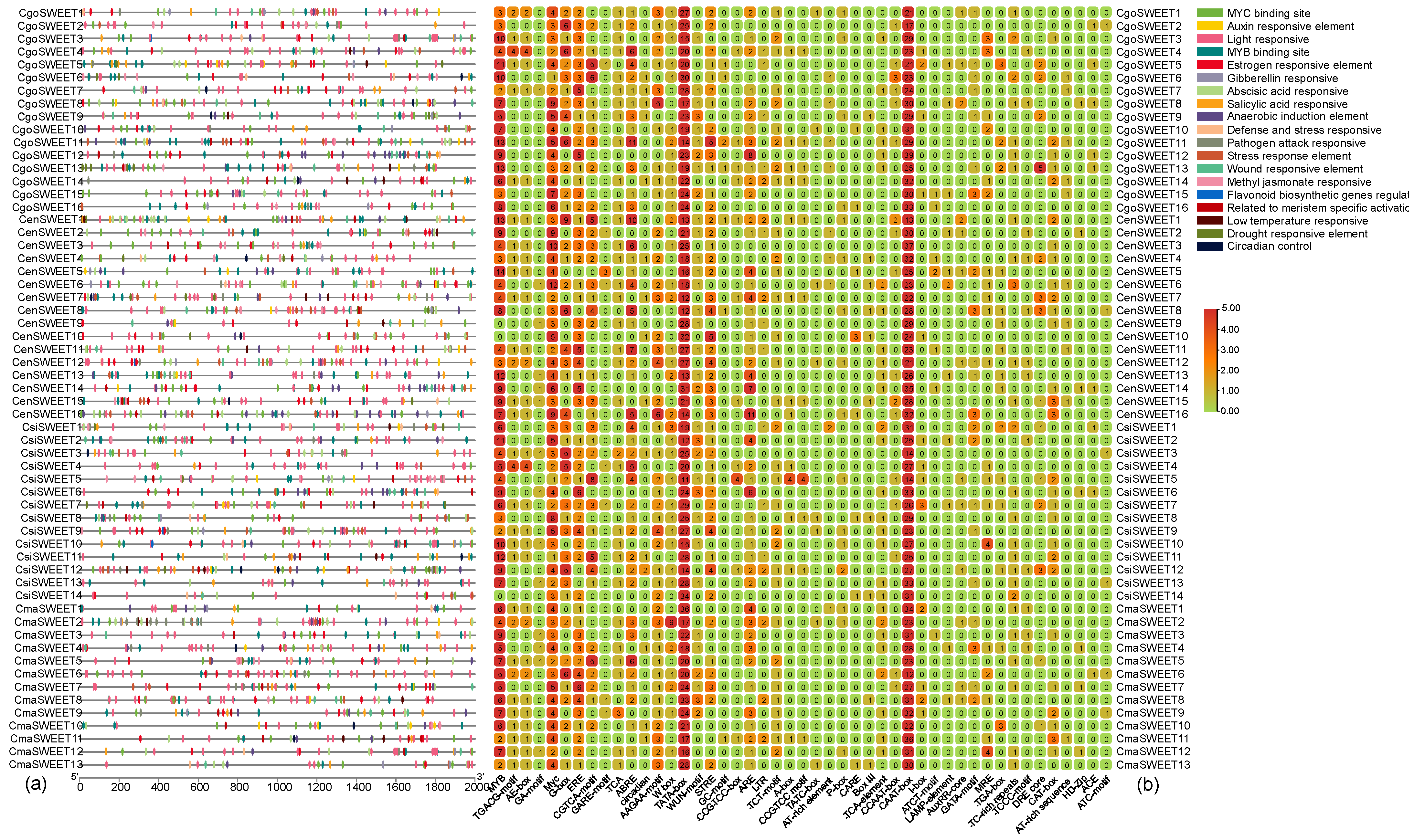
2.3. Gene Structure, Protein Motif, and Domains of SWEET Members in Cymbidium
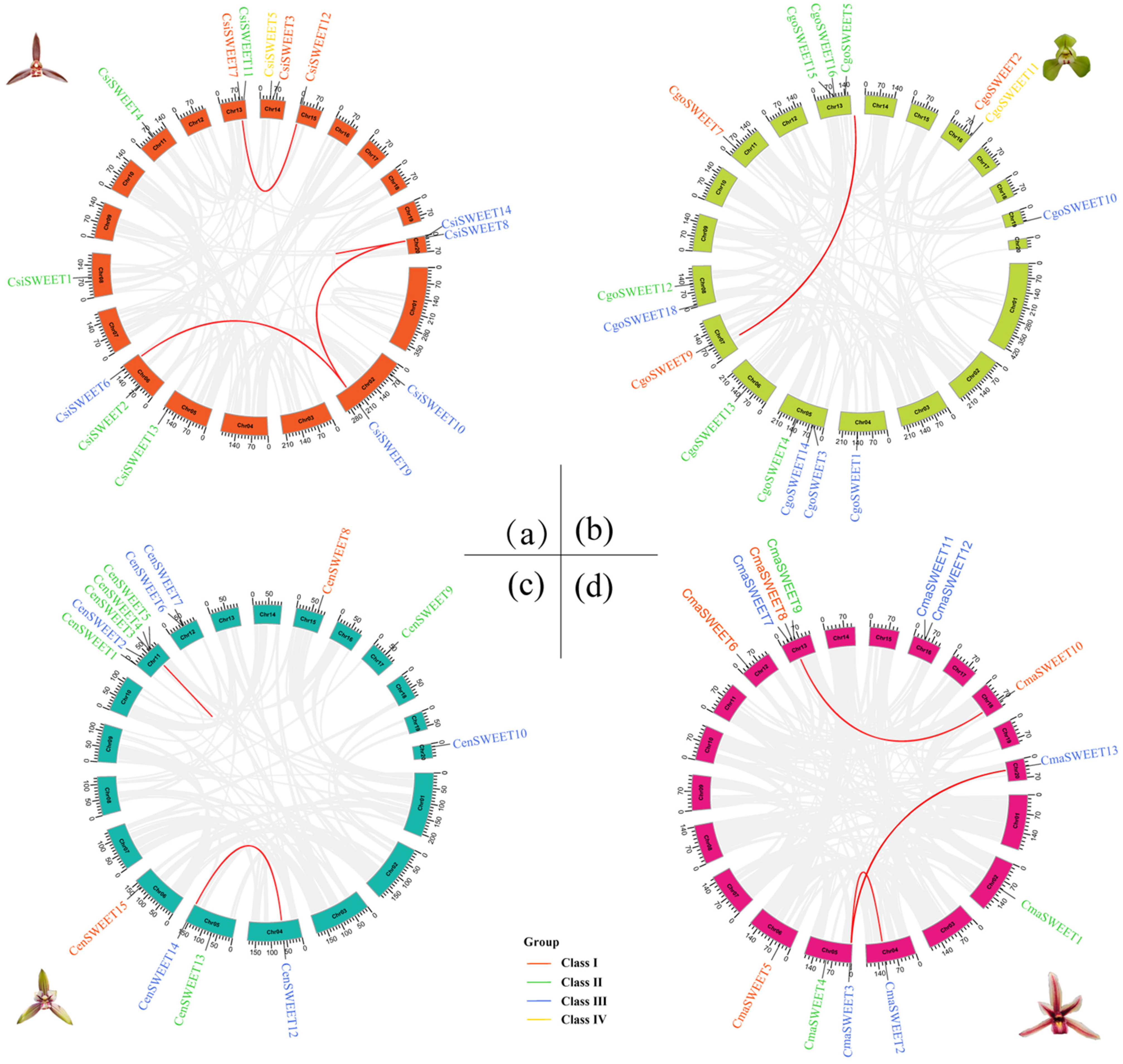
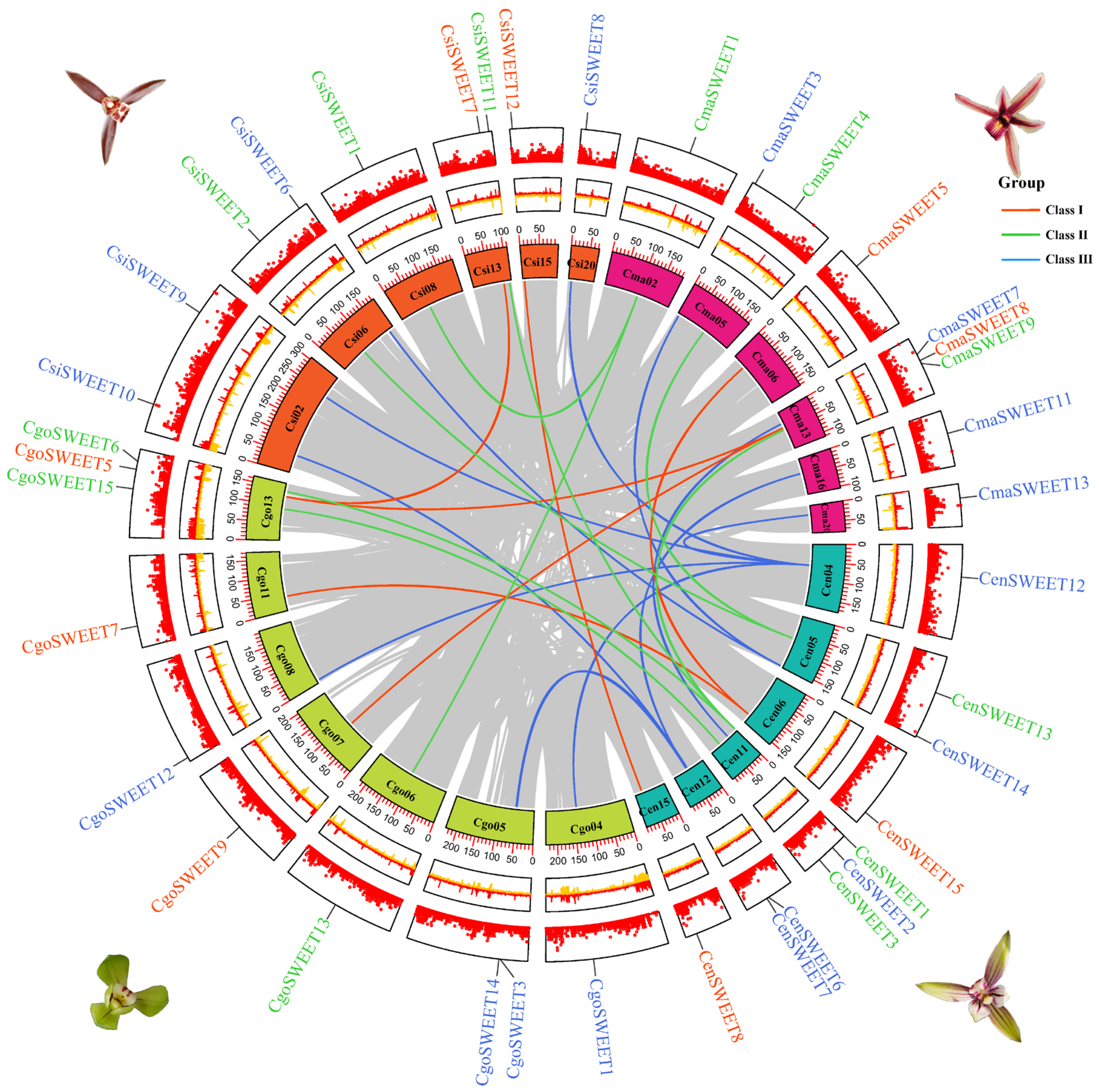
2.4. Duplication and Collinearity of SWEET Genes in Cymbidium
2.5. Analysis of Cis-Acting Elements Analysis of SWEET Genes in Cymbidium
2.6. Expression Patterns of SWEET Genes in Cymbidium
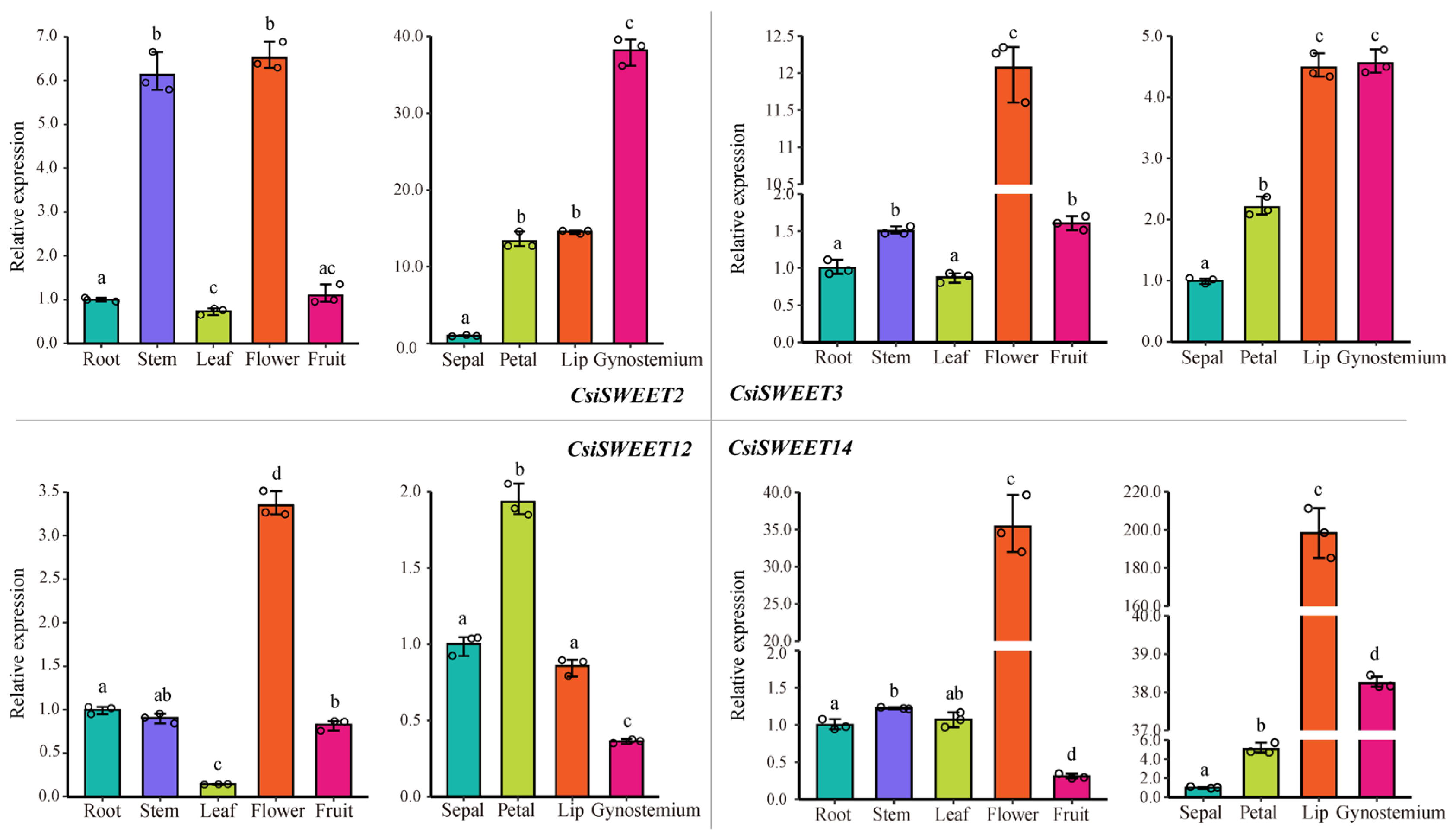
2.7. Evaluation of CsiSWEET Gene Expression Using qRT–PCR Analysis
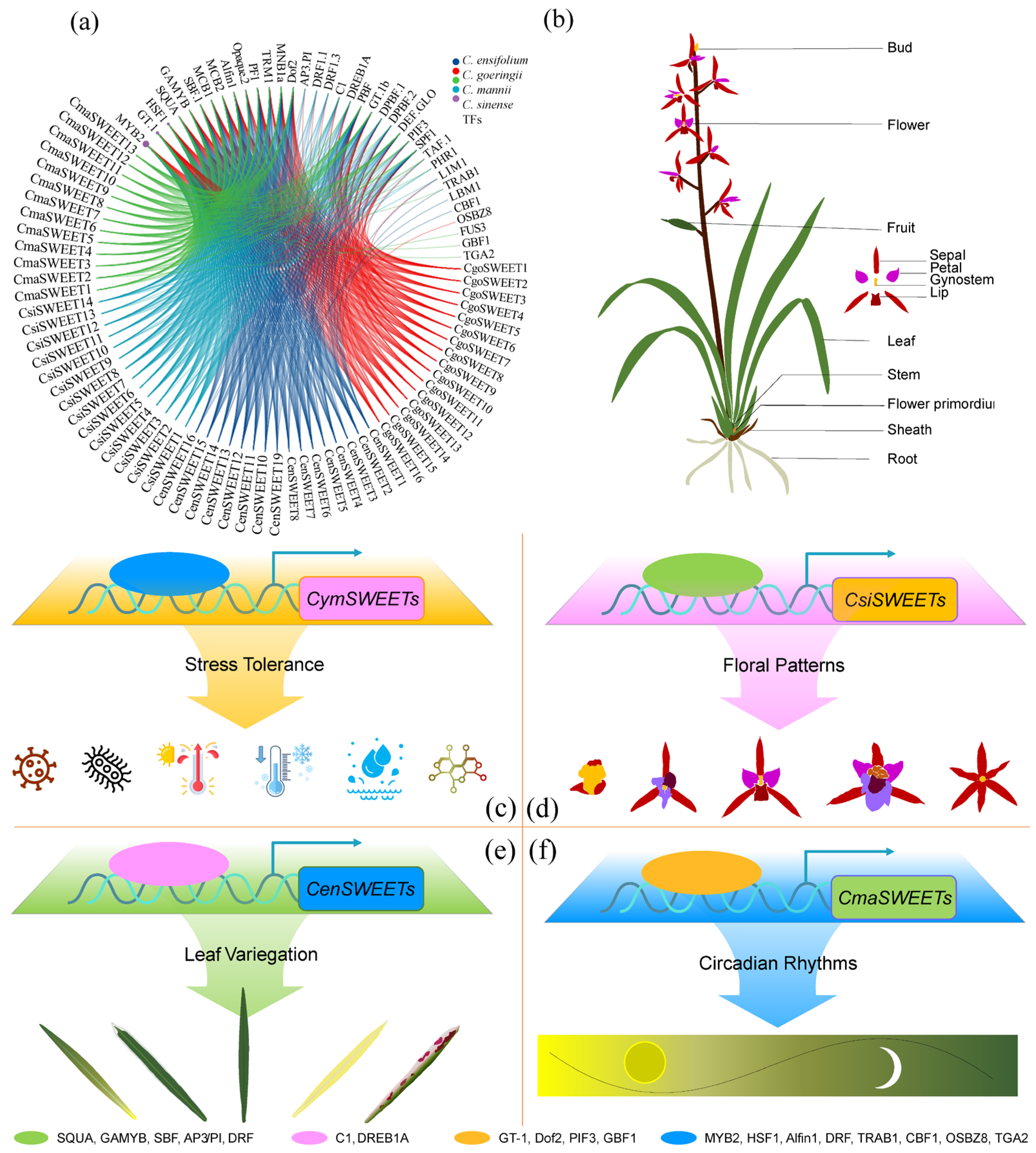
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Identification of the SWEET Gene Family in Four Cymbidium Species
4.3. Homology, Collinearity, Synteny and Duplication Analysis
4.4. Selection Pressure, Divergence Time, and Ka/Ks Calculation
4.5. Multiple Sequence Alignment and Phylogenetic Analysis
4.6. Conserved Motif, Gene Structure, and Promoter Region Analysis
4.7. Expression Pattern Analysis of CymSWEET Genes
4.8. RNA Isolation and qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.J.; Wang, G.L.; Ma, J.; Xu, Z.S.; Wang, F.; Xiong, A.S. Transcript profiling of sucrose synthase genes involved in sucrose metabolism among four carrot (Daucus carota L.) cultivars reveals distinct patterns. BMC Plant Biol. 2018, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Delgado, D.; Ñústez-López, C.-E.; Narváez-Cuenca, C.-E.; Restrepo-Sánchez, L.-P.; Melo, S.E.; Sarmiento, F.; Kushalappa, A.C.; Mosquera-Vásquez, T. Natural variation of sucrose, glucose and fructose contents in Colombian genotypes of Solanum tuberosum Group Phureja at harvest. J. Sci. Food Agric. 2016, 96, 4288–4294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zou, L.; Ren, C.; Ren, F.; Wang, Y.; Fan, P.; Li, S.; Liang, Z. VvSWEET10 Mediates Sugar Accumulation in Grapes. Genes 2019, 10, 225. [Google Scholar] [CrossRef]
- Ji, Y.; Nuñez Ocaña, D.; Choe, D.; Larsen, D.H.; Marcelis, L.F.M.; Heuvelink, E. Far-red radiation stimulates dry mass partitioning to fruits by increasing fruit sink strength in tomato. New Phytol. 2020, 228, 1914–1925. [Google Scholar] [CrossRef]
- Cho, L.-H.; Pasriga, R.; Yoon, J.; Jeon, J.-S.; An, G. Roles of Sugars in Controlling Flowering Time. J. Plant Biol. 2018, 61, 121–130. [Google Scholar] [CrossRef]
- Tew, N.E.; Memmott, J.; Vaughan, I.P.; Bird, S.; Stone, G.N.; Potts, S.G.; Baldock, K.C.R. Quantifying nectar production by flowering plants in urban and rural landscapes. J. Ecol. 2021, 109, 1747–1757. [Google Scholar] [CrossRef]
- Doidy, J.; Grace, E.; Kühn, C.; Simon-Plas, F.; Casieri, L.; Wipf, D. Sugar transporters in plants and in their interactions with fungi. Trends Plant Sci. 2012, 17, 413–422. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.; Yu, F.; Tang, J.; Shan, X.; Bao, K.; Yu, L.; Wang, H.; Fei, Z.; Li, J. Genome-wide characterization and expression profiling of SWEET genes in cabbage (Brassica oleracea var. capitata L.) reveal their roles in chilling and clubroot disease responses. BMC Genom. 2019, 20, 93. [Google Scholar] [CrossRef]
- Anjali, A.; Fatima, U.; Manu, M.S.; Ramasamy, S.; Senthil-Kumar, M. Structure and regulation of SWEET transporters in plants: An update. Plant Physiol. Biochem. 2020, 156, 1–6. [Google Scholar] [CrossRef]
- Chen, L.Q.; Hou, B.H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.Q.; Guo, W.J.; Kim, J.G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, S. Rice MtN3/saliva/SWEET family genes and their homologs in cellular organisms. Mol. Plant 2013, 6, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Osakabe, Y. Sugar compartmentation as an environmental stress adaptation strategy in plants. Semin. Cell Dev. Biol. 2018, 83, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Qu, X.Q.; Hou, B.H.; Sosso, D.; Osorio, S.; Fernie, A.R.; Frommer, W.B. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 2012, 335, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Das, S.; Jagadis Gupta, K.; Ranjan, A.; Foyer, C.H.; Thakur, J.K. Physiological implications of SWEETs in plants and their potential applications in improving source-sink relationships for enhanced yield. Plant Biotechnol. J. 2023, 21, 1528–1541. [Google Scholar] [CrossRef]
- Gamas, P.; de Carvalho Niebel, F.; Lescure, N.; Cullimore, J.V. Use of a subtractive hybridization approach to identify new Medicago truncatula genes induced during root nodule development. MPMI-Mol. Plant Microbe Interact. 1996, 9, 233–242. [Google Scholar] [CrossRef]
- Artero, R.D.; Terol-Alcayde, J.; Paricio, N.; Ring, J.; Bargues, M.; Torres, A.; Perez-Alonso, M. Saliva, a new Drosophila gene expressed in the embryonic salivary glands with homologues in plants and vertebrates. Mech. Dev. 1998, 75, 159–162. [Google Scholar] [CrossRef]
- Xue, X.; Wang, J.; Shukla, D.; Cheung, L.S.; Chen, L.-Q. When SWEETs Turn Tweens: Updates and Perspectives. Annu. Rev. Plant Biol. 2022, 73, 379–403. [Google Scholar] [CrossRef]
- Tränkner, M.; Tavakol, E.; Jákli, B. Functioning of potassium and magnesium in photosynthesis, photosynthate translocation and photoprotection. Physiol. Plant. 2018, 163, 414–431. [Google Scholar] [CrossRef]
- Lu, C.; Ye, J.; Chang, Y.; Mi, Z.; Liu, S.; Wang, D.; Wang, Z.; Niu, J. Genome-wide identification and expression patterns of the SWEET gene family in Bletilla striata and its responses to low temperature and oxidative stress. Int. J. Mol. Sci. 2022, 23, 10057. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, L.; Li, T.; Ruan, Y.; Zhang, A.; Dong, X.; Zhu, Y.; Li, C.; Fan, J. Genome-Wide Investigation and Characterization of SWEET Gene Family with Focus on Their Evolution and Expression during Hormone and Abiotic Stress Response in Maize. Genes 2022, 13, 1682. [Google Scholar] [CrossRef]
- Kumar, P.V.; Mallikarjuna, M.G.; Jha, S.K.; Mahato, A.; Lal, S.K.; Yathish, K.; Lohithaswa, H.C.; Chinnusamy, V. Unravelling structural, functional, evolutionary and genetic basis of SWEET transporters regulating abiotic stress tolerance in maize. Int. J. Biol. Macromol. 2023, 229, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Shi, X.; Qin, S.; Dong, J.; Shi, H.; Wang, Y.; Zhang, Y. Genome-wide identification, characterization and transcriptional profile of the SWEET gene family in Dendrobium officinale. BMC Genom. 2023, 24, 378. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Kou, Y.; Duan, M.; Feng, B.; Yu, X.; Jia, R.; Zhao, X.; Ge, H.; Yang, S. Genome-Wide Identification of the Rose SWEET Gene Family and Their Different Expression Profiles in Cold Response between Two Rose Species. Plants 2023, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.; Valliyodan, B.; Deshmukh, R.; Prince, S.; Nicander, B.; Zhao, M.; Sonah, H.; Song, L.; Lin, L.; Chaudhary, J. Soybean (Glycine max) SWEET gene family: Insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genom. 2015, 16, 520. [Google Scholar] [CrossRef]
- Li, W.; Ren, Z.; Wang, Z.; Sun, K.; Pei, X.; Liu, Y.; He, K.; Zhang, F.; Song, C.; Zhou, X.; et al. Evolution and Stress Responses of Gossypium hirsutum SWEET Genes. Int. J. Mol. Sci. 2018, 19, 769. [Google Scholar] [CrossRef]
- Tian, R.; Xu, J.; Xu, Z.; Li, J.; Li, H. Genome-Wide Identification and Expression Analysis of SWEET Gene Family in Strawberry. Horticulturae 2024, 10, 191. [Google Scholar] [CrossRef]
- Gautam, T.; Saripalli, G.; Gahlaut, V.; Kumar, A.; Sharma, P.K.; Balyan, H.S.; Gupta, P.K. Further studies on sugar transporter (SWEET) genes in wheat (Triticum aestivum L.). Mol. Biol. Rep. 2019, 46, 2327–2353. [Google Scholar] [CrossRef]
- Eom, J.-S.; Chen, L.-Q.; Sosso, D.; Julius, B.T.; Lin, I.; Qu, X.-Q.; Braun, D.M.; Frommer, W.B. SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 2015, 25, 53–62. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, X.; Li, Y.; Ke, S.; Yin, W.; Lan, S.; Liu, Z. Advances and prospects of orchid research and industrialization. Hortic. Res.-Engl. 2022, 9, uhac220. [Google Scholar] [CrossRef]
- Pal, R.; Meena, N.; Pant, R.; Dayamma, M. Cymbidium: Botany, production, and uses. Orchid. Phytochem. Biol. Hortic. Fundam. Appl. 2019, 1–37. [Google Scholar] [CrossRef]
- Yang, F.X.; Gao, J.; Wei, Y.L.; Ren, R.; Zhang, G.Q.; Lu, C.Q.; Jin, J.P.; Ai, Y.; Wang, Y.Q.; Chen, L.J.; et al. The genome of Cymbidium sinense revealed the evolution of orchid traits. Plant Biotechnol. J. 2021, 19, 2501–2516. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Li, Z.; Sun, W.H.; Chen, J.; Zhang, D.Y.; Ma, L.; Zhang, Q.H.; Chen, M.K.; Zheng, Q.D.; Liu, J.F.; et al. The Cymbidium genome reveals the evolution of unique morphological traits. Hortic. Res.-Engl. 2021, 8, 264. [Google Scholar] [CrossRef]
- Chung, O.; Kim, J.; Bolser, D.; Kim, H.M.; Jun, J.H.; Choi, J.P.; Jang, H.D.; Cho, Y.S.; Bhak, J.; Kwak, M. A chromosome-scale genome assembly and annotation of the spring orchid (Cymbidium goeringii). Mol. Ecol. Resour. 2022, 22, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, G.-Z.; Huang, J.; Liu, D.-K.; Xue, F.; Chen, X.-L.; Chen, S.-Q.; Liu, C.-G.; Liu, H.; Ma, H.; et al. The Cymbidium goeringii genome provides insight into organ development and adaptive evolution in orchids. Ornam. Plant Res. 2021, 1, 10. [Google Scholar] [CrossRef]
- Fan, W.; He, Z.-S.; Zhe, M.; Feng, J.-Q.; Zhang, L.; Huang, Y.; Liu, F.; Huang, J.-L.; Ya, J.-D.; Zhang, S.-B.; et al. High-quality Cymbidium mannii genome and multifaceted regulation of crassulacean acid metabolism in epiphytes. Plant Commun. 2023, 4, 100564. [Google Scholar] [CrossRef]
- Hew, C.S.; Wong, Y.S. Chinese Cymbidium Orchid: History of Chinese Cymbidium. World Sci. 2023, 1–15. [Google Scholar] [CrossRef]
- Yang, F.; Guo, Y.; Li, J.; Lu, C.; Wei, Y.; Gao, J.; Xie, Q.; Jin, J.; Zhu, G. Genome-wide association analysis identified molecular markers and candidate genes for flower traits in Chinese orchid (Cymbidium sinense). Hortic. Res.-Engl. 2023, 10, uhad206. [Google Scholar] [CrossRef]
- Wang, H.-Z.; Lu, J.-J.; Hu, X.; Liu, J.-J. Genetic variation and cultivar identification in Cymbidium ensifolium. Plant Syst. Evol. 2011, 293, 101–110. [Google Scholar] [CrossRef]
- Cao, H.; Li, H.; Chen, X.; Zhang, Y.; Lu, L.; Li, S.; Tao, X.; Zhu, W.; Wang, J.; Ma, L. Insight into the molecular mechanisms of leaf coloration in Cymbidium ensifolium. Front. Genet. 2022, 13, 923082. [Google Scholar] [CrossRef]
- Ning, H.-J.; Gui, F.-F.; Tian, E.-W.; Yang, L.-Y. The novel developed microsatellite markers revealed potential hybridization among Cymbidium species and the interspecies sub-division of C. goeringii and C. ensifolium. BMC Plant Biol. 2023, 23, 492. [Google Scholar] [CrossRef]
- Wu, J.; Ma, H.; Xu, X.; Qiao, N.; Guo, S.; Liu, F.; Zhang, D.; Zhou, L. Mycorrhizas alter nitrogen acquisition by the terrestrial orchid Cymbidium goeringii. Ann. Bot. 2013, 111, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.X.; Zhu, G.F.; Wei, Y.L.; Gao, J.; Liang, G.; Peng, L.Y.; Lu, C.Q.; Jin, J.P. Low-temperature-induced changes in the transcriptome reveal a major role of CgSVP genes in regulating flowering of Cymbidium goeringii. BMC Genom. 2019, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Lee, Y.; Gao, J. Ex situ symbiotic seed germination, isolation and identification of effective symbiotic fungus in Cymbidium mannii (Orchidaceae). Chin. J. Plant Ecol. 2012, 36, 859–869. [Google Scholar] [CrossRef]
- Zhang, L.S.; Chen, F.; Zhang, G.Q.; Zhang, Y.Q.; Niu, S.; Xiong, J.S.; Lin, Z.G.; Cheng, Z.M.; Liu, Z.J. Origin and mechanism of crassulacean acid metabolism in orchids as implied by comparative transcriptomics and genomics of the carbon fixation pathway. Plant J. 2016, 86, 175–185. [Google Scholar] [CrossRef]
- Mirdita, M.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Nobukuni, M.; Mochizuki, H.; Okada, S.; Kameyama, N.; Tanaka, A.; Yamamoto, H.; Amano, T.; Seki, T.; Sakai, N. The C-Terminal Region of Serotonin Transporter Is Important for Its Trafficking and Glycosylation. J. Pharmacol. Sci. 2009, 111, 392–404. [Google Scholar] [CrossRef]
- Fujita, S.; Sato, D.; Kasai, H.; Ohashi, M.; Tsukue, S.; Takekoshi, Y.; Gomi, K.; Shintani, T. The C-terminal region of the yeast monocarboxylate transporter Jen1 acts as a glucose signal–responding degron recognized by the α-arrestin Rod1. J. Biol. Chem. 2018, 293, 10926–10936. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, L.; Xia, W.; Liang, L.; Bai, X.; Li, L.; Xu, L.; Liu, L. Mycorrhizal Compatibility and Germination-Promoting Activity of Tulasnella Species in Two Species of Orchid (Cymbidium mannii and Epidendrum radicans). Horticulturae 2021, 7, 472. [Google Scholar] [CrossRef]
- Frank Baker, R.; Leach, K.A.; Braun, D.M. SWEET as Sugar: New Sucrose Effluxers in Plants. Mol. Plant 2012, 5, 766–768. [Google Scholar] [CrossRef]
- Zhu, Y.; Tian, Y.; Han, S.; Wang, J.; Liu, Y.; Yin, J. Structure, evolution, and roles of SWEET proteins in growth and stress responses in plants. Int. J. Biol. Macromol. 2024, 263, 130441. [Google Scholar] [CrossRef]
- Wendel, J.F.; Jackson, S.A.; Meyers, B.C.; Wing, R.A. Evolution of plant genome architecture. Genome Biol. 2016, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.C.; Purugganan, M.D. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 15682–15687. [Google Scholar] [CrossRef] [PubMed]
- Conant, G.C.; Wolfe, K.H. Turning a hobby into a job: How duplicated genes find new functions. Nat. Rev. Genet. 2008, 9, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Carretero-Paulet, L.; Fares, M.A. Evolutionary dynamics and functional specialization of plant paralogs formed by whole and small-scale genome duplications. Mol. Biol. Evol. 2012, 29, 3541–3551. [Google Scholar] [CrossRef]
- Tao, Y.; Cheung, L.S.; Li, S.; Eom, J.-S.; Chen, L.-Q.; Xu, Y.; Perry, K.; Frommer, W.B.; Feng, L. Structure of a eukaryotic SWEET transporter in a homotrimeric complex. Nature 2015, 527, 259–263. [Google Scholar] [CrossRef]
- Zhang, Y.; Hao, L.; Wang, N.; Bai, X.; Zhang, Y. Transcriptome-wide identification and expression profiling of the SWEET family in Bletilla striata and regulation analysis with non-coding RNAs. Ind. Crops Prod. 2023, 201, 116876. [Google Scholar] [CrossRef]
- Yin, Q.; Zhu, L.; Du, P.; Fan, C.; Wang, J.; Zhang, B.; Li, H. Comprehensive analysis of SWEET family genes in Eucalyptus (Eucalyptus grandis). Biotechnol. Biotechnol. Equip. 2020, 34, 595–604. [Google Scholar] [CrossRef]
- Yuan, M.; Zhao, J.; Huang, R.; Li, X.; Xiao, J.; Wang, S. Rice MtN3/saliva/SWEET gene family: Evolution, expression profiling, and sugar transport. J. Integr. Plant Biol. 2014, 56, 559–570. [Google Scholar] [CrossRef]
- Xu, Y.; Tao, Y.; Cheung, L.S.; Fan, C.; Chen, L.-Q.; Xu, S.; Perry, K.; Frommer, W.B.; Feng, L. Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature 2014, 515, 448–452. [Google Scholar] [CrossRef]
- Wang, T.; Song, Z.; Meng, W.; Li, L. Identification, characterization, and expression of the SWEET gene family in Phalaenopsis equestris and Dendrobium officinale. Biol. Plant. 2018, 62, 24–32. [Google Scholar] [CrossRef]
- Balakirev, E.S.; Ayala, F.J. Pseudogenes: Are they “junk” or functional DNA? Annu. Rev. Genet. 2003, 37, 123–151. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Shen, Q.; Li, Y.; Zhou, D.; Zhang, Z.; Akbar, S.; Wang, Z.; Zhang, J. Functional characterization and analysis of transcriptional regulation of sugar transporter SWEET13c in sugarcane Saccharum spontaneum. BMC Plant Biol. 2022, 22, 363. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zhang, M.; Zhang, Z.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.; Pan, Y.; Zhou, G.; Liu, S.; et al. Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef]
- Kim, P.; Xue, C.Y.; Song, H.D.; Gao, Y.; Feng, L.; Li, Y.; Xuan, Y.H. Tissue-specific activation of DOF11 promotes rice resistance to sheath blight disease and increases grain weight via activation of SWEET14. Plant Biotechnol. J. 2021, 19, 409–411. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, T.; Li, X.; Song, C.-P.; Zhu, J.-K.; Chen, L.; Zhao, Y. Phosphorylation of SWEET sucrose transporters regulates plant root:shoot ratio under drought. Nat. Plants 2022, 8, 68–77. [Google Scholar] [CrossRef]
- Messeguer, X.; Escudero, R.; Farré, D.; Núñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef]
- Yang, A.; Dai, X.; Zhang, W.-H. A R2R3-type MYB gene, OsMYB2, is involved in salt, cold, and dehydration tolerance in rice. J. Exp. Bot. 2012, 63, 2541–2556. [Google Scholar] [CrossRef]
- Liu, N.; Ding, Y.; Fromm, M.; Avramova, Z. Different gene-specific mechanisms determine the ‘revised-response’ memory transcription patterns of a subset of A. thaliana dehydration stress responding genes. Nucleic Acids Res. 2014, 42, 5556–5566. [Google Scholar] [CrossRef]
- Kaplan-Levy, R.N.; Brewer, P.B.; Quon, T.; Smyth, D.R. The trihelix family of transcription factors—Light, stress and development. Trends Plant Sci. 2012, 17, 163–171. [Google Scholar] [CrossRef]
- Zhu, J.; Cao, X.; Deng, X. Epigenetic and transcription factors synergistically promote the high temperature response in plants. Trends Biochem. Sci. 2023, 48, 788–800. [Google Scholar] [CrossRef]
- Andrási, N.; Pettkó-Szandtner, A.; Szabados, L. Diversity of plant heat shock factors: Regulation, interactions, and functions. J. Exp. Bot. 2021, 72, 1558–1575. [Google Scholar] [CrossRef] [PubMed]
- Krizek, B.A.; Fletcher, J.C. Molecular mechanisms of flower development: An armchair guide. Nat. Rev. Genet. 2005, 6, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, B.; Yu, H. Molecular genetic insights into orchid reproductive development. J. Exp. Bot. 2022, 73, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wu, H.; Zhu, H.; Huang, C.; Liu, C.; Chang, Y.; Kong, Z.; Zhou, Z.; Wang, G.; Lin, Y.; et al. Determining factors, regulation system, and domestication of anthocyanin biosynthesis in rice leaves. New Phytol. 2019, 223, 705–721. [Google Scholar] [CrossRef]
- Wang, W.-B.; He, X.-F.; Yan, X.-M.; Ma, B.; Lu, C.-F.; Wu, J.; Zheng, Y.; Wang, W.-H.; Xue, W.-B.; Tian, X.-C.; et al. Chromosome-scale genome assembly and insights into the metabolome and gene regulation of leaf color transition in an important oak species, Quercus dentata. New Phytol. 2023, 238, 2016–2032. [Google Scholar] [CrossRef]
- Tyagi, A.K.; Gaur, T. Light Regulation of Nuclear Photosynthetic Genes in Higher Plants. Crit. Rev. Plant Sci. 2003, 22, 417–452. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, R.; Zeng, X.-Y.; Lee, S.; Ye, L.-H.; Tian, S.-L.; Zhang, Y.-J.; Busch, W.; Zhou, W.-B.; Zhu, X.-G.; et al. GLK transcription factors accompany ELONGATED HYPOCOTYL5 to orchestrate light-induced seedling development in Arabidopsis. Plant Physiol. 2024, 194, 2400–2421. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar Gustavo, A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.e.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Cavalcanti, A.; Chen, F.-C.; Bouman, P.; Li, W.-H. Extent of Gene Duplication in the Genomes of Drosophila, Nematode, and Yeast. Mol. Biol. Evol. 2002, 19, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.; Wang, J.; Wong, G.K.; Yu, J. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.-H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37 (Suppl. S2), W202–W208. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Cramer, P. AlphaFold2 and the future of structural biology. Nat. Struct. Mol. Biol. 2021, 28, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Janson, G.; Zhang, C.; Prado, M.G.; Paiardini, A. PyMod 2.0: Improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics 2016, 33, 444–446. [Google Scholar] [CrossRef] [PubMed]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Higo, H. PLACE: A database of plant cis -acting regulatory DNA elements. Nucleic Acids Res. 1998, 26, 358–359. [Google Scholar] [CrossRef] [PubMed]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef]
- Li, D.; Mei, H.; Shen, Y.; Su, S.; Zhang, W.; Wang, J.; Zu, M.; Chen, W. ECharts: A declarative framework for rapid construction of web-based visualization. Vis. Inform. 2018, 2, 136–146. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Li, J.; Jin, J.; Gao, J.; Xie, Q.; Lu, C.; Zhu, G.; Yang, F. Genome-Wide Characterization, Comparative Analysis, and Expression Profiling of SWEET Genes Family in Four Cymbidium Species (Orchidaceae). Int. J. Mol. Sci. 2025, 26, 3946. https://doi.org/10.3390/ijms26093946
Wei Y, Li J, Jin J, Gao J, Xie Q, Lu C, Zhu G, Yang F. Genome-Wide Characterization, Comparative Analysis, and Expression Profiling of SWEET Genes Family in Four Cymbidium Species (Orchidaceae). International Journal of Molecular Sciences. 2025; 26(9):3946. https://doi.org/10.3390/ijms26093946
Chicago/Turabian StyleWei, Yonglu, Jie Li, Jianpeng Jin, Jie Gao, Qi Xie, Chuqiao Lu, Genfa Zhu, and Fengxi Yang. 2025. "Genome-Wide Characterization, Comparative Analysis, and Expression Profiling of SWEET Genes Family in Four Cymbidium Species (Orchidaceae)" International Journal of Molecular Sciences 26, no. 9: 3946. https://doi.org/10.3390/ijms26093946
APA StyleWei, Y., Li, J., Jin, J., Gao, J., Xie, Q., Lu, C., Zhu, G., & Yang, F. (2025). Genome-Wide Characterization, Comparative Analysis, and Expression Profiling of SWEET Genes Family in Four Cymbidium Species (Orchidaceae). International Journal of Molecular Sciences, 26(9), 3946. https://doi.org/10.3390/ijms26093946