
Targeting DNA Damage Repair to Enhance Antitumor Immunity in Radiotherapy: Mechanisms and Opportunities


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. DNA Damage Repair in Radiotherapy
2.1. DNA Damage in Radiotherapy
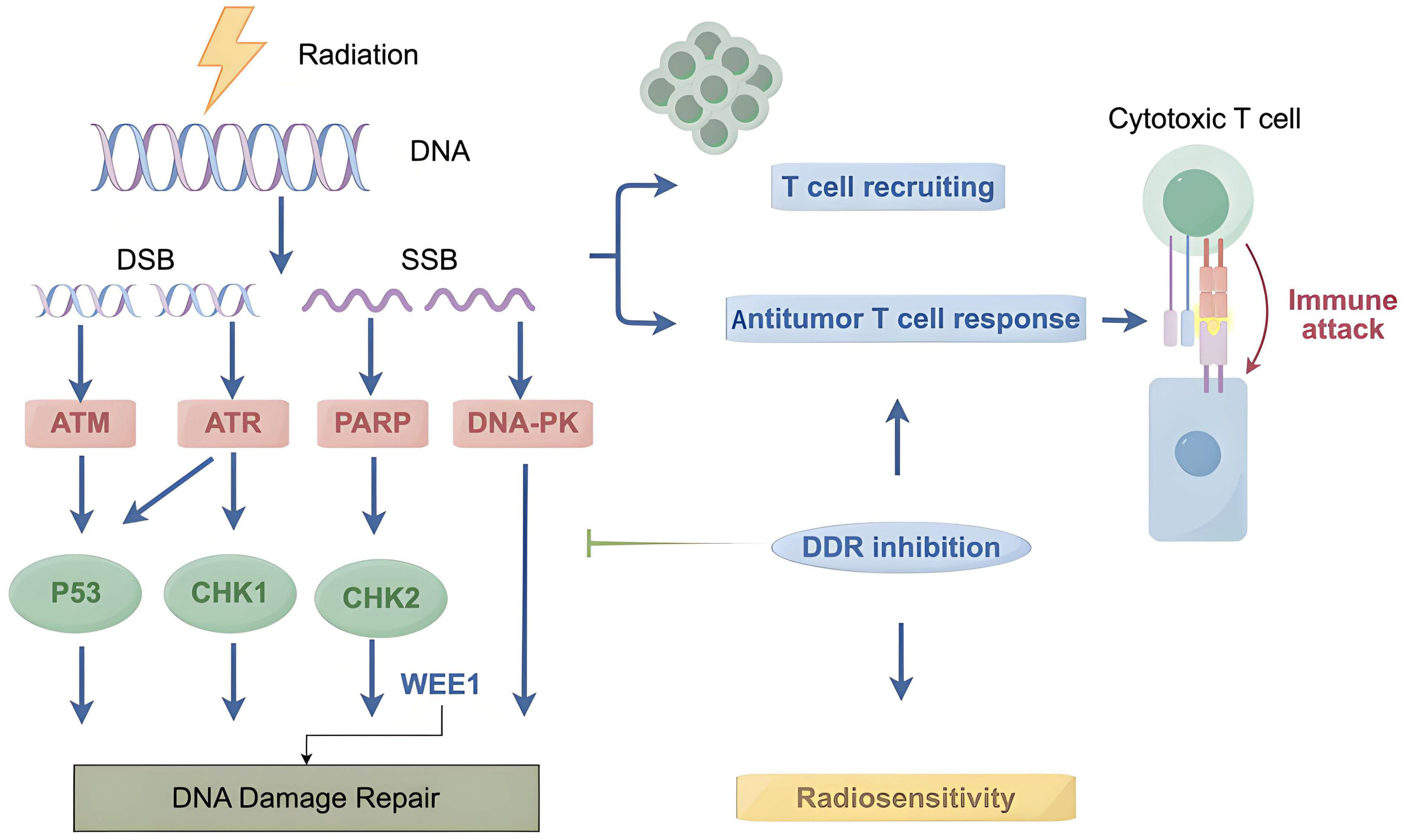
2.2. DNA Damage Sensors and Early Responders
2.3. DNA Damage Repair Pathways
2.4. Cellular Response to Radiotherapy
3. Immune Response in Radiotherapy
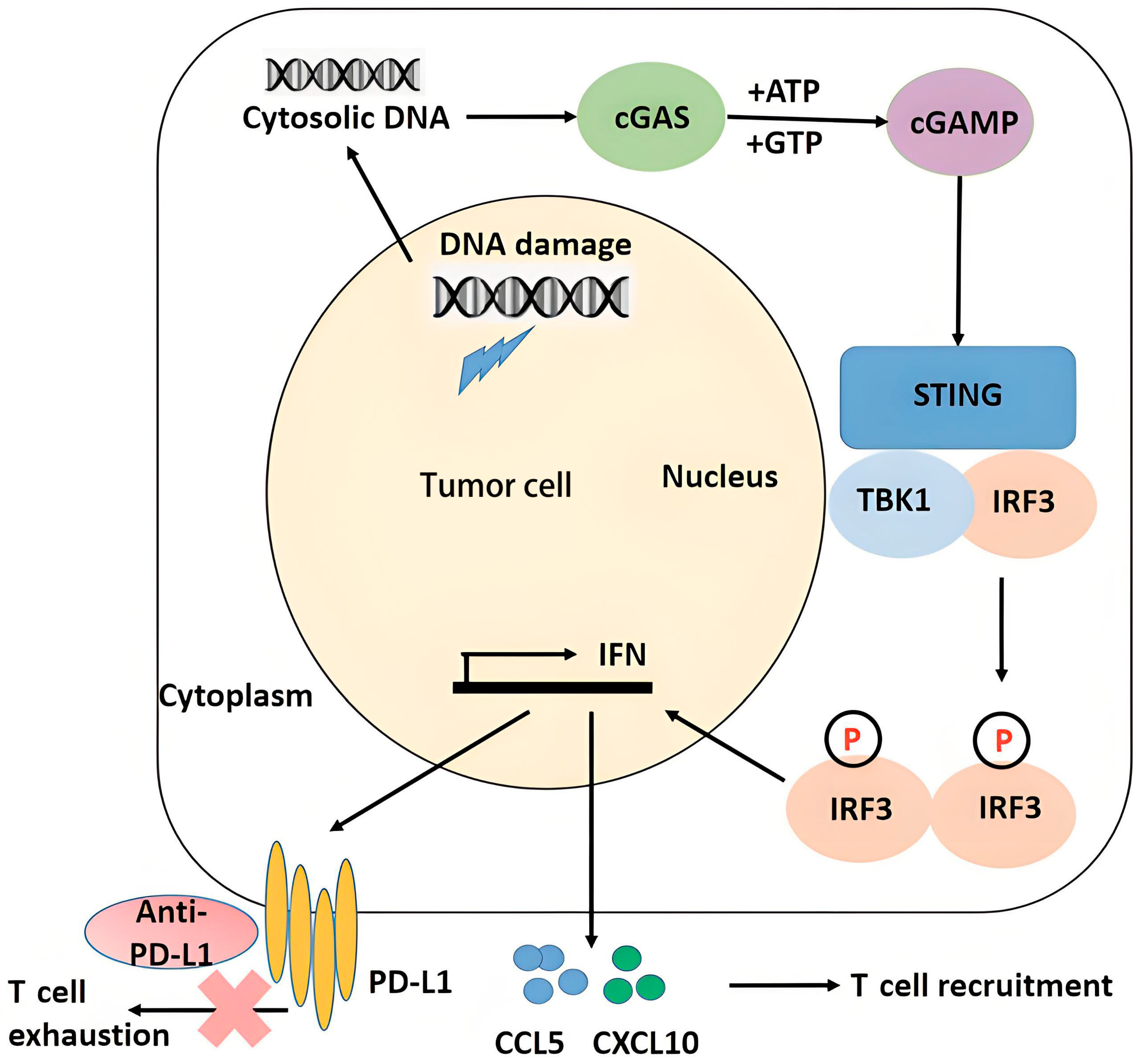
3.1. Radiation and Innate Immune Signaling
3.2. Radiation and Adaptive Immune Signaling
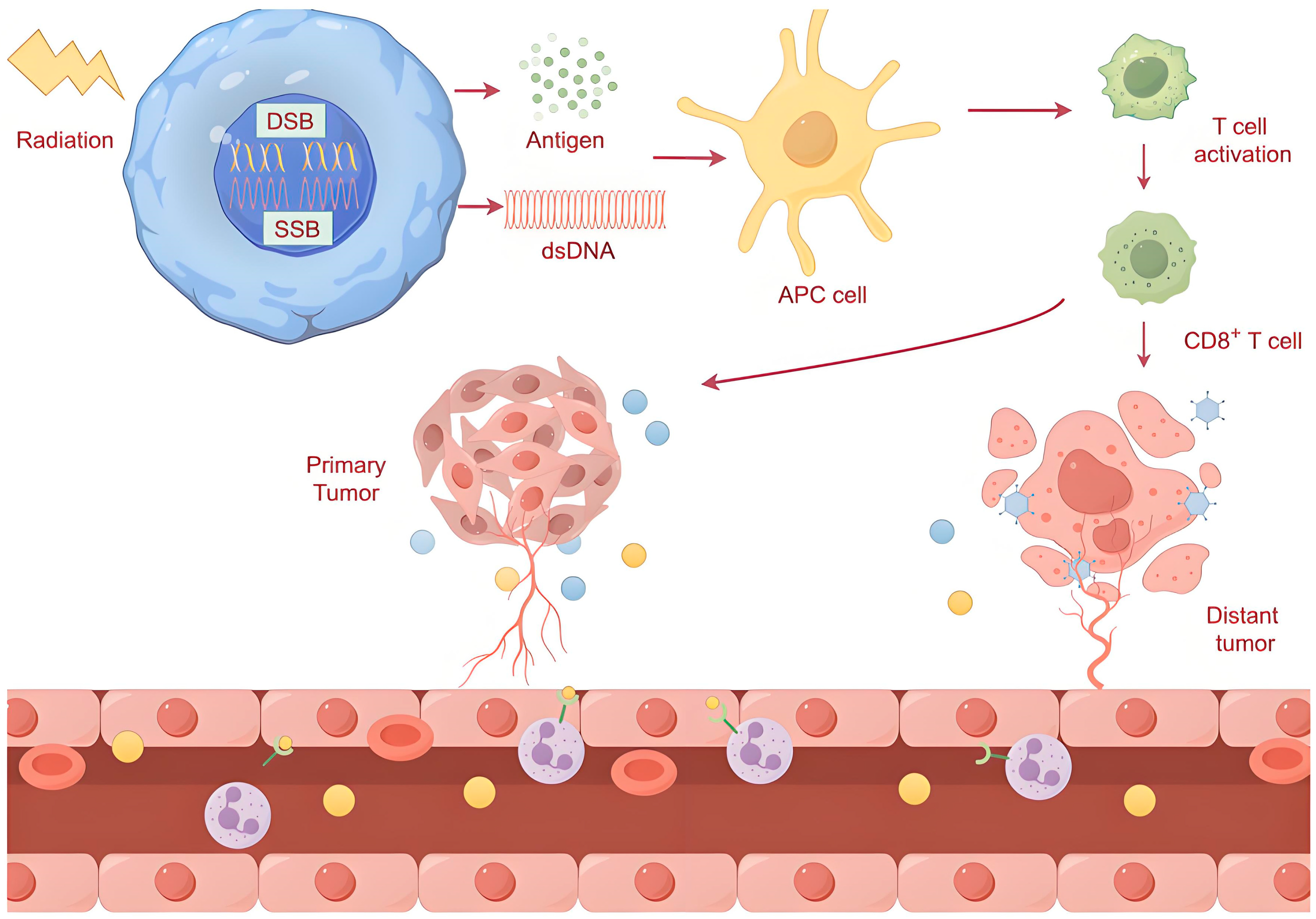
3.3. Radiotherapy and Abscopal Effects
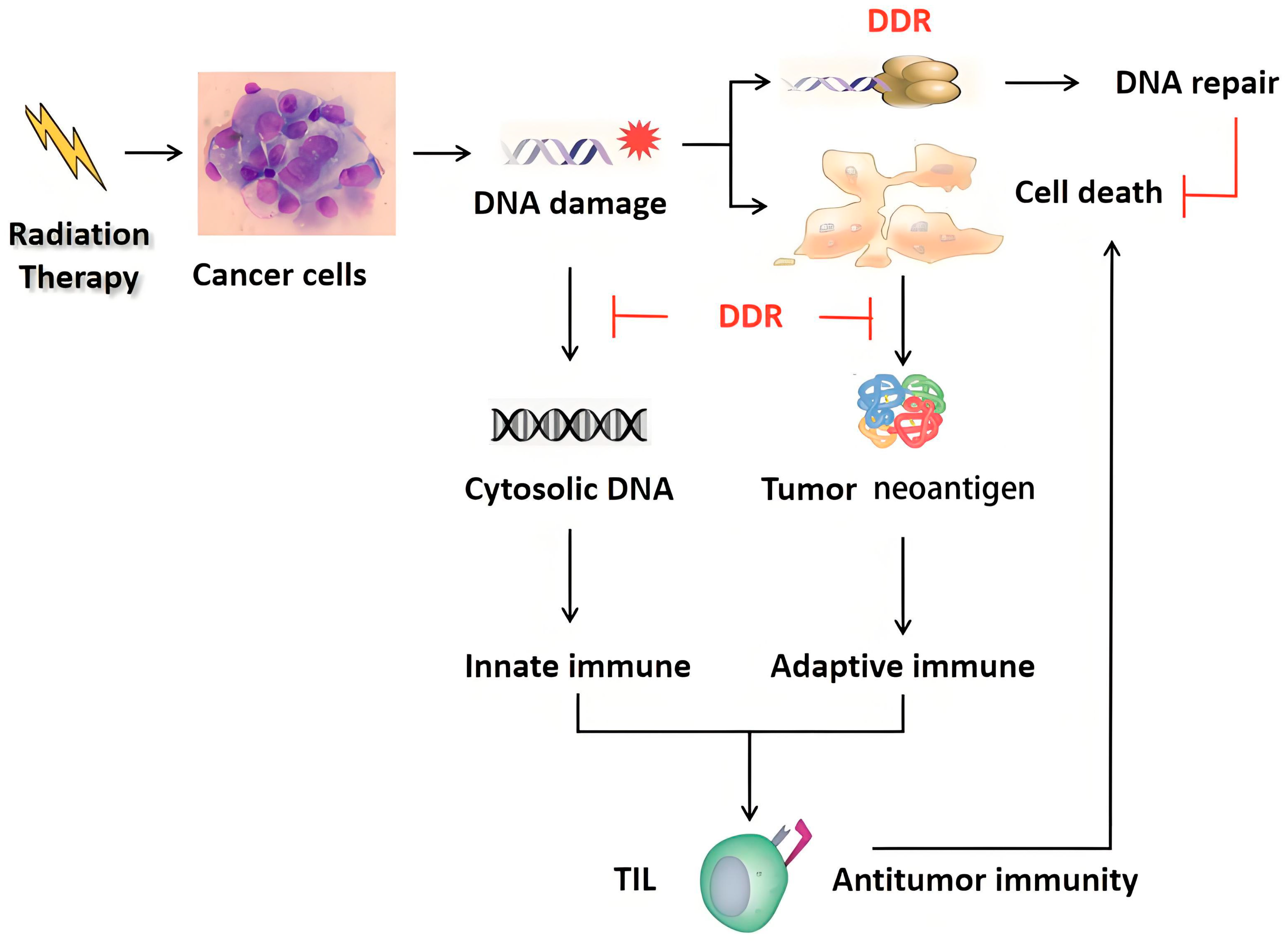
4. DDR and Immunotherapy
4.1. Predictive Function of the DDR in Immunotherapy
4.2. Targeting the DDR Enhances the Effect of Immunotherapy
5. Targeting the DDR Enhances Antitumor Immunity in Radiotherapy
5.1. PARP
5.2. ATM/ATR
5.3. WEE1
5.4. CHK1
5.5. DNA-PKcs
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kumari, S.; Mukherjee, S.; Sinha, D.; Abdisalaam, S.; Krishnan, S.; Asaithamby, A. Immunomodulatory effects of radiotherapy. Int. J. Mol. Sci. 2020, 21, 8151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Jia, K.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; He, Y.; Zhou, C. Alterations of DNA damage repair in cancer: From mechanisms to applications. Ann. Transl. Med. 2020, 8, 1685. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; Zenke, F.T.; Curtin, N.J. DNA damage response inhibitors in cancer therapy: Lessons from the past, current status and future implications. Nat. Rev. Drug Discov. 2025, 24, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.J.; Nanavaty, N.S.; Devanaboyina, M.; Stanbery, L.; Hamouda, D.; Edelman, G.; Dworkin, L.; Nemunaitis, J.J. The abscopal effect of radiation therapy. Future Oncol. 2021, 17, 1683–1694. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar]
- Qiu, H.; Shao, Z.; Wen, X.; Qu, D.; Liu, Z.; Chen, Z.; Zhang, X.; Ding, X.; Zhang, L. HMGB1/TREM2 positive feedback loop drives the development of radioresistance and immune escape of glioblastoma by regulating TLR4/Akt signaling. J. Transl. Med. 2024, 22, 688. [Google Scholar] [CrossRef]
- Deng, S.; Vlatkovic, T.; Li, M.; Zhan, T.; Veldwijk, M.R.; Herskind, C. Targeting the DNA Damage Response and DNA Repair Pathways to Enhance Radiosensitivity in Colorectal Cancer. Cancers 2022, 14, 4874. [Google Scholar]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.; Ozawa, H. A comprehensive review of current therapeutic strategies in cancers targeting DNA damage response mechanisms in head and neck squamous cell cancer. Biochim. Biophys. Acta Rev. Cancer 2025, 1880, 189255. [Google Scholar] [CrossRef]
- Dagar, G.; Gupta, A.; Shankar, A.; Chauhan, R.; Macha, M.A.; Bhat, A.A.; Das, D.; Goyal, R.; Bhoriwal, S.; Pandita, R.K.; et al. The future of cancer treatment: Combining radiotherapy with immunotherapy. Front. Mol. Biosci. 2024, 11, 1409300. [Google Scholar] [CrossRef]
- Yang, Y.; Xiong, L.; Li, M.; Jiang, P.; Wang, J.; Li, C. Advances in radiotherapy and immunity in hepatocellular carcinoma. J. Transl. Med. 2023, 21, 526. [Google Scholar] [CrossRef]
- Spigel, D.R.; Faivre-Finn, C.; Gray, J.E.; Vicente, D.; Planchard, D.; Paz-Ares, L.; Vansteenkiste, J.F.; Garassino, M.C.; Hui, R.; Quantin, X.; et al. Five-Year Survival Outcomes From the PACIFIC Trial: Durvalumab After Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 1301–1311. [Google Scholar] [CrossRef]
- Meng, X.; Yu, J.M. Advances of radiation oncology in China. Chin. Med. J. (Engl.) 2009, 122, 2231–2235. [Google Scholar]
- Riches, L.C.; Lynch, A.M.; Gooderham, N.J. Early events in the mammalian response to DNA double-strand breaks. Mutagenesis 2008, 23, 331–339. [Google Scholar] [CrossRef]
- Deriano, L.; Guipaud, O.; Merle-Béral, H.; Binet, J.L.; Ricoul, M.; Potocki-Veronese, G.; Favaudon, V.; Maciorowski, Z.; Muller, C.; Salles, B.; et al. Human chronic lymphocytic leukemia B cells can escape DNA damage-induced apoptosis through the nonhomologous end-joining DNA repair pathway. Blood 2005, 105, 4776–4783. [Google Scholar] [CrossRef]
- Wilson, C.R.; Davidson, S.E.; Margison, G.P.; Jackson, S.P.; Hendry, J.H.; West, C.M. Expression of Ku70 correlates with survival in carcinoma of the cervix. Br. J. Cancer 2000, 83, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Li, L.; Kim, D.; Wen, B.; Deng, X.; Gutin, P.H.; Ling, C.C.; Li, G.C. Adenovirus-mediated expression of a dominant negative Ku70 fragment radiosensitizes human tumor cells under aerobic and hypoxic conditions. Cancer Res. 2007, 67, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Palmer, J.D.; Siedow, M.; Haque, S.J.; Chakravarti, A. Overcoming Radiation Resistance in Gliomas by Targeting Metabolism and DNA Repair Pathways. Int. J. Mol. Sci. 2022, 23, 2246. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J. Biol. Chem. 2010, 285, 1097–1104. [Google Scholar] [CrossRef]
- Dupré, A.; Boyer-Chatenet, L.; Gautier, J. Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat. Struct. Mol. Biol. 2006, 13, 451–457. [Google Scholar] [CrossRef]
- Horejsí, Z.; Falck, J.; Bakkenist, C.J.; Kastan, M.B.; Lukas, J.; Bartek, J. Distinct functional domains of Nbs1 modulate the timing and magnitude of ATM activation after low doses of ionizing radiation. Oncogene 2004, 23, 3122–3127. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Scarbrough, P.M.; Weber, R.P.; Iversen, E.S.; Brhane, Y.; Amos, C.I.; Kraft, P.; Hung, R.J.; Sellers, T.A.; Witte, J.S.; Pharoah, P.; et al. A Cross-Cancer Genetic Association Analysis of the DNA Repair and DNA Damage Signaling Pathways for Lung, Ovary, Prostate, Breast, and Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2016, 25, 193–200. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Kass, E.M.; Jasin, M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010, 584, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef]
- Nagasawa, H.; Little, J.B.; Inkret, W.C.; Carpenter, S.; Raju, M.R.; Chen, D.J.; Strniste, G.F. Response of X-ray-sensitive CHO mutant cells (xrs-6c) to radiation. II. Relationship between cell survival and the induction of chromosomal damage with low doses of alpha particles. Radiat. Res. 1991, 126, 280–288. [Google Scholar] [CrossRef]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef]
- Dosani, M.; Schrader, K.A.; Nichol, A.; Sun, S.; Shenkier, T.; Lohn, Z.; Aubertin, G.; Tyldesley, S. Severe Late Toxicity After Adjuvant Breast Radiotherapy in a Patient with a Germline Ataxia Telangiectasia Mutated Gene: Future Treatment Decisions. Cureus 2017, 9, e1458. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Lin, X.; Liu, Z.; Dong, X.; Wang, K.; Sun, Y.; Zhang, H.; Wang, F.; Chen, Y.; Ling, J.; Guo, Y.; et al. Radiotherapy enhances the anti-tumor effect of CAR-NK cells for hepatocellular carcinoma. J. Transl. Med. 2024, 22, 929. [Google Scholar] [CrossRef]
- Mukherjee, S.; Abdisalaam, S.; Bhattacharya, S.; Srinivasan, K.; Sinha, D.; Asaithamby, A. Mechanistic link between DNA damage sensing, repairing and signaling factors and immune signaling. Adv. Protein. Chem. Struct. Biol. 2019, 115, 297–324. [Google Scholar]
- Ashrafizadeh, M.; Farhood, B.; Eleojo Musa, A.; Taeb, S.; Rezaeyan, A.; Najafi, M. Abscopal effect in radioimmunotherapy. Int. Immunopharmacol. 2020, 85, 106663. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Korpics, M.C.; Katipally, R.R.; Bestvina, C.M.; Pitroda, S.P.; Patel, J.D.; Luke, J.J.; Chmura, S.J.; Juloori, A. Safety of combined ablative radiotherapy and immune checkpoint inhibitors in three phase I trials. Eur. J. Cancer 2024, 209, 114264. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chu, X.; Tanzhu, G.; Zhou, R. Optimal timing and sequence of combining stereotactic radiosurgery with immune checkpoint inhibitors in treating brain metastases: Clinical evidence and mechanistic basis. J. Transl. Med. 2023, 21, 244. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef]
- Chen, Y.A.; Shen, Y.L.; Hsia, H.Y.; Tiang, Y.P.; Sung, T.L.; Chen, L.Y. Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat. Struct. Mol. Biol. 2017, 24, 1124–1131. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, Z.; Xue, Y.; Ning, R.; Guo, X.; Sun, Y.; Zhang, Q. Carbon ion irradiation exerts antitumor activity by inducing cGAS-STING activation and immune response in prostate cancer-bearing mice. Cancer Med. 2024, 13, e6950. [Google Scholar] [CrossRef]
- Xia, L.; Liu, Y.; Wang, Y. PD-1/PD-L1 Blockade Therapy in Advanced Non-Small-Cell Lung Cancer: Current Status and Future Directions. Oncologist 2019, 24 (Suppl. S1), S31–S41. [Google Scholar] [CrossRef]
- Karapetyan, L.; Iheagwara, U.K.; Olson, A.C.; Chmura, S.J.; Skinner, H.K.; Luke, J.J. Radiation dose, schedule, and novel systemic targets for radio-immunotherapy combinations. J. Natl. Cancer Inst. 2023, 115, 1278–1293. [Google Scholar] [CrossRef]
- Chiu, H.W.; Lee, H.L.; Lee, H.H.; Lu, H.W.; Lin, K.Y.; Lin, Y.F.; Lin, C.H. AIM2 promotes irradiation resistance, migration ability and PD-L1 expression through STAT1/NF-κB activation in oral squamous cell carcinoma. J. Transl. Med. 2024, 22, 13. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, C.; Li, H.B.; Tong, J.; Ouyang, X.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lynch, C.; Pitroda, S.P.; Piffkó, A.; Yang, K.; Huser, A.K.; Liang, H.L.; Weichselbaum, R.R. Radiotherapy and immunology. J. Exp. Med. 2024, 221, e20232101. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, M.; Patin, E.C.; Pedersen, M.; Wilkins, A.; Dillon, M.T.; Melcher, A.A.; Harrington, K.J. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 2020, 20, 203–217. [Google Scholar] [CrossRef]
- Takeshima, T.; Chamoto, K.; Wakita, D.; Ohkuri, T.; Togashi, Y.; Shirato, H.; Kitamura, H.; Nishimura, T. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: Its potentiation by combination with Th1 cell therapy. Cancer Res. 2010, 70, 2697–2706. [Google Scholar] [CrossRef]
- Manda, K.; Glasow, A.; Paape, D.; Hildebrandt, G. Effects of ionizing radiation on the immune system with special emphasis on the interaction of dendritic and T cells. Front. Oncol. 2012, 2, 102. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Vermeer, D.W.; Spanos, W.C.; Vermeer, P.D.; Bruns, A.M.; Lee, K.M.; Lee, J.H. Radiation-induced loss of cell surface CD47 enhances immune-mediated clearance of human papillomavirus-positive cancer. Int. J. Cancer 2013, 133, 120–129. [Google Scholar] [CrossRef]
- Perez, C.A.; Fu, A.; Onishko, H.; Hallahan, D.E.; Geng, L. Radiation induces an antitumour immune response to mouse melanoma. Int. J. Radiat. Biol. 2009, 85, 1126–1136. [Google Scholar]
- Wang, R.; Zhou, T.; Liu, W.; Zuo, L. Molecular mechanism of bystander effects and related abscopal/cohort effects in cancer therapy. Oncotarget 2018, 9, 18637–18647. [Google Scholar] [CrossRef]
- Calveley, V.L.; Khan, M.A.; Yeung, I.W.; Vandyk, J.; Hill, R.P. Partial volume rat lung irradiation: Temporal fluctuations of in-field and out-of-field DNA damage and inflammatory cytokines following irradiation. Int. J. Radiat. Biol. 2005, 81, 887–899. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; de Andrade, L.F.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K.; Potes, Y.; Mirtavoos-Mahyari, H.; Motevaseli, E.; Shabeeb, D.; Musa, A.E.; Najafi, M.; Farhood, B. Boosting immune system against cancer by melatonin: A mechanistic viewpoint. Life Sci. 2019, 238, 116960. [Google Scholar] [CrossRef] [PubMed]
- Daguenet, E.; Louati, S.; Wozny, A.S.; Vial, N.; Gras, M.; Guy, J.B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef]
- Tesei, A.; Arienti, C.; Bossi, G.; Santi, S.; De Santis, I.; Bevilacqua, A.; Zanoni, M.; Pignatta, S.; Cortesi, M.; Zamagni, A.; et al. TP53 drives abscopal effect by secretion of senescence-associated molecular signals in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2021, 40, 89. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, Q.; Wang, R.; Li, Y.; Sun, Y.; Yang, L. Targeting DNA Damage Repair for Immune Checkpoint Inhibition: Mechanisms and Potential Clinical Applications. Front. Oncol. 2021, 11, 648687. [Google Scholar] [CrossRef]
- Germano, G.; Amirouchene-Angelozzi, N.; Rospo, G.; Bardelli, A. The Clinical Impact of the Genomic Landscape of Mismatch Repair-Deficient Cancers. Cancer Discov. 2018, 8, 1518–1528. [Google Scholar]
- He, Y.; Zhang, L.; Zhou, R.; Wang, Y.; Chen, H. The role of DNA mismatch repair in immunotherapy of human cancer. Int. J. Biol. Sci. 2022, 18, 2821–2832. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Wang, Z.; Li, A.; Lu, Y.; Han, M.; Ruan, M.; Wang, C.; Zhang, X.; Zhu, C.; Shen, K.; Dong, L.; et al. Association of tumor immune infiltration and prognosis with homologous recombination repair genes mutations in early triple-negative breast cancer. Front. Immunol. 2024, 15, 1407837. [Google Scholar] [CrossRef]
- van Wilpe, S.; Simnica, D.; Slootbeek, P.; van Ee, T.; Pamidimarri Naga, S.; Gorris, M.A.J.; van der Woude, L.L.; Sultan, S.; Koornstra, R.H.T.; van Oort, I.M.; et al. Homologous recombination repair deficient prostate cancer represents an immunologically distinct subtype. Oncoimmunology 2022, 11, 2094133. [Google Scholar] [CrossRef] [PubMed]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- He, M.; Jiang, H.; Li, S.; Xue, M.; Wang, H.; Zheng, C.; Tong, J. The crosstalk between DNA-damage responses and innate immunity. Int. Immunopharmacol. 2024, 140, 112768. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Rivero Belenchón, I.; Congregado Ruiz, C.B.; Saez, C.; Osman García, I.; Medina López, R.A. Parp Inhibitors and Radiotherapy: A New Combination for Prostate Cancer (Systematic Review). Int. J. Mol. Sci. 2023, 24, 12978. [Google Scholar] [CrossRef] [PubMed]
- Slade, D. PARP and PARG Inhibitors in Cancer Treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, J.; Hildebrand, L.S.; Kuhlmann, L.; Fietkau, R.; Distel, L.V. Effective radiosensitization of HNSCC cell lines by DNA-PKcs Inhibitor AZD7648 and PARP Inhibitors Talazoparib and Niraparib. Int. J. Mol. Sci. 2024, 25, 5629. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.M.; Ngoi, N.Y.L.; Peng, G.; Tan, D.S.P.; Yap, T.A. Development of poly(ADP-ribose) polymerase inhibitor and immunotherapy combinations: Progress, pitfalls, and promises. Trends Cancer 2021, 7, 958–970. [Google Scholar] [CrossRef]
- Luo, Y.; Nie, M.; Chen, Y.; Huang, X.; Cui, X.; Lin, W.; Xu, L.; Li, E.; Cheng, Y. Clinical Application of PARP1 Inhibitors and Challenges in Cancer Therapy. Curr. Cancer Drug Targets 2025. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, Y.; Lin, M.; Li, Z.; Shen, Z.; Yin, S.; Zheng, Y.; Zou, Y.; Zhang, Y.; Zhan, Y.; et al. Targeting ATM enhances radiation sensitivity of colorectal cancer by potentiating radiation-induced cell death and antitumor immunity. J. Adv. Res. 2024. [Google Scholar] [CrossRef]
- Meidenbauer, J.; Wachter, M.; Schulz, S.R.; Mostafa, N.; Zülch, L.; Frey, B.; Fietkau, R.; Gaipl, U.S.; Jost, T. Inhibition of ATM or ATR in combination with hypo-fractionated radiotherapy leads to a different immunophenotype on transcript and protein level in HNSCC. Front. Oncol. 2024, 14, 1460150. [Google Scholar] [CrossRef]
- Kwon, M.; Kim, G.; Kim, R.; Kim, K.T.; Kim, S.T.; Smith, S.; Mortimer, P.G.S.; Hong, J.Y.; Loembé, A.B.; Irurzun-Arana, I.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J. Immunother. Cancer 2022, 10, e005041. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Chan, G.K.; Gamper, A.M. Targeting the DNA Damage Response for Cancer Therapy by Inhibiting the Kinase Wee1. Front. Oncol. 2022, 12, 828684. [Google Scholar] [CrossRef]
- Al-Jamaei, A.H.; de Visscher, J.G.A.M.; Subramanyam, V.R.; Forouzanfar, T.; Sminia, P.; Doulabi, B.Z.; Helder, M.N. WEE1 kinase inhibitor MK-1775 sensitizes oral tongue squamous cell carcinoma cells to radiation irrespective of TP53 status. Oral Dis. 2023, 29, 2640–2649. [Google Scholar] [CrossRef] [PubMed]
- PosthumaDeBoer, J.; Würdinger, T.; Graat, H.C.; van Beusechem, V.W.; Helder, M.N.; van Royen, B.J.; Kaspers, G.J. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer 2011, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Falchook, G.S.; Wang, J.S.; Imedio, E.R.; Kumar, S.; Miah, K.; Mugundu, G.M.; Jones, S.F.; Spigel, D.R.; Hamilton, E.P. Open-Label, Multicenter, Phase I Study to Assess Safety and Tolerability of Adavosertib Plus Durvalumab in Patients with Advanced Solid Tumors. Target Oncol. 2024, 20, 127–138. [Google Scholar] [CrossRef] [PubMed]
- González Besteiro, M.A.; Gottifredi, V. The fork and the kinase: A DNA replication tale from a CHK1 perspective. Mutat. Res. Rev. Mutat. Res. 2015, 763, 168–180. [Google Scholar] [CrossRef]
- Güster, J.D.; Weissleder, S.V.; Busch, C.J.; Kriegs, M.; Petersen, C.; Knecht, R.; Dikomey, E.; Rieckmann, T. The inhibition of PARP but not EGFR results in the radiosensitization of HPV/p16-positive HNSCC cell lines. Radiother. Oncol. 2014, 113, 345–351. [Google Scholar] [CrossRef]
- Patel, R.; Barker, H.E.; Kyula, J.; McLaughlin, M.; Dillon, M.T.; Schick, U.; Hafsi, H.; Thompson, A.; Khoo, V.; Harrington, K.; et al. An orally bioavailable Chk1 inhibitor, CCT244747, sensitizes bladder and head and neck cancer cell lines to radiation. Radiother. Oncol. 2017, 122, 470–475. [Google Scholar]
- Barker, H.E.; Patel, R.; McLaughlin, M.; Schick, U.; Zaidi, S.; Nutting, C.M.; Newbold, K.L.; Bhide, S.; Harrington, K.J. CHK1 Inhibition Radiosensitizes Head and Neck Cancers to Paclitaxel-Based Chemoradiotherapy. Mol. Cancer Ther. 2016, 15, 2042–2054. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity Through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef]
- Sen, T.; Della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination Treatment of the Oral Chk1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef]
- Lohberger, B.; Barna, S.; Glänzer, D.; Eck, N.; Leithner, A.; Georg, D. DNA-PKcs Inhibition Sensitizes Human Chondrosarcoma Cells to Carbon Ion Irradiation via Cell Cycle Arrest and Telomere Capping Disruption. Int. J. Mol. Sci. 2024, 25, 6179. [Google Scholar] [CrossRef]
- Qian, J.; Liao, G.; Chen, M.; Peng, R.W.; Yan, X.; Du, J.; Huang, R.; Pan, M.; Lin, Y.; Gong, X.; et al. Advancing cancer therapy: New frontiers in targeting DNA damage response. Front. Pharmacol. 2024, 15, 1474337. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Wei, W.; Yuan, X.; Guo, E.; Peng, P.; Wang, J.; Sun, W. Targeting DNA Damage Repair to Enhance Antitumor Immunity in Radiotherapy: Mechanisms and Opportunities. Int. J. Mol. Sci. 2025, 26, 3743. https://doi.org/10.3390/ijms26083743
Yang L, Wei W, Yuan X, Guo E, Peng P, Wang J, Sun W. Targeting DNA Damage Repair to Enhance Antitumor Immunity in Radiotherapy: Mechanisms and Opportunities. International Journal of Molecular Sciences. 2025; 26(8):3743. https://doi.org/10.3390/ijms26083743
Chicago/Turabian StyleYang, Lin, Wenjie Wei, Xun Yuan, Ergang Guo, Ping Peng, Jing Wang, and Wei Sun. 2025. "Targeting DNA Damage Repair to Enhance Antitumor Immunity in Radiotherapy: Mechanisms and Opportunities" International Journal of Molecular Sciences 26, no. 8: 3743. https://doi.org/10.3390/ijms26083743
APA StyleYang, L., Wei, W., Yuan, X., Guo, E., Peng, P., Wang, J., & Sun, W. (2025). Targeting DNA Damage Repair to Enhance Antitumor Immunity in Radiotherapy: Mechanisms and Opportunities. International Journal of Molecular Sciences, 26(8), 3743. https://doi.org/10.3390/ijms26083743