
Extracellular Vesicles in Renal Inflammatory Diseases: Revealing Mechanisms of Extracellular Vesicle-Mediated Macrophage Regulation
Abstract

1. Introduction
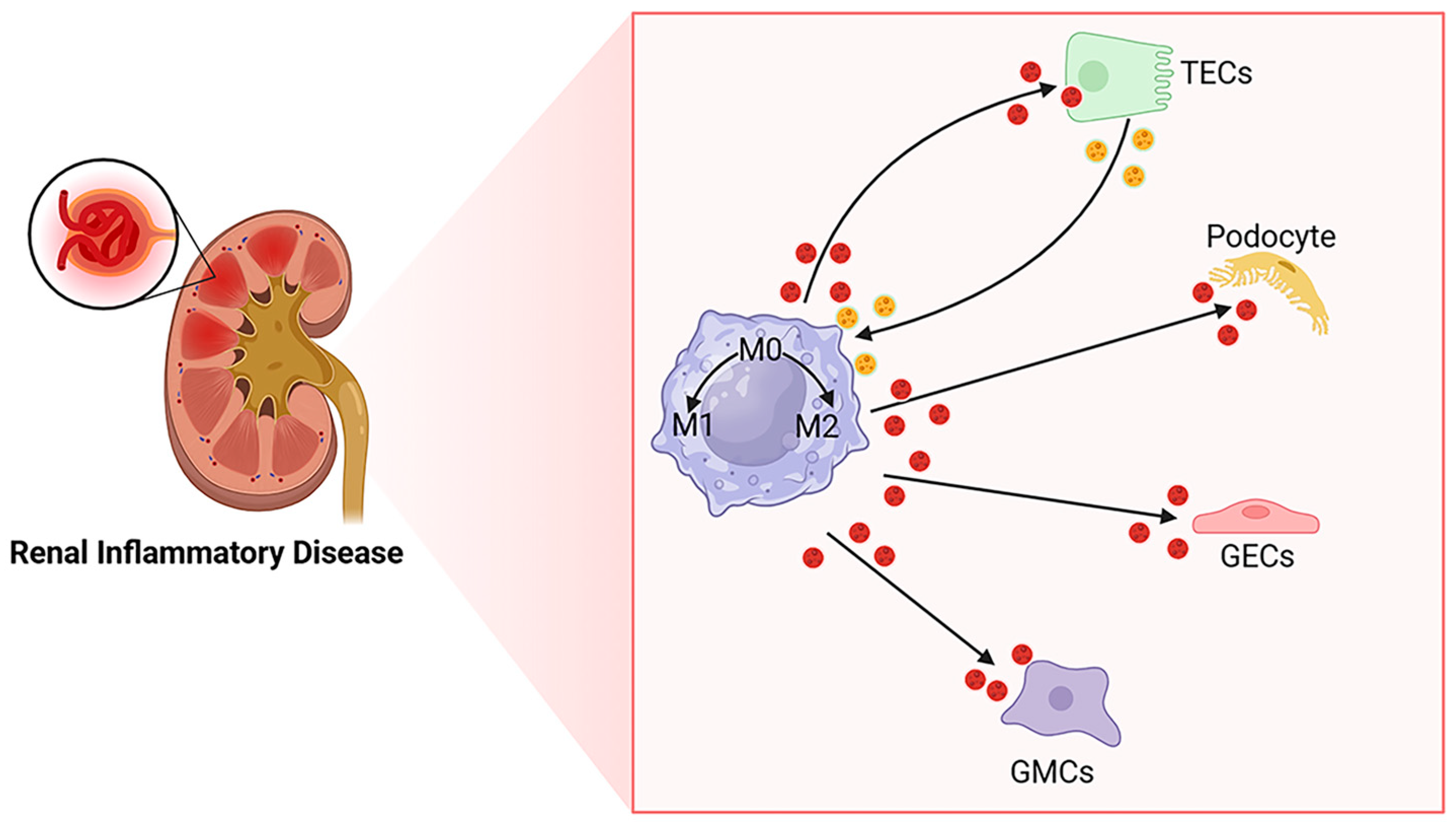
2. Macrophages and EVs in the Kidney
2.1. Origin of Macrophages in the Kidney
2.2. Plasticity and Polarization of Macrophages in the Kidney
2.3. EVs and Kidney Disease
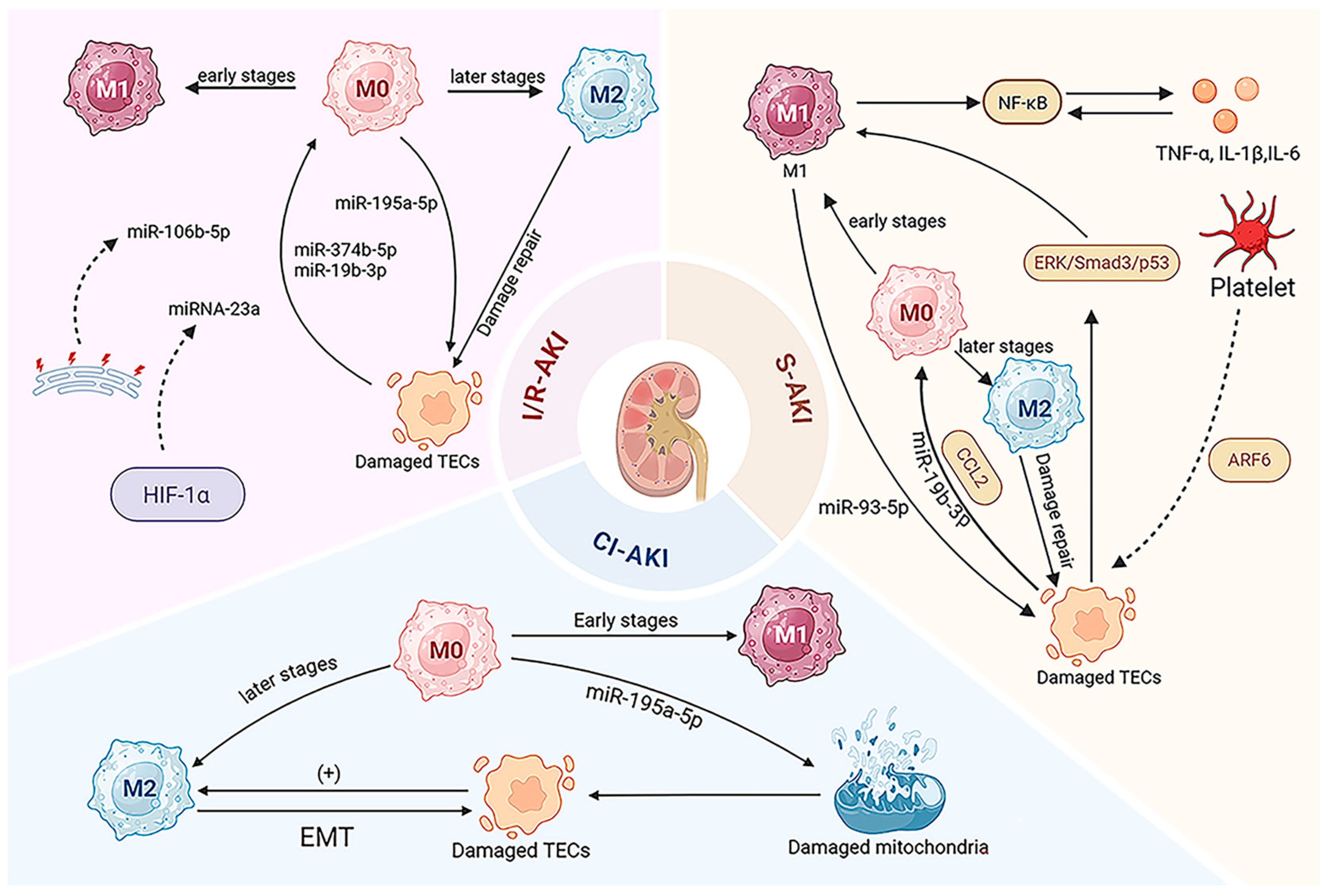
3. EV-Mediated Macrophage Regulation in AKI
3.1. Ischemia/Reperfusion AKI (I/R-AKI)
3.2. Sepsis-Induced AKI (S-AKI)
3.3. Cisplatin-Induced AKI (CI-AKI)
3.4. EVs as Diagnostic Biomarkers for AKI
3.5. AKI Therapeutic Strategies Targeting EV-Regulated Macrophages
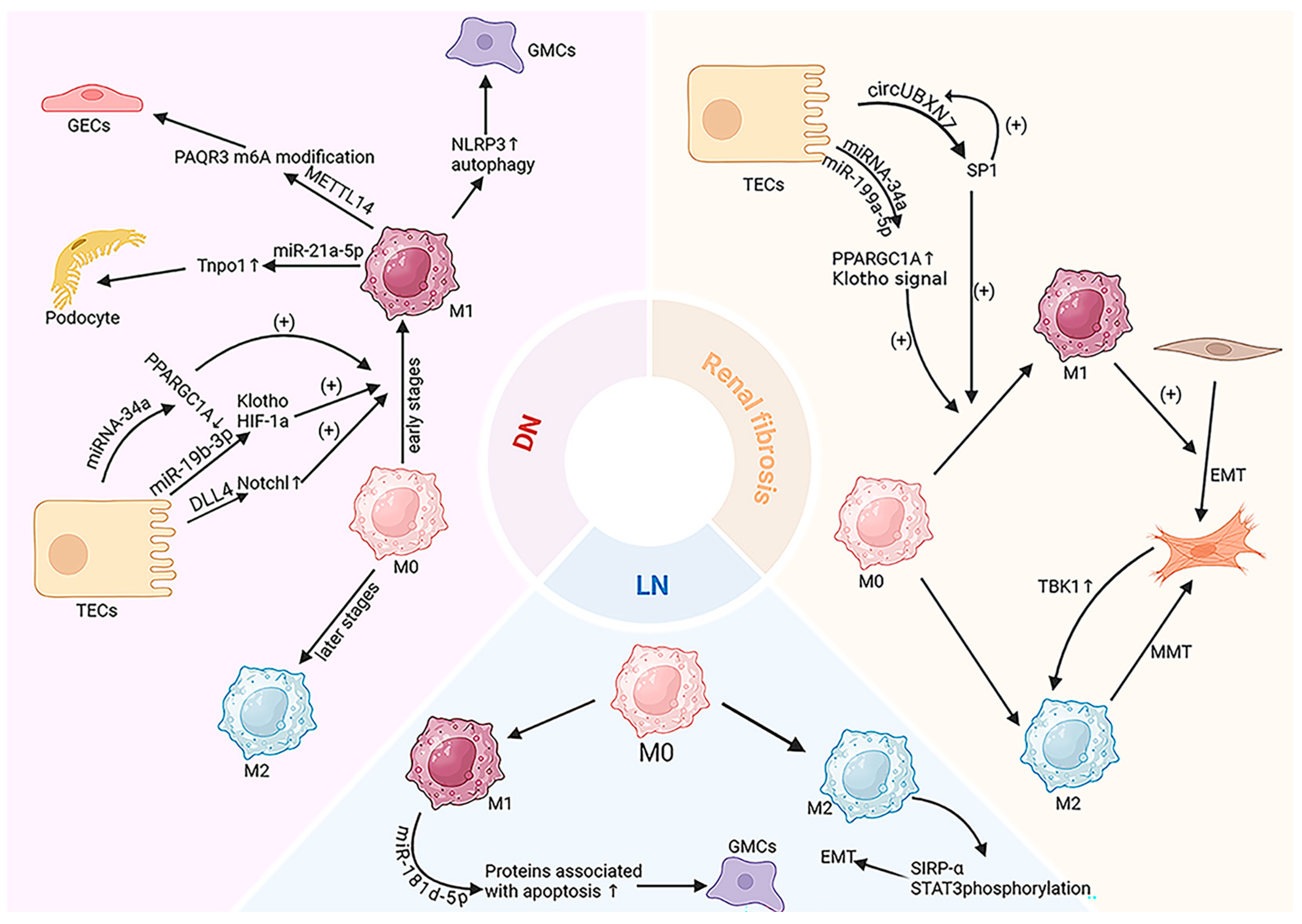
4. EV-Mediated Macrophage Regulation in CKD
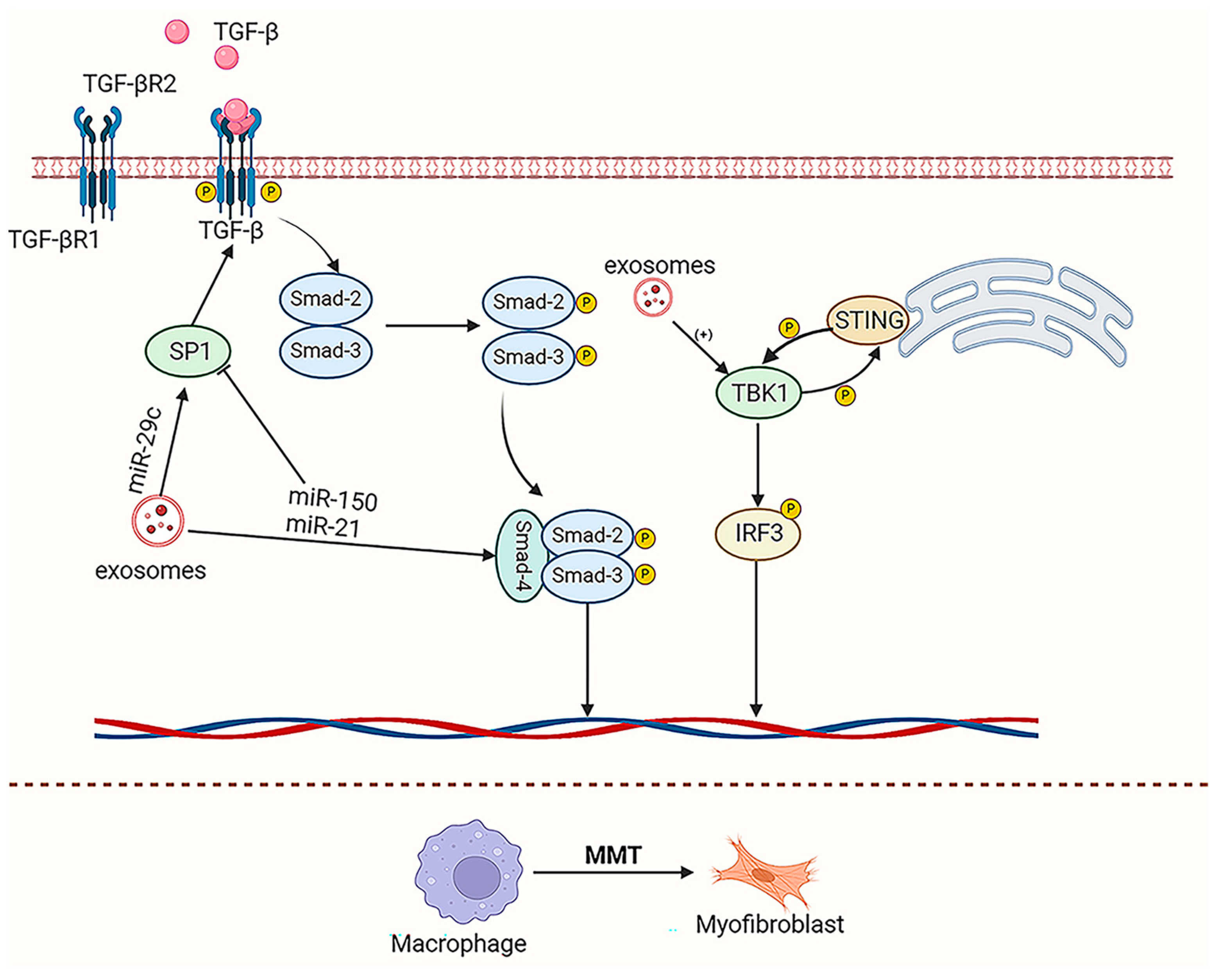
4.1. Kidney Fibrosis
4.2. Diabetic Nephropathy (DN)
4.3. Lupus Nephritis (LN)
4.4. EVs as Diagnostic Biomarkers for CKD
4.5. CKD Therapeutic Strategies Targeting EV-Regulated Macrophages
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luyckx, V.A.; Tonelli, M.; Stanifer, J.W. The Global Burden of Kidney Disease and the Sustainable Development Goals. Bull. World Health Organ. 2018, 96, 414–422D. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P. Epidemiology of Chronic Kidney Disease: An Update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef]
- Anders, H.-J.; Kitching, A.R.; Leung, N.; Romagnani, P. Glomerulonephritis: Immunopathogenesis and Immunotherapy. Nat. Rev. Immunol. 2023, 23, 453–471. [Google Scholar] [CrossRef]
- Das, F.; Bera, A.; Ghosh-Choudhury, N.; Sataranatarajan, K.; Kamat, A.; Kasinath, B.S.; Choudhury, G.G. High Glucose-Stimulated Enhancer of Zeste Homolog-2 (EZH2) Forces Suppression of Deptor to Cause Glomerular Mesangial Cell Pathology. Cell. Signal. 2021, 86, 110072. [Google Scholar] [CrossRef]
- GBD Chronic Kidney Disease Collaboration. Global, Regional, and National Burden of Chronic Kidney Disease, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Tang, P.M.-K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: Versatile Players in Renal Inflammation and Fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.-S.; Ruhrberg, C.; Cantley, L.G. Distinct Macrophage Phenotypes Contribute to Kidney Injury and Repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal Information for Studies of Extracellular Vesicles (MISEV2023): From Basic to Advanced Approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef]
- Kumar, M.A.; Baba, S.K.; Sadida, H.Q.; Marzooqi, S.A.; Jerobin, J.; Altemani, F.H.; Algehainy, N.; Alanazi, M.A.; Abou-Samra, A.-B.; Kumar, R.; et al. Extracellular Vesicles as Tools and Targets in Therapy for Diseases. Signal Transduct. Target. Ther. 2024, 9, 27. [Google Scholar] [CrossRef]
- Barile, L.; Marbán, E. Injury Minimization after Myocardial Infarction: Focus on Extracellular Vesicles. Eur. Heart J. 2024, 45, 1602–1609. [Google Scholar] [CrossRef]
- Dou, G.; Tian, R.; Liu, X.; Yuan, P.; Ye, Q.; Liu, J.; Liu, S.; Zhou, J.; Deng, Z.; Chen, X.; et al. Chimeric Apoptotic Bodies Functionalized with Natural Membrane and Modular Delivery System for Inflammation Modulation. Sci. Adv. 2020, 6, eaba2987. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current Knowledge on Exosome Biogenesis and Release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Fussenegger, M. Shedding Light on Extracellular Vesicle Biogenesis and Bioengineering. Adv. Sci. 2020, 8, 2003505. [Google Scholar] [CrossRef]
- Koga, K.; Matsumoto, K.; Akiyoshi, T.; Kubo, M.; Yamanaka, N.; Tasaki, A.; Nakashima, H.; Nakamura, M.; Kuroki, S.; Tanaka, M.; et al. Purification, Characterization and Biological Significance of Tumor-Derived Exosomes. Anticancer Res. 2005, 25, 3703–3707. [Google Scholar]
- Raimondo, F.; Morosi, L.; Chinello, C.; Magni, F.; Pitto, M. Advances in Membranous Vesicle and Exosome Proteomics Improving Biological Understanding and Biomarker Discovery. Proteomics 2011, 11, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, D.; Song, Y.; He, R.; Wang, T. Research Progress in the Application of Exosomes in Immunotherapy. Front. Immunol. 2022, 13, 731516. [Google Scholar] [CrossRef]
- Joo, H.S.; Suh, J.H.; Lee, H.J.; Bang, E.S.; Lee, J.M. Current Knowledge and Future Perspectives on Mesenchymal Stem Cell-Derived Exosomes as a New Therapeutic Agent. Int. J. Mol. Sci. 2020, 21, 727. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, Biologic Function and Clinical Potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The Exosome Journey: From Biogenesis to Uptake and Intracellular Signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Li, Z.-L.; Lv, L.-L.; Tang, T.-T.; Wang, B.; Feng, Y.; Zhou, L.-T.; Cao, J.-Y.; Tang, R.-N.; Wu, M.; Liu, H.; et al. HIF-1α Inducing Exosomal microRNA-23a Expression Mediates the Cross-Talk between Tubular Epithelial Cells and Macrophages in Tubulointerstitial Inflammation. Kidney Int. 2019, 95, 388–404. [Google Scholar] [CrossRef]
- Liu, J.-L.; Zhang, L.; Huang, Y.; Li, X.-H.; Liu, Y.-F.; Zhang, S.-M.; Zhao, Y.-E.; Chen, X.-J.; Liu, Y.; He, L.-Y.; et al. Epsin1-Mediated Exosomal Sorting of Dll4 Modulates the Tubular-Macrophage Crosstalk in Diabetic Nephropathy. Mol. Ther. 2023, 31, 1451–1467. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-L.; Feng, Y.; Wu, M.; Wang, B.; Li, Z.-L.; Zhong, X.; Wu, W.-J.; Chen, J.; Ni, H.-F.; Tang, T.-T.; et al. Exosomal miRNA-19b-3p of Tubular Epithelial Cells Promotes M1 Macrophage Activation in Kidney Injury. Cell Death Differ. 2020, 27, 210–226. [Google Scholar] [CrossRef]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bai, J.; Wang, Y.; Zhang, K.; Li, Y.; Qian, H.; Zhang, H.; Wang, L. Feruloylated Arabinoxylan from Wheat Bran Inhibited M1-Macrophage Activation and Enhanced M2-Macrophage Polarization. Int. J. Biol. Macromol. 2022, 194, 993–1001. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Marquez-Expósito, L.; Rodrigues-Diez, R.; Sanz, A.B.; Guiteras, R.; Doladé, N.; Rubio-Soto, I.; Manonelles, A.; Codina, S.; Ortiz, A.; et al. Molecular Mechanisms of Kidney Injury and Repair. Int. J. Mol. Sci. 2022, 23, 1542. [Google Scholar] [CrossRef]
- Chen, T.; Cao, Q.; Wang, Y.; Harris, D.C.H. M2 Macrophages in Kidney Disease: Biology, Therapies, and Perspectives. Kidney Int. 2019, 95, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Wang, S.; Hu, Y.; Wang, L.; Jiang, X.; Zhang, J.; Liu, X.; Guo, X.; Luo, Z.; Zhu, C.; et al. Precisely Regulating M2 Subtype Macrophages for Renal Fibrosis Resolution. ACS Nano 2023, 17, 22508–22526. [Google Scholar] [CrossRef]
- Thongboonkerd, V. Roles for Exosome in Various Kidney Diseases and Disorders. Front. Pharmacol. 2019, 10, 1655. [Google Scholar] [CrossRef]
- Dear, J.W.; Street, J.M.; Bailey, M.A. Urinary Exosomes: A Reservoir for Biomarker Discovery and Potential Mediators of Intrarenal Signalling. Proteomics 2013, 13, 1572–1580. [Google Scholar] [CrossRef]
- Gu, W.; Gong, L.; Wu, X.; Yao, X. Hypoxic TAM-Derived Exosomal miR-155-5p Promotes RCC Progression through HuR-Dependent IGF1R/AKT/PI3K Pathway. Cell Death Discov. 2021, 7, 147. [Google Scholar] [CrossRef]
- Lv, L.-L.; Feng, Y.; Wen, Y.; Wu, W.-J.; Ni, H.-F.; Li, Z.-L.; Zhou, L.-T.; Wang, B.; Zhang, J.-D.; Crowley, S.D.; et al. Exosomal CCL2 from Tubular Epithelial Cells Is Critical for Albumin-Induced Tubulointerstitial Inflammation. J. Am. Soc. Nephrol. 2018, 29, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, H.; Zhou, L.; Li, C.; Lu, G.; Wang, L. Macrophage-derived Exosomal miRNA-155 Promotes Tubular Injury in Ischemia-induced Acute Kidney Injury. Int. J. Mol. Med. 2022, 50, 116. [Google Scholar] [CrossRef]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.-W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-J.; Yang, J.-J.; Lu, Y.-B.; Liu, Z.-Y.; Wang, X.-X. Mesenchymal Stem Cell-Derived Exosomes: Toward Cell-Free Therapeutic Strategies in Regenerative Medicine. World J. Stem Cells 2020, 12, 814–840. [Google Scholar] [CrossRef] [PubMed]
- Arabpour, M.; Saghazadeh, A.; Rezaei, N. Anti-Inflammatory and M2 Macrophage Polarization-Promoting Effect of Mesenchymal Stem Cell-Derived Exosomes. Int. Immunopharmacol. 2021, 97, 107823. [Google Scholar] [CrossRef]
- Lafrance, J.-P.; Miller, D.R. Acute Kidney Injury Associates with Increased Long-Term Mortality. J. Am. Soc. Nephrol. 2010, 21, 345–352. [Google Scholar] [CrossRef]
- Murugan, R.; Kellum, J.A. Acute Kidney Injury: What’s the Prognosis? Nat. Rev. Nephrol. 2011, 7, 209–217. [Google Scholar] [CrossRef]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute Renal Failure in Critically Ill Patients: A Multinational, Multicenter Study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef]
- Chen, H.; Liu, N.; Zhuang, S. Macrophages in Renal Injury, Repair, Fibrosis Following Acute Kidney Injury and Targeted Therapy. Front. Immunol. 2022, 13, 934299. [Google Scholar] [CrossRef]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute Kidney Injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Choi, M.E. Autophagy in Kidney Disease. Annu. Rev. Physiol. 2020, 82, 297–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiong, M.; Fan, Y.; Liu, C.; Wang, Q.; Yang, D.; Yuan, Y.; Huang, Y.; Wang, S.; Zhang, Y.; et al. Mecp2 Protects Kidney from Ischemia-Reperfusion Injury through Transcriptional Repressing IL-6/STAT3 Signaling. Theranostics 2022, 12, 3896–3910. [Google Scholar] [CrossRef] [PubMed]
- Han, H.I.; Skvarca, L.B.; Espiritu, E.B.; Davidson, A.J.; Hukriede, N.A. The Role of Macrophages during Acute Kidney Injury: Destruction and Repair. Pediatr. Nephrol. 2019, 34, 561–569. [Google Scholar] [CrossRef]
- Ornellas, F.M.; Ornellas, D.S.; Martini, S.V.; Castiglione, R.C.; Ventura, G.M.; Rocco, P.R.; Gutfilen, B.; de Souza, S.A.; Takiya, C.M.; Morales, M.M. Bone Marrow-Derived Mononuclear Cell Therapy Accelerates Renal Ischemia-Reperfusion Injury Recovery by Modulating Inflammatory, Antioxidant and Apoptotic Related Molecules. Cell. Physiol. Biochem. 2017, 41, 1736–1752. [Google Scholar] [CrossRef]
- Han, S.J.; Lee, H.T. Mechanisms and Therapeutic Targets of Ischemic Acute Kidney Injury. Kidney Res. Clin. Pract. 2019, 38, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Liu, J.; Zhang, F.; Wang, Y.; Qin, Y.; Zhou, Z.; Qiu, J.; Fan, Y. CCR2 Positive Exosome Released by Mesenchymal Stem Cells Suppresses Macrophage Functions and Alleviates Ischemia/Reperfusion-Induced Renal Injury. Stem Cells Int. 2016, 2016, 1240301. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Zuk, A. Ischemic Acute Renal Failure: An. Inflammatory Disease? Kidney Int. 2004, 66, 480–485. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular Pathophysiology of Ischemic Acute Kidney Injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Kinsey, G.R.; Okusa, M.D. Role of Leukocytes in the Pathogenesis of Acute Kidney Injury. Crit. Care 2012, 16, 214. [Google Scholar] [CrossRef]
- Yuan, L.; Yang, J.; Liu, F.; Li, L.; Liu, J.; Chen, Y.; Cheng, J.; Lu, Y.; Yuan, Y. Macrophage-Derived Exosomal miR-195a-5p Impairs Tubular Epithelial Cells Mitochondria in Acute Kidney Injury Mice. FASEB J. 2023, 37, e22691. [Google Scholar] [CrossRef]
- Ding, C.; Zheng, J.; Wang, B.; Li, Y.; Xiang, H.; Dou, M.; Qiao, Y.; Tian, P.; Ding, X.; Xue, W. Exosomal MicroRNA-374b-5p From Tubular Epithelial Cells Promoted M1 Macrophages Activation and Worsened Renal Ischemia/Reperfusion Injury. Front. Cell Dev. Biol. 2020, 8, 587693. [Google Scholar] [CrossRef]
- Li, X.; Zhong, Y.; Yue, R.; Xie, J.; Zhang, Y.; Lin, Y.; Li, H.; Xu, Y.; Zheng, D. Inhibition of MiR-106b-5p Mediated by Exosomes Mitigates Acute Kidney Injury by Modulating Transmissible Endoplasmic Reticulum Stress and M1 Macrophage Polarization. J. Cell. Mol. Med. 2023, 27, 2876–2889. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Kerchberger, V.E.; Ware, L.B. The Role of Circulating Cell-Free Hemoglobin in Sepsis-Associated Acute Kidney Injury. Semin. Nephrol. 2020, 40, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Acute Dialysis Quality Initiative Consensus XIII Work Group Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2016, 27, 371–379. [Google Scholar] [CrossRef]
- Zarbock, A.; Nadim, M.K.; Pickkers, P.; Gomez, H.; Bell, S.; Joannidis, M.; Kashani, K.; Koyner, J.L.; Pannu, N.; Meersch, M.; et al. Sepsis-Associated Acute Kidney Injury: Consensus Report of the 28th Acute Disease Quality Initiative Workgroup. Nat. Rev. Nephrol. 2023, 19, 401–417. [Google Scholar] [CrossRef]
- Ergin, B.; Kapucu, A.; Demirci-Tansel, C.; Ince, C. The Renal Microcirculation in Sepsis. Nephrol. Dial. Transplant. 2015, 30, 169–177. [Google Scholar] [CrossRef]
- Ricci, Z.; Ronco, C. Pathogenesis of Acute Kidney Injury during Sepsis. Curr. Drug Targets 2009, 10, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Karl, I.E. The Pathophysiology and Treatment of Sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef]
- Khajevand-Khazaei, M.-R.; Mohseni-Moghaddam, P.; Hosseini, M.; Gholami, L.; Baluchnejadmojarad, T.; Roghani, M. Rutin, a Quercetin Glycoside, Alleviates Acute Endotoxemic Kidney Injury in C57BL/6 Mice via Suppression of Inflammation and up-Regulation of Antioxidants and SIRT1. Eur. J. Pharmacol. 2018, 833, 307–313. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and Target Genes of Rel/NF-kappaB Transcription Factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Liu, S.F.; Malik, A.B. NF-Kappa B Activation as a Pathological Mechanism of Septic Shock and Inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef]
- Jia, P.; Xu, S.; Wang, X.; Wu, X.; Ren, T.; Zou, Z.; Zeng, Q.; Shen, B.; Ding, X. Chemokine CCL2 from Proximal Tubular Epithelial Cells Contributes to Sepsis-Induced Acute Kidney Injury. Am. J. Physiol. Renal Physiol. 2022, 323, F107–F119. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, Y.; Mirzaaghasi, A.; Heo, J.; Kim, Y.N.; Shin, J.H.; Kim, S.; Kim, N.H.; Cho, E.S.; In Yook, J.; et al. Exosome-Based Delivery of Super-Repressor IκBα Relieves Sepsis-Associated Organ Damage and Mortality. Sci. Adv. 2020, 6, eaaz6980. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Ravichandran, K.; Wang, Q.; Ljubanovic, D.; Edelstein, C.L. NF-κB Transcriptional Inhibition Ameliorates Cisplatin-Induced Acute Kidney Injury (AKI). Toxicol. Lett. 2016, 240, 105–113. [Google Scholar] [CrossRef]
- Goh, F.G.; Midwood, K.S. Intrinsic Danger: Activation of Toll-like Receptors in Rheumatoid Arthritis. Rheumatology 2012, 51, 7–23. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB Proteins: Implications in Cancer and Inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lu, Y. Omega-3 Fatty Acid-Derived Resolvins and Protectins in Inflammation Resolution and Leukocyte Functions: Targeting Novel Lipid Mediator Pathways in Mitigation of Acute Kidney Injury. Front. Immunol. 2013, 4, 13. [Google Scholar] [CrossRef]
- Li, X.; Mu, G.; Song, C.; Zhou, L.; He, L.; Jin, Q.; Lu, Z. Role of M2 Macrophages in Sepsis-Induced Acute Kidney Injury. Shock 2018, 50, 233–239. [Google Scholar] [CrossRef]
- Zhang, R.; Zhu, Y.; Li, Y.; Liu, W.; Yin, L.; Yin, S.; Ji, C.; Hu, Y.; Wang, Q.; Zhou, X.; et al. Human Umbilical Cord Mesenchymal Stem Cell Exosomes Alleviate Sepsis-Associated Acute Kidney Injury via Regulating microRNA-146b Expression. Biotechnol. Lett. 2020, 42, 669–679. [Google Scholar] [CrossRef]
- Lu, X.; Jiang, G.; Gao, Y.; Chen, Q.; Sun, S.; Mao, W.; Zhang, N.; Zhu, Z.; Wang, D.; Zhang, G.; et al. Platelet-Derived Extracellular Vesicles Aggravate Septic Acute Kidney Injury via Delivering ARF6. Int. J. Biol. Sci. 2023, 19, 5055–5073. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Fang, J.-T.; Zhang, N.-S.; Qin, L.-J.; Zhuang, Y.-Y.; Wang, W.-W.; Zhu, H.-P.; Zhang, Y.-J.; Xia, P.; Zhang, Y. Caspase-1-Inhibitor AC-YVAD-CMK Inhibits Pyroptosis and Ameliorates Acute Kidney Injury in a Model of Sepsis. Biomed. Res. Int. 2021, 2021, 6636621. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Vilaysane, A.; Lau, A.; Stahl, M.; Morampudi, V.; Bondzi-Simpson, A.; Platnich, J.M.; Bracey, N.A.; French, M.-C.; Beck, P.L.; et al. NLRP3 Regulates a Non-Canonical Platform for Caspase-8 Activation during Epithelial Cell Apoptosis. Cell Death Differ. 2016, 23, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Chung, H.; Lau, A.; Platnich, J.M.; Beck, P.L.; Benediktsson, H.; Duff, H.J.; Jenne, C.N.; Muruve, D.A. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J. Am. Soc. Nephrol. 2018, 29, 1165–1181. [Google Scholar] [CrossRef]
- Li, C.; Wang, W.; Xie, S.-S.; Ma, W.-X.; Fan, Q.-W.; Chen, Y.; He, Y.; Wang, J.-N.; Yang, Q.; Li, H.; et al. The Programmed Cell Death of Macrophages, Endothelial Cells, and Tubular Epithelial Cells in Sepsis-AKI. Front. Med. 2021, 8, 796724. [Google Scholar] [CrossRef]
- Tonnus, W.; Meyer, C.; Paliege, A.; Belavgeni, A.; von Mässenhausen, A.; Bornstein, S.R.; Hugo, C.; Becker, J.U.; Linkermann, A. The Pathological Features of Regulated Necrosis. J. Pathol. 2019, 247, 697–707. [Google Scholar] [CrossRef]
- Sun, Q.; Hu, Z.; Huang, W.; Liu, X.; Wu, X.; Chang, W.; Tang, Y.; Peng, F.; Yang, Y. CircMLH3 Induces Mononuclear Macrophage Pyroptosis in Sepsis by Sponging miR-590-3p to Regulate TAK1 Expression. Int. J. Biol. Macromol. 2024, 263, 130179. [Google Scholar] [CrossRef]
- Xiang, H.; Xu, Z.; Zhang, C.; Xiong, J. Macrophage-Derived Exosomes Mediate Glomerular Endothelial Cell Dysfunction in Sepsis-Associated Acute Kidney Injury. Cell Biosci. 2023, 13, 46. [Google Scholar] [CrossRef]
- Juan, C.-X.; Mao, Y.; Cao, Q.; Chen, Y.; Zhou, L.-B.; Li, S.; Chen, H.; Chen, J.-H.; Zhou, G.-P.; Jin, R. Exosome-Mediated Pyroptosis of miR-93-TXNIP-NLRP3 Leads to Functional Difference between M1 and M2 Macrophages in Sepsis-Induced Acute Kidney Injury. J. Cell. Mol. Med. 2021, 25, 4786–4799. [Google Scholar] [CrossRef]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef]
- Yang, H.; Bi, Y.; Xue, L.; Wang, J.; Lu, Y.; Zhang, Z.; Chen, X.; Chu, Y.; Yang, R.; Wang, R.; et al. Multifaceted Modulation of SIRT1 in Cancer and Inflammation. Crit. Rev. Oncog. 2015, 20, 49–64. [Google Scholar] [CrossRef]
- Bai, X.-Z.; Zhang, J.-L.; Liu, Y.; Zhang, W.; Li, X.-Q.; Wang, K.-J.; Cao, M.-Y.; Zhang, J.-N.; Han, F.; Shi, J.-H.; et al. MicroRNA-138 Aggravates Inflammatory Responses of Macrophages by Targeting SIRT1 and Regulating the NF-κB and AKT Pathways. Cell. Physiol. Biochem. 2018, 49, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Zhang, W.; Li, J.; Zhang, J.; Li, L.; Liu, M.; Yin, W.; Bai, X. Inhibition of SIRT1 by microRNA-9, the Key Point in Process of LPS-Induced Severe Inflammation. Arch. Biochem. Biophys. 2019, 666, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhang, W.; Cui, B.; Gao, J.; Liu, Q.; Yao, M.; Ning, H.; Xing, L. Functional Delivery of lncRNA TUG1 by Endothelial Progenitor Cells Derived Extracellular Vesicles Confers Anti-Inflammatory Macrophage Polarization in Sepsis via Impairing miR-9-5p-Targeted SIRT1 inhibition. Cell Death Dis. 2021, 12, 1056. [Google Scholar]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular Mechanisms of Cisplatin-Induced Nephrotoxicity: A Balance on the Knife Edge between Renoprotection and Tumor Toxicity. J. Biomed. Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, Z. Cisplatin Nephrotoxicity: Mechanisms and Renoprotective Strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Safirstein, R.; Dong, Z. Cisplatin Nephrotoxicity: New Insights and Therapeutic Implications. Nat. Rev. Nephrol. 2023, 19, 53–72. [Google Scholar] [CrossRef]
- Wang, S.-W.; Xu, Y.; Weng, Y.-Y.; Fan, X.-Y.; Bai, Y.-F.; Zheng, X.-Y.; Lou, L.-J.; Zhang, F. Astilbin Ameliorates Cisplatin-Induced Nephrotoxicity through Reducing Oxidative Stress and Inflammation. Food Chem. Toxicol. 2018, 114, 227–236. [Google Scholar] [CrossRef]
- Huang, T.-Y.; Chien, M.-S.; Su, W.-T. Therapeutic Potential of Pretreatment with Exosomes Derived from Stem Cells from the Apical Papilla against Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 5721. [Google Scholar] [CrossRef]
- Wada, Y.; Iyoda, M.; Matsumoto, K.; Suzuki, T.; Tachibana, S.; Kanazawa, N.; Honda, H. Reno-Protective Effect of IL-34 Inhibition on Cisplatin-Induced Nephrotoxicity in Mice. PLoS ONE 2021, 16, e0245340. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, Z.; He, L.; Wang, Y. CXCL16 Regulates Cisplatin-Induced Acute Kidney Injury. Oncotarget 2016, 7, 31652–31662. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, Y.; Harris, D.C.H. Pathogenic and Protective Role of Macrophages in Kidney Disease. Am. J. Physiol. Renal Physiol. 2013, 305, F3–F11. [Google Scholar] [CrossRef]
- Yu, C.-C.; Chien, C.-T.; Chang, T.-C. M2 Macrophage Polarization Modulates Epithelial-Mesenchymal Transition in Cisplatin-Induced Tubulointerstitial Fibrosis. Biomedicine 2016, 6, 5. [Google Scholar] [CrossRef]
- Gao, J.; Deng, Q.; Yu, J.; Wang, C.; Wei, W. Role of Renal Tubular Epithelial Cells and Macrophages in Cisplatin-Induced Acute Renal Injury. Life Sci. 2024, 339, 122450. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jin, H.; Cai, H.; Zhu, X.; Yang, Y.; Wu, J.; Qi, C.; Shao, X.; Li, J.; et al. Mitophagy Alleviates Cisplatin-Induced Renal Tubular Epithelial Cell Ferroptosis through ROS/HO-1/GPX4 Axis. Int. J. Biol. Sci. 2023, 19, 1192–1210. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy Protects the Proximal Tubule from Degeneration and Acute Ischemic Injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yuan, L.; Yang, J.; Liu, F.; Liu, S.; Li, L.; Liao, G.; Tang, X.; Cheng, J.; Liu, J.; et al. Autophagy-Deficient Macrophages Exacerbate Cisplatin-Induced Mitochondrial Dysfunction and Kidney Injury via miR-195a-5p-SIRT3 Axis. Nat. Commun. 2024, 15, 4383. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-R.; Ye, L.; An, N.; Wu, C.-Y.; Wu, H.-L.; Li, H.-Y.; Huang, Y.-H.; Ye, Q.-R.; Liu, M.-D.; Yang, L.-W.; et al. Macrophage Autophagy Protects against Acute Kidney Injury by Inhibiting Renal Inflammation through the Degradation of TARM1. Autophagy 2024, 21, 120–140. [Google Scholar] [CrossRef]
- Yang, L.; Xing, G.; Wang, L.; Wu, Y.; Li, S.; Xu, G.; He, Q.; Chen, J.; Chen, M.; Liu, X.; et al. Acute Kidney Injury in China: A Cross-Sectional Survey. Lancet 2015, 386, 1465–1471. [Google Scholar] [CrossRef]
- Hu, J.; Liu, S.; Jia, P.; Xu, X.; Song, N.; Zhang, T.; Chen, R.; Ding, X. Protection of Remote Ischemic Preconditioning against Acute Kidney Injury: A Systematic Review and Meta-Analysis. Crit. Care 2016, 20, 111. [Google Scholar] [CrossRef]
- Yang, Y.; Song, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; Dong, Z. Renoprotective Approaches and Strategies in Acute Kidney Injury. Pharmacol. Ther. 2016, 163, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Boukouris, S.; Mathivanan, S. Exosomes in Bodily Fluids Are a Highly Stable Resource of Disease Biomarkers. Proteom. Clin. Appl. 2015, 9, 358–367. [Google Scholar] [CrossRef]
- Jiang, X.-C.; Gao, J.-Q. Exosomes as Novel Bio-Carriers for Gene and Drug Delivery. Int. J. Pharm. 2017, 521, 167–175. [Google Scholar] [CrossRef]
- Xie, X.; Yang, X.; Wu, J.; Tang, S.; Yang, L.; Fei, X.; Wang, M. Exosome from Indoleamine 2,3-Dioxygenase-Overexpressing Bone Marrow Mesenchymal Stem Cells Accelerates Repair Process of Ischemia/Reperfusion-Induced Acute Kidney Injury by Regulating Macrophages Polarization. Stem Cell Res. Ther. 2022, 13, 367. [Google Scholar] [CrossRef]
- Huang, J.; Cao, H.; Cui, B.; Ma, X.; Gao, L.; Yu, C.; Shen, F.; Yang, X.; Liu, N.; Qiu, A.; et al. Mesenchymal Stem Cells-Derived Exosomes Ameliorate Ischemia/Reperfusion Induced Acute Kidney Injury in a Porcine Model. Front. Cell Dev. Biol. 2022, 10, 899869. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Li, X.; Zhou, X.; Li, Y.; Yu, H.; Peng, X.; Bai, X.; Zhang, C.; Feng, Z.; Mei, Y.; et al. Extracellular Vesicles Secreted from Mesenchymal Stem Cells Ameliorate Renal Ischemia Reperfusion Injury by Delivering miR-100-5p Targeting FKBP5/AKT Axis. Sci. Rep. 2024, 14, 6720. [Google Scholar] [CrossRef]
- Li, X.; Liao, J.; Su, X.; Li, W.; Bi, Z.; Wang, J.; Su, Q.; Huang, H.; Wei, Y.; Gao, Y.; et al. Human Urine-Derived Stem Cells Protect against Renal Ischemia/Reperfusion Injury in a Rat Model via Exosomal miR-146a-5p Which Targets IRAK1. Theranostics 2020, 10, 9561–9578. [Google Scholar] [CrossRef]
- Ti, D.; Hao, H.; Tong, C.; Liu, J.; Dong, L.; Zheng, J.; Zhao, Y.; Liu, H.; Fu, X.; Han, W. LPS-Preconditioned Mesenchymal Stromal Cells Modify Macrophage Polarization for Resolution of Chronic Inflammation via Exosome-Shuttled Let-7b. J. Transl. Med. 2015, 13, 308. [Google Scholar] [CrossRef]
- Yao, M.; Cui, B.; Zhang, W.; Ma, W.; Zhao, G.; Xing, L. Exosomal miR-21 Secreted by IL-1β-Primed-Mesenchymal Stem Cells Induces Macrophage M2 Polarization and Ameliorates Sepsis. Life Sci. 2021, 264, 118658. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, Y.; Zheng, X.; Wang, H.; Kang, X.; Tang, J.; Qu, L.; Shao, X.; Wang, S.; Li, S.; et al. Human Amniotic Epithelial Cells Ameliorate Kidney Damage in Ischemia-Reperfusion Mouse Model of Acute Kidney Injury. Stem Cell Res. Ther. 2020, 11, 410. [Google Scholar] [CrossRef]
- Sun, J.; Sun, X.; Chen, J.; Liao, X.; He, Y.; Wang, J.; Chen, R.; Hu, S.; Qiu, C. microRNA-27b Shuttled by Mesenchymal Stem Cell-Derived Exosomes Prevents Sepsis by Targeting JMJD3 and Downregulating NF-κB Signaling Pathway. Stem Cell Res. Ther. 2021, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, S.; Zheng, B.; Liu, D.; Wan, F.; Ma, Y.; Wang, J.; Gao, Z.; Shan, Z. miR-30c-5p Reduces Renal Ischemia-Reperfusion Involving Macrophage. Med. Sci. Monit. 2019, 25, 4362–4369. [Google Scholar] [CrossRef]
- Yu, W.; Zeng, H.; Chen, J.; Fu, S.; Huang, Q.; Xu, Y.; Xu, A.; Lan, H.-Y.; Tang, Y. miR-20a-5p Is Enriched in Hypoxia-Derived Tubular Exosomes and Protects against Acute Tubular Injury. Clin. Sci. 2020, 134, 2223–2234. [Google Scholar] [CrossRef]
- Rendra, E.; Uhlig, S.; Moskal, I.; Thielemann, C.; Klüter, H.; Bieback, K. Adipose Stromal Cell-Derived Secretome Attenuates Cisplatin-Induced Injury In Vitro Surpassing the Intricate Interplay between Proximal Tubular Epithelial Cells and Macrophages. Cells 2024, 13, 121. [Google Scholar] [CrossRef]
- Tang, T.-T.; Wang, B.; Wu, M.; Li, Z.-L.; Feng, Y.; Cao, J.-Y.; Yin, D.; Liu, H.; Tang, R.-N.; Crowley, S.D.; et al. Extracellular Vesicle-Encapsulated IL-10 as Novel Nanotherapeutics against Ischemic AKI. Sci. Adv. 2020, 6, eaaz0748. [Google Scholar] [CrossRef] [PubMed]
- Chesnaye, N.C.; Ortiz, A.; Zoccali, C.; Stel, V.S.; Jager, K.J. The Impact of Population Ageing on the Burden of Chronic Kidney Disease. Nat. Rev. Nephrol. 2024, 20, 569–585. [Google Scholar] [CrossRef]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- August, P. Chronic Kidney Disease—Another Step Forward. N. Engl. J. Med. 2023, 388, 179–180. [Google Scholar] [CrossRef]
- Harris, R.C.; Neilson, E.G. Toward a Unified Theory of Renal Progression. Annu. Rev. Med. 2006, 57, 365–380. [Google Scholar] [CrossRef]
- Miguel, V.; Shaw, I.W.; Kramann, R. Metabolism at the Crossroads of Inflammation and Fibrosis in Chronic Kidney Disease. Nat. Rev. Nephrol. 2024, 21, 39–56. [Google Scholar] [CrossRef]
- Ammirati, A.L. Chronic Kidney Disease. Rev. Assoc. Med. Bras. 2020, 66 (Suppl. 1), S03–S09. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Miao, N.; Xu, J.; Gan, X.; Xu, D.; Zhou, L.; Xue, H.; Zhang, W.; Lu, L. Metformin Prevents Renal Fibrosis in Mice with Unilateral Ureteral Obstruction and Inhibits Ang II-Induced ECM Production in Renal Fibroblasts. Int. J. Mol. Sci. 2016, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Menon, M.C.; Wang, W.; Fu, J.; Yi, Z.; Sun, Z.; Liu, J.; Li, Z.; Mou, L.; Banu, K.; et al. HCK Induces Macrophage Activation to Promote Renal Inflammation and Fibrosis via Suppression of Autophagy. Nat. Commun. 2023, 14, 4297. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master Regulators of Inflammation and Fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Lovett, D.H. Gelatinase A (MMP-2) Is Necessary and Sufficient for Renal Tubular Cell Epithelial-Mesenchymal Transformation. Am. J. Pathol. 2003, 162, 1937–1949. [Google Scholar] [CrossRef]
- Fu, H.; Gu, Y.-H.; Tan, J.; Yang, Y.-N.; Wang, G.-H. CircACTR2 in Macrophages Promotes Renal Fibrosis by Activating Macrophage Inflammation and Epithelial-Mesenchymal Transition of Renal Tubular Epithelial Cells. Cell. Mol. Life Sci. 2022, 79, 253. [Google Scholar] [CrossRef]
- Lin, D.-W.; Yang, T.-M.; Ho, C.; Shih, Y.-H.; Lin, C.-L.; Hsu, Y.-C. Targeting Macrophages: Therapeutic Approaches in Diabetic Kidney Disease. Int. J. Mol. Sci. 2024, 25, 4350. [Google Scholar] [CrossRef]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages Promote Renal Fibrosis through Direct and Indirect Mechanisms. Kidney Int. Suppl. 2014, 4, 34–38. [Google Scholar] [CrossRef]
- Pan, B.; Liu, G.; Jiang, Z.; Zheng, D. Regulation of Renal Fibrosis by Macrophage Polarization. Cell. Physiol. Biochem. 2015, 35, 1062–1069. [Google Scholar] [CrossRef]
- Wei, J.; Xu, Z.; Yan, X. The Role of the Macrophage-to-Myofibroblast Transition in Renal Fibrosis. Front. Immunol. 2022, 13, 934377. [Google Scholar] [CrossRef]
- Ban, J.-Q.; Ao, L.-H.; He, X.; Zhao, H.; Li, J. Advances in Macrophage-Myofibroblast Transformation in Fibrotic Diseases. Front. Immunol. 2024, 15, 1461919. [Google Scholar] [CrossRef]
- Nakazawa, D.; Masuda, S.; Nishibata, Y.; Watanabe-Kusunoki, K.; Tomaru, U.; Ishizu, A. Neutrophils and NETs in Kidney Disease. Nat. Rev. Nephrol. 2025, 1–16. [Google Scholar] [CrossRef]
- Yu, W.; Song, J.; Chen, S.; Nie, J.; Zhou, C.; Huang, J.; Liang, H. Myofibroblast-Derived Exosomes Enhance Macrophages to Myofibroblasts Transition and Kidney Fibrosis. Ren. Fail. 2024, 46, 2334406. [Google Scholar] [CrossRef]
- Lin, Z.; Lv, D.; Liao, X.; Peng, R.; Liu, H.; Wu, T.; Wu, K.; Sun, Y.; Zhang, Z. CircUBXN7 Promotes Macrophage Infiltration and Renal Fibrosis Associated with the IGF2BP2-Dependent SP1 mRNA Stability in Diabetic Kidney Disease. Front. Immunol. 2023, 14, 1226962. [Google Scholar] [CrossRef]
- Magayr, T.A.; Song, X.; Streets, A.J.; Vergoz, L.; Chang, L.; Valluru, M.K.; Yap, H.L.; Lannoy, M.; Haghighi, A.; Simms, R.J.; et al. Global microRNA Profiling in Human Urinary Exosomes Reveals Novel Disease Biomarkers and Cellular Pathways for Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 2020, 98, 420–435. [Google Scholar] [CrossRef]
- Zheng, C.; Xie, L.; Qin, H.; Liu, X.; Chen, X.; Lv, F.; Wang, L.; Zhu, X.; Xu, J. The Role of Extracellular Vesicles in Systemic Lupus Erythematosus. Front. Cell Dev. Biol. 2022, 10, 835566. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zeng, Y.; Chu, L.; Gong, T.; Li, S.; Yang, M. Renal Tissue-Derived Exosomal miRNA-34a in Diabetic Nephropathy Induces Renal Tubular Cell Fibrosis by Promoting the Polarization of M1 Macrophages. IET Nanobiotechnol. 2024, 2024, 5702517. [Google Scholar] [CrossRef]
- Zhong, Q.; Zeng, J.; Li, Y.; Zhang, H.; Lin, T.; Song, T. Senescent Renal Tubular Cells Derived Extracellular Vesicles Transported miR-20a and miR-21 Induced Macrophage-to-Myofibroblast Transition in Renal Fibrosis after Ischemia Reperfusion Injury. Int. J. Biol. Sci. 2025, 21, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, W.; Yu, W.; Rao, T.; Li, H.; Ruan, Y.; Yuan, R.; Li, C.; Ning, J.; Li, S.; et al. Exosomal miR-21 from Tubular Cells Contributes to Renal Fibrosis by Activating Fibroblasts via Targeting PTEN in Obstructed Kidneys. Theranostics 2021, 11, 8660–8673. [Google Scholar] [CrossRef]
- Quaglia, M.; Dellepiane, S.; Guglielmetti, G.; Merlotti, G.; Castellano, G.; Cantaluppi, V. Extracellular Vesicles as Mediators of Cellular Crosstalk Between Immune System and Kidney Graft. Front. Immunol. 2020, 11, 74. [Google Scholar] [CrossRef]
- Kim, J.; Ha, S.; Son, M.; Kim, D.; Kim, M.-J.; Kim, B.; Kim, D.; Chung, H.Y.; Chung, K.W. TLR7 Activation by miR-21 Promotes Renal Fibrosis by Activating the pro-Inflammatory Signaling Pathway in Tubule Epithelial Cells. Cell Commun. Signal. 2023, 21, 215. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Wang, Y.; Chen, Y.; Jin, L.; Shi, Y.; Liu, C.; Fu, C.; Cao, Y. Exosomes Derived from TREM-2 Knocked-out Macrophages Alleviated Renal Fibrosis via HSPa1b/AKT Pathway. Am. J. Physiol. Renal Physiol. 2025, 328, F131–F151. [Google Scholar] [CrossRef]
- Song, A.; Wang, M.; Xie, K.; Lu, J.; Zhao, B.; Wu, W.; Qian, C.; Hong, W.; Gu, L. Exosomal Let-7b-5p Deriving from Parietal Epithelial Cells Attenuate Renal Fibrosis through Suppression of TGFβR1 and ARID3a in Obstructive Kidney Disease. FASEB J. 2024, 38, e70085. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Tang, T.-T.; Lu, X.-Y.; Ni, W.-J.; Yin, D.; Zhang, Y.-L.; Jiang, W.; Zhang, Y.; Li, Z.-L.; Wen, Y.; et al. Macrophage-Derived Exosomes Promote Telomere Fragility and Senescence in Tubular Epithelial Cells by Delivering miR-155. Cell Commun. Signal. 2024, 22, 357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, J.; Lei, S.; Ren, H.; Zou, Y.; Bai, L.; Zhang, R.; Xu, H.; Li, L.; Zhao, Y.; et al. Combining Glomerular Basement Membrane and Tubular Basement Membrane Assessment Improves the Prediction of Diabetic End-Stage Renal Disease. J. Diabetes 2021, 13, 572–584. [Google Scholar] [CrossRef]
- Yang, L.; Zheng, S.; Epstein, P.N. Metallothionein Over-Expression in Podocytes Reduces Adriamycin Nephrotoxicity. Free Radic. Res. 2009, 43, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Han, L.; Cai, X.; Zhou, H. Exosomal microRNAs: Potential Nanotherapeutic Targets for Diabetic Kidney Disease. Nanomedicine 2023, 18, 1669–1680. [Google Scholar] [CrossRef]
- Tang, S.C.W.; Yiu, W.H. Innate Immunity in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef]
- Mora, C.; Navarro, J.F. Inflammation and Pathogenesis of Diabetic Nephropathy. Metabolism 2004, 53, 265–266, author reply 266–267. [Google Scholar] [CrossRef]
- Li, J.-M.; Li, X.; Chan, L.W.C.; Hu, R.; Zheng, T.; Li, H.; Yang, S. Lipotoxicity-Polarised Macrophage-Derived Exosomes Regulate Mitochondrial Fitness through Miro1-Mediated Mitophagy Inhibition and Contribute to Type 2 Diabetes Development in Mice. Diabetologia 2023, 66, 2368–2386. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zheng, Z.; Xue, M.; Zhang, S.; Hu, F.; Li, Y.; Yang, Y.; Zou, M.; Li, S.; Wang, L.; et al. Extracellular Vesicles from Albumin-Induced Tubular Epithelial Cells Promote the M1 Macrophage Phenotype by Targeting Klotho. Mol. Ther. 2019, 27, 1452–1466. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, J.; Zheng, Z.; Tao, Y.; Zhang, S.; Zou, M.; Yang, Y.; Xue, M.; Hu, F.; Li, Y.; et al. Tubular Epithelial Cell-Derived Extracellular Vesicles Induce Macrophage Glycolysis by Stabilizing HIF-1α in Diabetic Kidney Disease. Mol. Med. 2022, 28, 95. [Google Scholar] [CrossRef]
- Jiang, W.-J.; Xu, C.-T.; Du, C.-L.; Dong, J.-H.; Xu, S.-B.; Hu, B.-F.; Feng, R.; Zang, D.-D.; Meng, X.-M.; Huang, C.; et al. Tubular Epithelial Cell-to-Macrophage Communication Forms a Negative Feedback Loop via Extracellular Vesicle Transfer to Promote Renal Inflammation and Apoptosis in Diabetic Nephropathy. Theranostics 2022, 12, 324–339. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Zhu, Y. METTL14 Derived from Exosomes of M1 Macrophages Promotes High Glucose-Induced Apoptosis, Inflammation and Oxidative Stress in Glomerular Endothelial Cells by Mediating PAQR3 m6A Modification. Clin. Exp. Nephrol. 2024, 28, 1221–1231. [Google Scholar] [CrossRef]
- Ma, T.; Li, X.; Zhu, Y.; Yu, S.; Liu, T.; Zhang, X.; Chen, D.; Du, S.; Chen, T.; Chen, S.; et al. Excessive Activation of Notch Signaling in Macrophages Promote Kidney Inflammation, Fibrosis, and Necroptosis. Front. Immunol. 2022, 13, 835879. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Zhao, M.; Wu, Y.; Xu, Y.; Li, X.; Fu, L.; Han, L.; Zhou, W.; Hu, Q.; et al. Macrophage-Derived Exosomes Promote Activation of NLRP3 Inflammasome and Autophagy Deficiency of Mesangial Cells in Diabetic Nephropathy. Life Sci. 2023, 330, 121991. [Google Scholar] [CrossRef]
- Zhu, M.; Sun, X.; Qi, X.; Xia, L.; Wu, Y. Exosomes from High Glucose-Treated Macrophages Activate Macrophages Andinduce Inflammatory Responses via NF-κB Signaling Pathway in Vitro and in Vivo. Int. Immunopharmacol. 2020, 84, 106551. [Google Scholar] [CrossRef] [PubMed]
- Medica, D.; Franzin, R.; Stasi, A.; Castellano, G.; Migliori, M.; Panichi, V.; Figliolini, F.; Gesualdo, L.; Camussi, G.; Cantaluppi, V. Extracellular Vesicles Derived from Endothelial Progenitor Cells Protect Human Glomerular Endothelial Cells and Podocytes from Complement- and Cytokine-Mediated Injury. Cells 2021, 10, 1675. [Google Scholar] [CrossRef]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte Depletion Causes Glomerulosclerosis: Diphtheria Toxin-Induced Podocyte Depletion in Rats Expressing Human Diphtheria Toxin Receptor Transgene. J. Am. Soc. Nephrol. 2005, 16, 2941–2952. [Google Scholar] [CrossRef]
- Ding, X.; Jing, N.; Shen, A.; Guo, F.; Song, Y.; Pan, M.; Ma, X.; Zhao, L.; Zhang, H.; Wu, L.; et al. MiR-21-5p in Macrophage-Derived Extracellular Vesicles Affects Podocyte Pyroptosis in Diabetic Nephropathy by Regulating A20. J. Endocrinol. Investig. 2021, 44, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Zheng, H.; Yang, Y.; Ni, H. GABA Alleviates High Glucose-Induced Podocyte Injury through Dynamically Altering the Expression of Macrophage M1/M2-Derived Exosomal miR-21a-5p/miR-25-3p. Biochem. Biophys. Res. Commun. 2022, 618, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Saxena, R.; Zhao, M.-H.; Parodis, I.; Salmon, J.E.; Mohan, C. Lupus Nephritis. Nat. Rev. Dis. Primers 2020, 6, 7. [Google Scholar] [CrossRef]
- Richoz, N.; Tuong, Z.K.; Loudon, K.W.; Patiño-Martínez, E.; Ferdinand, J.R.; Portet, A.; Bashant, K.R.; Thevenon, E.; Rucci, F.; Hoyler, T.; et al. Distinct Pathogenic Roles for Resident and Monocyte-Derived Macrophages in Lupus Nephritis. JCI Insight 2022, 7, e159751. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Boström, E.A.; Menke, J.; Rabacal, W.A.; Morel, L.; Wada, T.; Kelley, V.R. Aberrant Macrophages Mediate Defective Kidney Repair That Triggers Nephritis in Lupus-Susceptible Mice. J. Immunol. 2012, 188, 4568–4580. [Google Scholar] [CrossRef]
- Li, W.; Yao, C.; Guo, H.; Ni, X.; Zhu, R.; Wang, Y.; Yu, B.; Feng, X.; Gu, Z.; Da, Z. Macrophages Communicate with Mesangial Cells through the CXCL12/DPP4 Axis in Lupus Nephritis Pathogenesis. Cell Death Dis. 2024, 15, 344. [Google Scholar] [CrossRef]
- Sung, S.-S.J.; Fu, S.M. Interactions among Glomerulus Infiltrating Macrophages and Intrinsic Cells via Cytokines in Chronic Lupus Glomerulonephritis. J. Autoimmun. 2020, 106, 102331. [Google Scholar] [CrossRef]
- Wang, X.; Jia, P.; Ren, T.; Zou, Z.; Xu, S.; Zhang, Y.; Shi, Y.; Bao, S.; Li, Y.; Fang, Y.; et al. MicroRNA-382 Promotes M2-Like Macrophage via the SIRP-α/STAT3 Signaling Pathway in Aristolochic Acid-Induced Renal Fibrosis. Front. Immunol. 2022, 13, 864984. [Google Scholar] [CrossRef]
- Chai, F.; Chang, X.; Lin, Y.; Pang, X.; Luo, S.; Huang, H.; Qin, L.; Lan, Y.; Zeng, Y.; Wang, C. Effect of M0 Macrophage-Derived Exosome miR-181d-5p Targeting BCL-2 to Regulate NLRP3/Caspase-1/GSDMD Pathway on Human Renal Mesangial Cells Pyroptosis. Gene 2024, 908, 148289. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, L.; Chen, X.; Liu, J.; Nie, A.; Chen, X. Bone Marrow Mesenchymal Stem Cell-Derived Exosomes Improve Renal Fibrosis by Reducing the Polarisation of M1 and M2 Macrophages through the Activation of EP2 Receptors. IET Nanobiotechnol. 2022, 16, 14–24. [Google Scholar] [CrossRef]
- Gu, H.; Li, J.; Ni, Y. Sinomenine Improves Renal Fibrosis by Regulating Mesenchymal Stem Cell-Derived Exosomes and Affecting Autophagy Levels. Environ. Toxicol. 2023, 38, 2524–2537. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Li, P.; Chen, M.; Yu, Y.; Wan, Y.; Zhang, Z.; Ren, C.; Shen, L.; Liu, X.; He, D.; et al. Exosomes From Human Umbilical Cord Stem Cells Suppress Macrophage-to-Myofibroblast Transition, Alleviating Renal Fibrosis. Inflammation 2024, 47, 2094–2107. [Google Scholar] [CrossRef]
- Zhang, Y.; Le, X.; Zheng, S.; Zhang, K.; He, J.; Liu, M.; Tu, C.; Rao, W.; Du, H.; Ouyang, Y.; et al. MicroRNA-146a-5p-Modified Human Umbilical Cord Mesenchymal Stem Cells Enhance Protection against Diabetic Nephropathy in Rats through Facilitating M2 Macrophage Polarization. Stem Cell Res. Ther. 2022, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Yin, Y.; Zhao, J.; Hu, R.; Zhang, H.; Hu, J.; Ren, R.; Zhang, Y.; Wang, A.; Lyu, Z.; et al. Exosomes Derived from Umbilical Cord-Derived Mesenchymal Stem Cells Exposed to Diabetic Microenvironment Enhance M2 Macrophage Polarization and Protect against Diabetic Nephropathy. FASEB J. 2024, 38, e23798. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, Y.; Lu, Y.; Li, H.; Xiang, J.; Yang, D.; Wang, J.; Gao, X.; Wang, Y. Urinary Stem Cell-Derived Exocrine circRNA ATG7 Regulates the SOCS1/STAT3 Signaling Pathway through miR-4500, Inhibits M1 Macrophage Polarization, and Alleviates the Progression of Diabetes Nephropathy. Int. Urol. Nephrol. 2024, 56, 1449–1463. [Google Scholar] [CrossRef]
- Huang, H.; Liu, H.; Tang, J.; Xu, W.; Gan, H.; Fan, Q.; Zhang, W. M2 Macrophage-Derived Exosomal miR-25-3p Improves High Glucose-Induced Podocytes Injury through Activation Autophagy via Inhibiting DUSP1 Expression. IUBMB Life 2020, 72, 2651–2662. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, W.; Li, R.; Liu, Y. miRNA-93-5p in Exosomes Derived from M2 Macrophages Improves Lipopolysaccharide-Induced Podocyte Apoptosis by Targeting Toll-like Receptor 4. Bioengineered 2022, 13, 7683–7696. [Google Scholar] [CrossRef]
- Zhang, Z.; Niu, L.; Tang, X.; Feng, R.; Yao, G.; Chen, W.; Li, W.; Feng, X.; Chen, H.; Sun, L. Mesenchymal Stem Cells Prevent Podocyte Injury in Lupus-Prone B6.MRL-Faslpr Mice via Polarizing Macrophage into an Anti-Inflammatory Phenotype. Nephrol. Dial. Transplant. 2019, 34, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Johnson-Stephenson, T.K.; Wang, W.; Wang, Y.; Li, J.; Li, L.; Zen, K.; Chen, X.; Zhu, D. Mesenchymal Stem Cell-Derived Exosome-Educated Macrophages Alleviate Systemic Lupus Erythematosus by Promoting Efferocytosis and Recruitment of IL-17+ Regulatory T Cell. Stem Cell Res. Ther. 2022, 13, 484. [Google Scholar] [CrossRef]
- Luan, J.; Fu, J.; Chen, C.; Jiao, C.; Kong, W.; Zhang, Y.; Chang, Q.; Wang, Y.; Li, D.; Illei, G.G.; et al. LNA-Anti-miR-150 Ameliorated Kidney Injury of Lupus Nephritis by Inhibiting Renal Fibrosis and Macrophage Infiltration. Arthritis Res. Ther. 2019, 21, 276. [Google Scholar] [CrossRef]
- Campion, C.G.; Sanchez-Ferras, O.; Batchu, S.N. Potential Role of Serum and Urinary Biomarkers in Diagnosis and Prognosis of Diabetic Nephropathy. Can. J. Kidney Health Dis. 2017, 4, 2054358117705371. [Google Scholar] [CrossRef] [PubMed]
- Masaoutis, C.; Al Besher, S.; Koutroulis, I.; Theocharis, S. Exosomes in Nephropathies: A Rich Source of Novel Biomarkers. Dis. Markers 2020, 2020, 8897833. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.E.; Blaine, C.; Dawnay, A.; Devonald, M.A.J.; Ftouh, S.; Laing, C.; Latchem, S.; Lewington, A.; Milford, D.V.; Ostermann, M. The Definition of Acute Kidney Injury and Its Use in Practice. Kidney Int. 2015, 87, 62–73. [Google Scholar] [CrossRef]
- Pisitkun, T.; Shen, R.-F.; Knepper, M.A. Identification and Proteomic Profiling of Exosomes in Human Urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef]
- Salih, M.; Zietse, R.; Hoorn, E.J. Urinary Extracellular Vesicles and the Kidney: Biomarkers and Beyond. Am. J. Physiol. Renal Physiol. 2014, 306, F1251–F1259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Pisitkun, T.; Aponte, A.; Yuen, P.S.T.; Hoffert, J.D.; Yasuda, H.; Hu, X.; Chawla, L.; Shen, R.-F.; Knepper, M.A.; et al. Exosomal Fetuin-A Identified by Proteomics: A Novel Urinary Biomarker for Detecting Acute Kidney Injury. Kidney Int. 2006, 70, 1847–1857. [Google Scholar] [CrossRef]
- Ugarte, F.; Santapau, D.; Gallardo, V.; Garfias, C.; Yizmeyián, A.; Villanueva, S.; Sepúlveda, C.; Rocco, J.; Pasten, C.; Urquidi, C.; et al. Urinary Extracellular Vesicles as a Source of NGAL for Diabetic Kidney Disease Evaluation in Children and Adolescents With Type 1 Diabetes Mellitus. Front. Endocrinol. 2021, 12, 654269. [Google Scholar] [CrossRef]
- Khandpur, S.; Srivastava, M.; Sharma, R.; Asif, S.; Bhadauria, D.S.; Mishra, P.; Purty, A.J.; Tiwari, S. Association of Wilms Tumor-1 Protein in Urinary Exosomes with Kidney Injury: A Population-Based Cross-Sectional Study. Front. Med. 2023, 10, 1220309. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Zhu, X.-Y.; Eirin, A.; Nargesi, A.A.; Woollard, J.R.; Santelli, A.; Sun, I.O.; Textor, S.C.; Lerman, L.O. Early Podocyte Injury and Elevated Levels of Urinary Podocyte-Derived Extracellular Vesicles in Swine with Metabolic Syndrome: Role of Podocyte Mitochondria. Am. J. Physiol. Renal Physiol. 2019, 317, F12–F22. [Google Scholar] [CrossRef]
- Kwon, S.H.; Woollard, J.R.; Saad, A.; Garovic, V.D.; Zand, L.; Jordan, K.L.; Textor, S.C.; Lerman, L.O. Elevated Urinary Podocyte-Derived Extracellular Microvesicles in Renovascular Hypertensive Patients. Nephrol. Dial. Transplant. 2017, 32, 800–807. [Google Scholar] [CrossRef]
- Abe, H.; Sakurai, A.; Ono, H.; Hayashi, S.; Yoshimoto, S.; Ochi, A.; Ueda, S.; Nishimura, K.; Shibata, E.; Tamaki, M.; et al. Urinary Exosomal mRNA of WT1 as Diagnostic and Prognostic Biomarker for Diabetic Nephropathy. J. Med. Investig. 2018, 65, 208–215. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, W.; Liu, M.-L. Extracellular Vesicles and Lupus Nephritis—New Insights into Pathophysiology and Clinical Implications. J. Autoimmun. 2020, 115, 102540. [Google Scholar] [CrossRef]
- Lv, L.-L.; Cao, Y.-H.; Pan, M.-M.; Liu, H.; Tang, R.-N.; Ma, K.-L.; Chen, P.-S.; Liu, B.-C. CD2AP mRNA in Urinary Exosome as Biomarker of Kidney Disease. Clin. Chim. Acta 2014, 428, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Mohan, A.; Godbole, M.M.; Bhatia, E.; Gupta, A.; Sharma, R.K.; Tiwari, S. Wilm’s Tumor-1 Protein Levels in Urinary Exosomes from Diabetic Patients with or without Proteinuria. PLoS ONE 2013, 8, e60177. [Google Scholar] [CrossRef]
- Yu, Y.; Bai, F.; Qin, N.; Liu, W.; Sun, Q.; Zhou, Y.; Yang, J. Non-Proximal Renal Tubule-Derived Urinary Exosomal miR-200b as a Biomarker of Renal Fibrosis. Nephron 2018, 139, 269–282. [Google Scholar] [CrossRef]
- Lv, L.-L.; Cao, Y.-H.; Ni, H.-F.; Xu, M.; Liu, D.; Liu, H.; Chen, P.-S.; Liu, B.-C. MicroRNA-29c in Urinary Exosome/Microvesicle as a Biomarker of Renal Fibrosis. Am. J. Physiol. Renal Physiol. 2013, 305, F1220–F1227. [Google Scholar] [CrossRef]
- Solé, C.; Moliné, T.; Vidal, M.; Ordi-Ros, J.; Cortés-Hernández, J. An Exosomal Urinary miRNA Signature for Early Diagnosis of Renal Fibrosis in Lupus Nephritis. Cells 2019, 8, 773. [Google Scholar] [CrossRef]
- Panich, T.; Chancharoenthana, W.; Somparn, P.; Issara-Amphorn, J.; Hirankarn, N.; Leelahavanichkul, A. Urinary Exosomal Activating Transcriptional Factor 3 as the Early Diagnostic Biomarker for Sepsis-Induced Acute Kidney Injury. BMC Nephrol. 2017, 18, 10. [Google Scholar] [CrossRef]
- Sonoda, H.; Yokota-Ikeda, N.; Oshikawa, S.; Kanno, Y.; Yoshinaga, K.; Uchida, K.; Ueda, Y.; Kimiya, K.; Uezono, S.; Ueda, A.; et al. Decreased Abundance of Urinary Exosomal Aquaporin-1 in Renal Ischemia-Reperfusion Injury. Am. J. Physiol. Renal Physiol. 2009, 297, F1006–F1016. [Google Scholar] [CrossRef]
- Sonoda, H.; Lee, B.R.; Park, K.-H.; Nihalani, D.; Yoon, J.-H.; Ikeda, M.; Kwon, S.-H. miRNA Profiling of Urinary Exosomes to Assess the Progression of Acute Kidney Injury. Sci. Rep. 2019, 9, 4692. [Google Scholar] [CrossRef]
- Gudehithlu, K.P.; Hart, P.; Joshi, A.; Garcia-Gomez, I.; Cimbaluk, D.J.; Dunea, G.; Arruda, J.A.L.; Singh, A.K. Urine Exosomal Ceruloplasmin: A Potential Early Biomarker of Underlying Kidney Disease. Clin. Exp. Nephrol. 2019, 23, 1013–1021. [Google Scholar] [CrossRef]
- Benito-Martin, A.; Ucero, A.C.; Zubiri, I.; Posada-Ayala, M.; Fernandez-Fernandez, B.; Cannata-Ortiz, P.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Egido, J.; Alvarez-Llamas, G.; et al. Osteoprotegerin in Exosome-like Vesicles from Human Cultured Tubular Cells and Urine. PLoS ONE 2013, 8, e72387. [Google Scholar] [CrossRef]
- Sun, H.; Yao, W.; Tang, Y.; Zhuang, W.; Wu, D.; Huang, S.; Sheng, H. Urinary Exosomes as a Novel Biomarker for Evaluation of α-Lipoic Acid’s Protective Effect in Early Diabetic Nephropathy. J. Clin. Lab. Anal. 2017, 31, e22129. [Google Scholar] [CrossRef]
- Trnka, P.; Ivanova, L.; Hiatt, M.J.; Matsell, D.G. Urinary Biomarkers in Obstructive Nephropathy. Clin. J. Am. Soc. Nephrol. 2012, 7, 1567–1575. [Google Scholar] [CrossRef]
- Jia, Y.; Guan, M.; Zheng, Z.; Zhang, Q.; Tang, C.; Xu, W.; Xiao, Z.; Wang, L.; Xue, Y. miRNAs in Urine Extracellular Vesicles as Predictors of Early-Stage Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 7932765. [Google Scholar] [CrossRef]
- Xie, Y.; Jia, Y.; Cuihua, X.; Hu, F.; Xue, M.; Xue, Y. Urinary Exosomal MicroRNA Profiling in Incipient Type 2 Diabetic Kidney Disease. J. Diabetes Res. 2017, 2017, 6978984. [Google Scholar] [CrossRef]
- Solé, C.; Cortés-Hernández, J.; Felip, M.L.; Vidal, M.; Ordi-Ros, J. miR-29c in Urinary Exosomes as Predictor of Early Renal Fibrosis in Lupus Nephritis. Nephrol. Dial. Transplant. 2015, 30, 1488–1496. [Google Scholar] [CrossRef]
- Awdishu, L.; Le, A.; Amato, J.; Jani, V.; Bal, S.; Mills, R.H.; Carrillo-Terrazas, M.; Gonzalez, D.J.; Tolwani, A.; Acharya, A.; et al. Urinary Exosomes Identify Inflammatory Pathways in Vancomycin Associated Acute Kidney Injury. Int. J. Mol. Sci. 2021, 22, 2784. [Google Scholar] [CrossRef]
- Hoorn, E.J.; Pisitkun, T.; Zietse, R.; Gross, P.; Frokiaer, J.; Wang, N.S.; Gonzales, P.A.; Star, R.A.; Knepper, M.A. Prospects for Urinary Proteomics: Exosomes as a Source of Urinary Biomarkers. Nephrology 2005, 10, 283–290. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Xu, A.; Wei, L.; Pei, M.; Shen, T.; Xian, X.; Yang, K.; Fei, L.; Pan, Y.; et al. Future Embracing: Exosomes Driving a Revolutionary Approach to the Diagnosis and Treatment of Idiopathic Membranous Nephropathy. J. Nanobiotechnology 2024, 22, 472. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Wang, C. A Review of the Regulatory Mechanisms of Extracellular Vesicles-Mediated Intercellular Communication. Cell Commun. Signal. 2023, 21, 77. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.A.; Yoon, H.; Kim, M.Y.; Yoo, J.-K.; Ahn, S.-H.; Park, C.H.; Park, J.; Nam, B.Y.; Park, J.T.; et al. Exosome-Based Delivery of Super-Repressor IκBα Ameliorates Kidney Ischemia-Reperfusion Injury. Kidney Int. 2021, 100, 570–584. [Google Scholar] [CrossRef]
- Hu, Q.; Lyon, C.J.; Fletcher, J.K.; Tang, W.; Wan, M.; Hu, T.Y. Extracellular Vesicle Activities Regulating Macrophage- and Tissue-Mediated Injury and Repair Responses. Acta Pharm. Sin. B 2021, 11, 1493–1512. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as Therapeutic Drug Carriers and Delivery Vehicles across Biological Membranes: Current Perspectives and Future Challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Hu, X.; Shen, N.; Liu, A.; Wang, W.; Zhang, L.; Sui, Z.; Tang, Q.; Du, X.; Yang, N.; Ying, W.; et al. Bone Marrow Mesenchymal Stem Cell-Derived Exosomal miR-34c-5p Ameliorates RIF by Inhibiting the Core Fucosylation of Multiple Proteins. Mol. Ther. 2022, 30, 763–781. [Google Scholar] [CrossRef]
- Jin, C.; Wu, P.; Wu, W.; Chen, W.; Liu, W.; Zhu, Y.; Wu, Q.; Chen, B.; Ji, C.; Qian, H. Therapeutic Role of hucMSC-sEV-Enriched miR-13896 in Cisplatin-Induced Acute Kidney Injury through M2 Macrophage Polarization. Cell Biol. Toxicol. 2025, 41, 50. [Google Scholar] [CrossRef]
- Dixson, A.C.; Dawson, T.R.; Di Vizio, D.; Weaver, A.M. Context-Specific Regulation of Extracellular Vesicle Biogenesis and Cargo Selection. Nat. Rev. Mol. Cell Biol. 2023, 24, 454–476. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, W.; Zhang, H.; Zhang, F.; Chen, L.; Ma, L.; Larcher, L.M.; Chen, S.; Liu, N.; Zhao, Q.; et al. Progress, Opportunity, and Perspective on Exosome Isolation—Efforts for Efficient Exosome-Based Theranostics. Theranostics 2020, 10, 3684–3707. [Google Scholar] [CrossRef]
- Huang, W.; Zhu, X.-Y.; Lerman, A.; Lerman, L.O. Extracellular Vesicles as Theranostic Tools in Kidney Disease. Clin. J. Am. Soc. Nephrol. CJASN 2022, 17, 1418. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Wang, B.; Sun, H.; Ren, Y.; Zhang, H. Compounding Engineered Mesenchymal Stem Cell-Derived Exosomes: A Potential Rescue Strategy for Retinal Degeneration. Biomed. Pharmacother. 2024, 173, 116424. [Google Scholar] [CrossRef]
- Wang, Y.; Huo, Y.; Zhao, C.; Liu, H.; Shao, Y.; Zhu, C.; An, L.; Chen, X.; Chen, Z. Engineered Exosomes with Enhanced Stability and Delivery Efficiency for Glioblastoma Therapy. J. Control. Release 2024, 368, 170–183. [Google Scholar] [CrossRef]
- Donoso-Quezada, J.; Ayala-Mar, S.; González-Valdez, J. State-of-the-Art Exosome Loading and Functionalization Techniques for Enhanced Therapeutics: A Review. Crit. Rev. Biotechnol. 2020, 40, 804–820. [Google Scholar] [CrossRef]
- Rädler, J.; Gupta, D.; Zickler, A.; Andaloussi, S.E. Exploiting the Biogenesis of Extracellular Vesicles for Bioengineering and Therapeutic Cargo Loading. Mol. Ther. 2023, 31, 1231–1250. [Google Scholar] [CrossRef]
- Ma, Y.; Dong, S.; Grippin, A.J.; Teng, L.; Lee, A.S.; Kim, B.Y.S.; Jiang, W. Engineering Therapeutical Extracellular Vesicles for Clinical Translation. Trends Biotechnol. 2025, 43, 61–82. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, C.; Du, Y.; Yang, X.; Liu, M.; Yang, W.; Lei, G.; Wang, G. Exosomal Transfer of microRNA-590-3p between Renal Tubular Epithelial Cells after Renal Ischemia-Reperfusion Injury Regulates Autophagy by Targeting TRAF6. Chin. Med. J. 2022, 135, 2467–2477. [Google Scholar] [CrossRef]
- Wu, P.; Tang, Y.; Jin, C.; Wang, M.; Li, L.; Liu, Z.; Shi, H.; Sun, Z.; Hou, X.; Chen, W.; et al. Neutrophil Membrane Engineered HucMSC sEVs Alleviate Cisplatin-Induced AKI by Enhancing Cellular Uptake and Targeting. J. Nanobiotechnol. 2022, 20, 353. [Google Scholar] [CrossRef]
- Ceccotti, E.; Saccu, G.; Herrera Sanchez, M.B.; Bruno, S. Naïve or Engineered Extracellular Vesicles from Different Cell Sources: Therapeutic Tools for Kidney Diseases. Pharmaceutics 2023, 15, 1715. [Google Scholar] [CrossRef]
- Cao, Q.; Huang, C.; Chen, X.-M.; Pollock, C.A. Mesenchymal Stem Cell-Derived Exosomes: Toward Cell-Free Therapeutic Strategies in Chronic Kidney Disease. Front. Med. 2022, 9, 816656. [Google Scholar] [CrossRef]
- Tran, P.H.L.; Wang, T.; Yin, W.; Tran, T.T.D.; Nguyen, T.N.G.; Lee, B.-J.; Duan, W. Aspirin-Loaded Nanoexosomes as Cancer Therapeutics. Int. J. Pharm. 2019, 572, 118786. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, F.; Xiang, J.; Zhou, X.; Wu, B.; Fan, B.; Tang, H.; Liu, B.; Chen, L. Mesoporous Microneedles Enabled Localized Controllable Delivery of Stimulator of Interferon Gene Agonist Nanoexosomes for FLASH Radioimmunotherapy against Breast Cancer. ACS Appl. Mater. Interfaces 2024, 16, 58180–58190. [Google Scholar] [CrossRef]
- Tan, A.; Rajadas, J.; Seifalian, A.M. Exosomes as Nano-Theranostic Delivery Platforms for Gene Therapy. Adv. Drug Deliv. Rev. 2013, 65, 357–367. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | Product | Mechanism | Creature | References |
|---|---|---|---|---|
| MSCs | IDO | Regulation of macrophage polarization to M2-type promotes kidney self-repair in IRI | Mouse | [104] |
| Huc-MSCs | Unknown | Reduction in macrophage infiltration inhibits kidney inflammation, inhibits tubular cell apoptosis | Pig | [105] |
| Huc-MSCs | Unknown | Upregulation of miR-146b level reduces IRAK1 expression and inhibits NF-κB activity to suppress the secretion of inflammatory mediators by M1-type macrophages | Mouse | [46] |
| MSCs | CCR2 | Reduce the concentration of free CCL2 to inhibit its recruitment and activation of macrophages | Mouse | [70] |
| MSCs | miR-100-5p | Targeting the FKBP5/AKT axis promotes M1 to M2 macrophage transformation and inhibits TEC apoptosis | Human being | [106] |
| USCs | miR-146a-5p | Targeted downregulation of IRAK-1 inhibits M1 macrophage polarization and attenuates HK-2 cell apoptosis | Human being | [107] |
| USCs | let-7b-5p | Targeting TLR4/NF-κB/STAT3/AKT promotes M2-type macrophage polarization | Human being | [108] |
| MSCs | miR-21 | Targeting PDCD4 promotes M2 polarization | Mouse | [109] |
| hAECs | Multiple proteins | Reduction in macrophage infiltration to inhibit TNF-α production and ultimately suppress TNF α-induced inflammatory response and tubular cell apoptosis | Mouse | [110] |
| MSCs | miR-27b | Targeting the JMJD3/NF-κB/p6 axis to inhibit macrophage inflammation and reduce kidney injury | Mouse | [111] |
| Unknown | miR-30c-5p | Promotion of M1 macrophages transforming to M2 to attenuate kidney I/R injury | Rat | [112] |
| TECs | miR-20a-5p | Inhibition of macrophage infiltration prevents acute tubular injury | Mouse | [113] |
| ASCs | Unknown | Promotion of macrophage polarizing to M2-type to increase phagocytosis and inhibit TEC death | Human being | [114] |
| Macrophages (M2 > M1) | miR-93-5p | Regulation of miR-93/TXNIP signaling pathway inhibits TEC’s pyroptosis | Mouse | [79] |
| Macrophage | IL-10 | Inhibition of mTOR signaling induces mitochondrial autophagy and promotes M2 polarization | Mouse | [115] |
| SCAP | Biologically active compound | Inhibition of oxidative stress, inflammation, and apoptosis to protect TECs | Rat | [89] |
| Origin | Product | Mechanism | Creature | References |
|---|---|---|---|---|
| BMSCs | Unknown | Activate EP2 receptors and reduce the number of M1 and M2 macrophages to inhibit MMT process and alleviate renal fibrosis | UUO-Mouse | [170] |
| BMSCs | miR-204-5p | Regulation of the PI3K-AKT pathway affects autophagy, inhibits M1-type polarization, and promotes M2-type polarization | Mouse | [171] |
| Huc-MSCs | Unknown | Inhibition of kidney fibrosis by blocking the MMT process through the inhibition of ARNTL expression | UUO-Mouse | [172] |
| Huc-MSCs | miR-146a-5p | Targeting TRAF6 to promote M2-type macrophage polarization alleviates kidney impairment in DN | Human being | [173] |
| MSCs | miR-486-5p | Targeting PIK3R1 via the PI3K/Akt pathway promotes M2-type macrophage polarization and prevents diabetic nephropathy | Mouse | [174] |
| USCs | circRNA ATG7 | Regulation of the SOCS1/STAT3 pathway by miR-4500 promotes macrophage polarization from M1 to M2 to inhibit DN progression | Human being | [175] |
| M2-type macrophage | miR-93-5p | Activation of autophagy by inhibiting DUSP1 expression ameliorates HG-induced podocyte damage | Mouse | [176] |
| M2-type macrophage | miR-25-3p | Targeting TLR4 attenuates podocyte apoptosis or upregulates ATXN3 expression to promote podocyte proliferation | Mouse | [162,177] |
| M1-type macrophage | miR-21a-5p | Inhibition of Tnpo1 expression in podocytes attenuates podocyte damage in HG | Mouse | [162] |
| Huc-MSCs | Unknown | Alleviation of podocyte injury in LN by promoting M2-type macrophage polarization | Human being | [178] |
| BMSCs | miR-16 | Attenuation of lupus nephritis by regulating PDCD4 pathway to induce M2-type macrophage polarization | Mouse | [179] |
| BMSCs | miR-21 | Alleviation of lupus nephritis by regulating PTEN pathway to induce M2-type macrophage polarization | Mouse | [179] |
| Unknown | LNA-anti-miR-150 | Inhibition of M1 macrophage infiltration by targeting SOCS1 to attenuate LN-induced kidney injury | Mouse | [180] |
| Kidney Disease | Urinary EV Product | Number Enrolled/Animal Model | Results | References |
|---|---|---|---|---|
| AKI | Fetuin-A | 3 healthy volunteers 3 ICU patients with CI-AKI 3 ICU patients without AKI | Patients in the ICU with CI-AKI show a significant increase in Fetuin-A levels. | [186] |
| AKI | ATF3 | S-AKI patients (n = 8) and healthy controls (n = 8) | Compared to healthy volunteers, S-AKI patients have higher levels of uATF3, which is negative in all the healthy volunteers. | [198] |
| AKI | Aquaporin-1 | Renal ischemia/reperfusion rat model | Urinary vesicle aquaporin-1 abundance reduction emerges. | [199] |
| AKI | miR-16 | Rat model of I/R-AKI | miR-16 expression is increased in UEs. | [200] |
| CKD | Ceruloplasmin | CKD (n = 15) and controls (n = 15) | Elevated urinary vesicle Ceruloplasmin was observed prior to proteinuria. | [201] |
| CKD | Osteoprotegerin | CKD (n = 14) and healthy controls (n = 4) | Urinary vesicle protein Osteoprotegerin levels are higher in patients with CKD than in healthy volunteers. | [202] |
| CKD | WT1 | Type-1 diabetes mellitus patients (n = 48) patients and healthy controls (n = 25) | The WT1 protein is predominantly present in UE of diabetic patients, with its expression levels increasing as kidney function declines. | [191,194] |
| CKD | CD63 | Microalbuminuria-stage DN patients (n = 62) and controls (n = 29) | During the early stages of DN, the content of EVs containing CD63 in urine significantly increased. | [203] |
| CKD | E-cadherin | 27 prevalent case-patients with posterior urethral valves and 20 age-matched controls | Patients excrete significantly lower levels of E-cadherin. | [204] |
| CKD | TGF-B1 | 27 prevalent case-patients with posterior urethral valves and 20 age-matched controls | Patients excrete significantly higher levels of TGF-B1. | [204] |
| CKD | N-cadherin | 27 prevalent case-patients with posterior urethral valves and 20 age-matched controls | Patients excrete significantly lower levels of N-cadherin. | [204] |
| CKD | L1CAM | 27 prevalent case-patients with posterior urethral valves and 20 age-matched controls | Patients excrete significantly higher levels of L1CAM. | [204] |
| CKD | miR-192 | 80 patients diagnosed with Type-2 diabetes (30 normoal buminuria, 30 microalbuminuria, and 20 macroalbuminuria) and 10 healthy controls | miR-192 is better than miR-194 and miR-215 in identifying DN in patients with normoal buminuria or microalbuminuria, which indicated that uEVs miR-192 might be able to diagnose DN earlier. | [205] |
| CKD | miR-15a-5p | 40 patients with type-2 diabetes mellitus (T2DM) patients, 20 patients with normoal buminuria, and 20 patients with macroalbuminuria | miR-15a-5p is downregulated in DN patients compared to T2DM patients. | [206] |
| CKD | miR-29c | 45 LN patients and 20 healthy/LN (n = 32), nonlupus CKD patients (n = 15) and healthy controls (n = 20) | In patients with LN, urinary vesicle miR-29c may serve as a potential biomarker for predicting disease progression. | [197,207] |
| CKD | miR-200b/miR-200 | 38 CKD patients with different degrees of renal fibrosis and in 12 normal individuals/32 CKD patients and 7 controls | The level of miR-200b in the CKD group is lower and negatively correlated with fibrosis | [208] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, J.; Xie, Z.; Kuang, X. Extracellular Vesicles in Renal Inflammatory Diseases: Revealing Mechanisms of Extracellular Vesicle-Mediated Macrophage Regulation. Int. J. Mol. Sci. 2025, 26, 3646. https://doi.org/10.3390/ijms26083646
Wei J, Xie Z, Kuang X. Extracellular Vesicles in Renal Inflammatory Diseases: Revealing Mechanisms of Extracellular Vesicle-Mediated Macrophage Regulation. International Journal of Molecular Sciences. 2025; 26(8):3646. https://doi.org/10.3390/ijms26083646
Chicago/Turabian StyleWei, Jiatai, Zijie Xie, and Xiaodong Kuang. 2025. "Extracellular Vesicles in Renal Inflammatory Diseases: Revealing Mechanisms of Extracellular Vesicle-Mediated Macrophage Regulation" International Journal of Molecular Sciences 26, no. 8: 3646. https://doi.org/10.3390/ijms26083646
APA StyleWei, J., Xie, Z., & Kuang, X. (2025). Extracellular Vesicles in Renal Inflammatory Diseases: Revealing Mechanisms of Extracellular Vesicle-Mediated Macrophage Regulation. International Journal of Molecular Sciences, 26(8), 3646. https://doi.org/10.3390/ijms26083646