
The Functions and Regulatory Mechanisms of Histone Modifications in Skeletal Muscle Development and Disease

Abstract
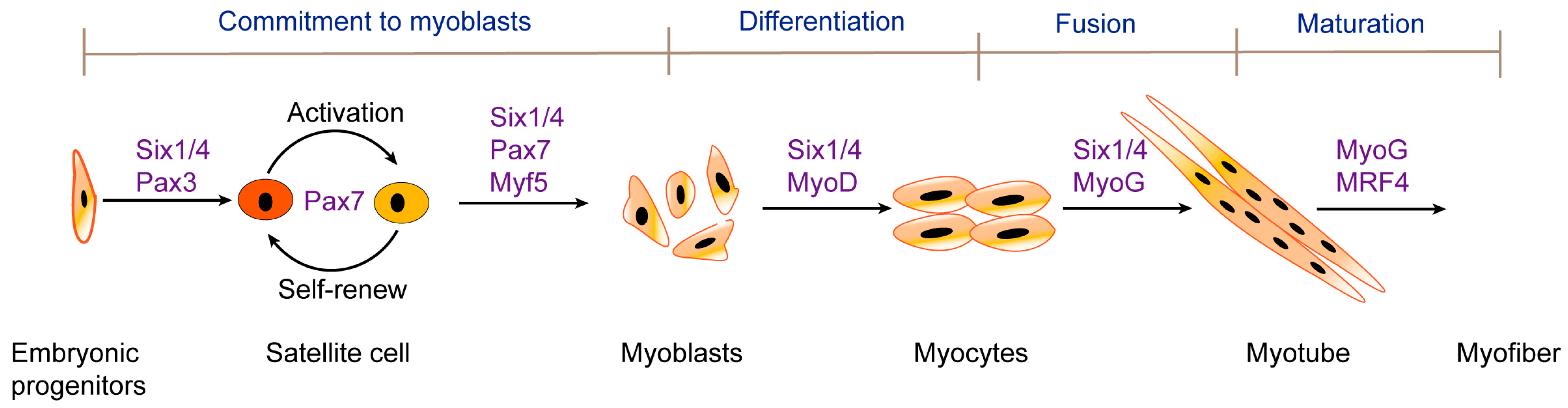
1. Introduction
2. Histone Modifications in Muscle Development and Regeneration
2.1. Role of Histone Methylation
2.1.1. H3K9 Methylation Modification
2.1.2. H3K27 Methylation Modification
2.1.3. H4K20 Methylation Modification
2.1.4. H3K4 Methylation Modification
2.1.5. H3K36 Methylation Modification
2.2. Role of Histone Acetylation
2.3. Role of Other Histone Modifications
3. Histone Modifications in Skeletal Muscle Atrophy
4. Histone Modifications in Skeletal Muscle Hypertrophy
5. Histone Modifications and Human Skeletal Muscle Diseases
5.1. Histone Modifications in DMD
5.2. Histone Modifications in FSHD
5.3. Histone Modifications in Sarcopenia
5.4. Histone Modifications in Other Skeletal-Muscle-Related Diseases
6. Challenges and Future Perspectives
- i.
- The diversity and complexity of histone modifications make it difficult to fully reveal their specific functions in different biological contexts. For example, the specific mechanisms of some newly discovered histone modifications, such as histone lactylation [134], have not been fully elucidated in skeletal muscle. The application of new technologies will result in breakthroughs in this field. The further development of high-throughput sequencing technology will help to reveal the complex regulatory network of histone modifications, especially dynamic changes at different developmental stages and pathological states. The application of CRISPR/Cas9 gene editing technology can precisely manipulate histone modification-related genes, aiding in the study of their specific functions in determining the fate of muscle cells.
- ii.
- Histone modifications may interact with other epigenetic mechanisms [203,204]. Several histone modifications were found to be involved in regulating miRNA expression in cancer or during its development [205]. In turn, some miRNAs can regulate the expression and enzymatic activity of histone-modifying enzymes in cancer [206]. MiRNA and histone modifications jointly regulate the development of and adaptive changes in skeletal muscle [203]. Histone modifications can also interact with DNA methylation. For example, histone modifications and DNA methylation act cooperatively in regulating symbiosis genes in the sea anemone Aiptasia [207]. However, existing research has mostly focused on a single type of modification and has overlooked the synergistic effects of different modifications, which may lead to a one-sided understanding of muscle development and disease mechanisms. Therefore, understanding the interaction between histone modifications and other epigenetic modifications such as ncRNA will be beneficial for a comprehensive understanding of the regulatory network of histone modifications.
- iii.
- There are multiple technological methods currently available for studying the function of histone modifications. For example, ChIP-seq and CUT&Tag can be used to locate the genomic regions of histone modifications. Mass spectrometry can accurately identify the types, sites, and combination patterns of histone modifications (acetylation, methylation, phosphorylation, etc.), making it particularly suitable for the discovery of unknown modifications. Single cell-ChIP-seq can be used to study histone modification heterogeneity at a single-cell resolution. Although the application of high-throughput sequencing technology provides a powerful tool for histone modification research, challenges remain in data analysis and interpretation. In complex biological systems in particular, further exploration is needed to accurately identify and validate the functional sites of histone modifications and their specific roles in gene regulatory networks.
- iv.
- Sex hormones are crucial for muscle physiological function and seem to be the most probable candidates in the regulation of key molecular pathways of skeletal muscle, with an impact on its mass and functionality [208,209]. Differences in the muscle transcriptome between males and females are largely related to testosterone and estradiol [210]. Testosterone is known to promote muscle protein synthesis and muscle regeneration through its androgen receptor [211]. However, it is currently unclear whether the regulation of the skeletal muscle’s physiological function by sex hormones is mediated by histone modifications. Therefore, an in-depth analysis of the impact of hormone differences on muscle histone modification can not only reveal the molecular mechanisms of differences in muscle development and function between sexes but also provide a valuable theoretical basis for sex-specific intervention strategies for the prevention and treatment of muscle-related diseases.
- v.
- Epigenetic drugs have shown broad prospects in the treatment of skeletal muscle diseases. For example, selective inhibitors targeting HDAC6, such as Tubastatin A, have been shown to stabilize microtubule networks, restore autophagy function, and improve muscle function in DMD mouse models [163]. In spinal muscular atrophy, the combination of the HDAC inhibitor LBH589 and antisense oligonucleotides (such as Nusirersen) significantly enhances the splicing of the SMN2 gene and the expression of the SMN protein [212], suggesting that combination therapy may improve therapeutic efficacy. Nevertheless, existing research has mostly been based on animal models, and determining how to effectively translate these findings into treatment strategies for human diseases remains an important research direction.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yin, L.; Li, N.; Jia, W.; Wang, N.; Liang, M.; Yang, X.; Du, G. Skeletal muscle atrophy: From mechanisms to treatments. Pharmacol. Res. 2021, 172, 105807. [Google Scholar] [CrossRef]
- Buckingham, M.; Bajard, L.; Chang, T.; Daubas, P.; Hadchouel, J.; Meilhac, S.; Montarras, D.; Rocancourt, D.; Relaix, F. The formation of skeletal muscle: From somite to limb. J. Anat. 2003, 202, 59–68. [Google Scholar] [CrossRef]
- Comai, G.; Tajbakhsh, S. Molecular and cellular regulation of skeletal myogenesis. Curr. Top. Dev. Biol. 2014, 110, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.C.; Nowicki, J.L. A new view of patterning domains in the vertebrate mesoderm. Dev. Cell 2003, 4, 159–165. [Google Scholar] [CrossRef]
- Shearman, R.M.; Burke, A.C. The lateral somitic frontier in ontogeny and phylogeny. J. Exp. Zool. B Mol. Dev. Evol. 2009, 312, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M. Skeletal muscle formation in vertebrates. Curr. Opin. Genet. Dev. 2001, 11, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Weintraub, H.; Davis, R.; Tapscott, S.; Thayer, M.; Krause, M.; Benezra, R.; Blackwell, T.K.; Turner, D.; Rupp, R.; Hollenberg, S.; et al. The myoD gene family: Nodal point during specification of the muscle cell lineage. Science 1991, 251, 761–766. [Google Scholar] [CrossRef]
- Moncaut, N.; Rigby, P.W.; Carvajal, J.J. Dial M(RF) for myogenesis. FEBS J. 2013, 280, 3980–3990. [Google Scholar] [CrossRef]
- Fong, A.P.; Tapscott, S.J. Skeletal muscle programming and re-programming. Curr. Opin. Genet. Dev. 2013, 23, 568–573. [Google Scholar] [CrossRef]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Nabeshima, Y.; Hanaoka, K.; Hayasaka, M.; Esumi, E.; Li, S.; Nonaka, I.; Nabeshima, Y. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993, 364, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Relaix, F. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 645–673. [Google Scholar] [CrossRef]
- Buckingham, M.; Rigby, P.W. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev. Cell 2014, 28, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Heanue, T.A.; Reshef, R.; Davis, R.J.; Mardon, G.; Oliver, G.; Tomarev, S.; Lassar, A.B.; Tabin, C.J. Synergistic regulation of vertebrate muscle development by Dach2, Eya2, and Six1, homologs of genes required for Drosophila eye formation. Genes Dev. 1999, 13, 3231–3243. [Google Scholar] [CrossRef]
- Grifone, R.; Demignon, J.; Houbron, C.; Souil, E.; Niro, C.; Seller, M.J.; Hamard, G.; Maire, P. Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development 2005, 132, 2235–2249. [Google Scholar] [CrossRef]
- Tremblay, P.; Dietrich, S.; Mericskay, M.; Schubert, F.R.; Li, Z.; Paulin, D. A crucial role for Pax3 in the development of the hypaxial musculature and the long-range migration of muscle precursors. Dev. Biol. 1998, 203, 49–61. [Google Scholar] [CrossRef]
- Lewis, S.E. Developmental analysis of lethal effects of homozygosity for the c25H deletion in the mouse. Dev. Biol. 1978, 65, 553–557. [Google Scholar] [CrossRef]
- Goulding, M.; Lumsden, A.; Paquette, A.J. Regulation of Pax-3 expression in the dermomyotome and its role in muscle development. Development 1994, 120, 957–971. [Google Scholar] [CrossRef]
- Bober, E.; Franz, T.; Arnold, H.H.; Gruss, P.; Tremblay, P. Pax-3 is required for the development of limb muscles: A possible role for the migration of dermomyotomal muscle progenitor cells. Development 1994, 120, 603–612. [Google Scholar] [CrossRef]
- Gunther, S.; Kim, J.; Kostin, S.; Lepper, C.; Fan, C.M.; Braun, T. Myf5-positive satellite cells contribute to Pax7-dependent long-term maintenance of adult muscle stem cells. Cell Stem Cell 2013, 13, 590–601. [Google Scholar] [CrossRef] [PubMed]
- von Maltzahn, J.; Jones, A.E.; Parks, R.J.; Rudnicki, M.A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. USA 2013, 110, 16474–16479. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 2005, 435, 948–953. [Google Scholar] [CrossRef]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef]
- Persson, P.B. Skeletal muscle satellite cells as myogenic progenitors for muscle homoeostasis, growth, regeneration and repair. Acta Physiol. 2015, 213, 537–538. [Google Scholar] [CrossRef]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef] [PubMed]
- Sambasivan, R.; Yao, R.; Kissenpfennig, A.; Van Wittenberghe, L.; Paldi, A.; Gayraud-Morel, B.; Guenou, H.; Malissen, B.; Tajbakhsh, S.; Galy, A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 2011, 138, 3647–3656. [Google Scholar] [CrossRef]
- Sandri, M. Signaling in muscle atrophy and hypertrophy. Physiology 2008, 23, 160–170. [Google Scholar] [CrossRef]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 330. [Google Scholar] [CrossRef]
- Damluji, A.A.; Alfaraidhy, M.; AlHajri, N.; Rohant, N.N.; Kumar, M.; Al Malouf, C.; Bahrainy, S.; Ji Kwak, M.; Batchelor, W.B.; Forman, D.E.; et al. Sarcopenia and Cardiovascular Diseases. Circulation 2023, 147, 1534–1553. [Google Scholar] [CrossRef]
- Sayer, A.A.; Cruz-Jentoft, A. Sarcopenia definition, diagnosis and treatment: Consensus is growing. Age Ageing 2022, 51, afac220. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Fitz-James, M.H.; Cavalli, G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 2022, 23, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Peng, J.; Jiang, S. The epigenetic regulation of embryonic myogenesis and adult muscle regeneration by histone methylation modification. Biochem. Biophys. Rep. 2016, 6, 209–219. [Google Scholar] [CrossRef]
- Barreiro, E.; Tajbakhsh, S. Epigenetic regulation of muscle development. J. Muscle Res. Cell Motil. 2017, 38, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Bharathy, N.; Ling, B.M.; Taneja, R. Epigenetic regulation of skeletal muscle development and differentiation. Subcell. Biochem. 2013, 61, 139–150. [Google Scholar] [CrossRef]
- Nitsch, S.; Zorro Shahidian, L.; Schneider, R. Histone acylations and chromatin dynamics: Concepts, challenges, and links to metabolism. EMBO Rep. 2021, 22, e52774. [Google Scholar] [CrossRef]
- Hagihara, H.; Shoji, H.; Otabi, H.; Toyoda, A.; Katoh, K.; Namihira, M.; Miyakawa, T. Protein lactylation induced by neural excitation. Cell Rep. 2021, 37, 109820. [Google Scholar] [CrossRef]
- Lepack, A.E.; Werner, C.T.; Stewart, A.F.; Fulton, S.L.; Zhong, P.; Farrelly, L.A.; Smith, A.C.W.; Ramakrishnan, A.; Lyu, Y.; Bastle, R.M.; et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 2020, 368, 197–201. [Google Scholar] [CrossRef]
- Farrelly, L.A.; Thompson, R.E.; Zhao, S.; Lepack, A.E.; Lyu, Y.; Bhanu, N.V.; Zhang, B.; Loh, Y.E.; Ramakrishnan, A.; Vadodaria, K.C.; et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 2019, 567, 535–539. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Moresi, V.; Marroncelli, N.; Coletti, D.; Adamo, S. Regulation of skeletal muscle development and homeostasis by gene imprinting, histone acetylation and microRNA. Biochim. Biophys. Acta 2015, 1849, 309–316. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Trievel, R.C.; Beach, B.M.; Dirk, L.M.; Houtz, R.L.; Hurley, J.H. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell 2002, 111, 91–103. [Google Scholar] [CrossRef]
- Mal, A.; Harter, M.L. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 1735–1739. [Google Scholar] [CrossRef]
- Zhang, C.L.; McKinsey, T.A.; Olson, E.N. Association of class II histone deacetylases with heterochromatin protein 1: Potential role for histone methylation in control of muscle differentiation. Mol. Cell Biol. 2002, 22, 7302–7312. [Google Scholar] [CrossRef] [PubMed]
- Mal, A.K. Histone methyltransferase Suv39h1 represses MyoD-stimulated myogenic differentiation. EMBO J. 2006, 25, 3323–3334. [Google Scholar] [CrossRef]
- Ait-Si-Ali, S.; Guasconi, V.; Fritsch, L.; Yahi, H.; Sekhri, R.; Naguibneva, I.; Robin, P.; Cabon, F.; Polesskaya, A.; Harel-Bellan, A. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004, 23, 605–615. [Google Scholar] [CrossRef]
- Lee, H.; Habas, R.; Abate-Shen, C. MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science 2004, 304, 1675–1678. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.M.; Bharathy, N.; Chung, T.K.; Kok, W.K.; Li, S.; Tan, Y.H.; Rao, V.K.; Gopinadhan, S.; Sartorelli, V.; Walsh, M.J.; et al. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jang, H.; Kim, H.; Kim, S.T.; Cho, E.J.; Youn, H.D. Histone demethylase LSD1 is required to induce skeletal muscle differentiation by regulating myogenic factors. Biochem. Biophys. Res. Commun. 2010, 401, 327–332. [Google Scholar] [CrossRef]
- Zhu, Q.; Liang, F.; Cai, S.; Luo, X.; Duo, T.; Liang, Z.; He, Z.; Chen, Y.; Mo, D. KDM4A regulates myogenesis by demethylating H3K9me3 of myogenic regulatory factors. Cell Death Dis. 2021, 12, 514. [Google Scholar] [CrossRef]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef]
- Peng, J.C.; Valouev, A.; Swigut, T.; Zhang, J.; Zhao, Y.; Sidow, A.; Wysocka, J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 2009, 139, 1290–1302. [Google Scholar] [CrossRef]
- Stojic, L.; Jasencakova, Z.; Prezioso, C.; Stutzer, A.; Bodega, B.; Pasini, D.; Klingberg, R.; Mozzetta, C.; Margueron, R.; Puri, P.L.; et al. Chromatin regulated interchange between polycomb repressive complex 2 (PRC2)-Ezh2 and PRC2-Ezh1 complexes controls myogenin activation in skeletal muscle cells. Epigenetics Chromatin 2011, 4, 16. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Seenundun, S.; Rampalli, S.; Liu, Q.C.; Aziz, A.; Palii, C.; Hong, S.; Blais, A.; Brand, M.; Ge, K.; Dilworth, F.J. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 2010, 29, 1401–1411. [Google Scholar] [CrossRef]
- Boonsanay, V.; Zhang, T.; Georgieva, A.; Kostin, S.; Qi, H.; Yuan, X.; Zhou, Y.; Braun, T. Regulation of Skeletal Muscle Stem Cell Quiescence by Suv4-20h1-Dependent Facultative Heterochromatin Formation. Cell Stem Cell 2016, 18, 229–242. [Google Scholar] [CrossRef]
- Tao, Y.; Neppl, R.L.; Huang, Z.P.; Chen, J.; Tang, R.H.; Cao, R.; Zhang, Y.; Jin, S.W.; Wang, D.Z. The histone methyltransferase Set7/9 promotes myoblast differentiation and myofibril assembly. J. Cell Biol. 2011, 194, 551–565. [Google Scholar] [CrossRef] [PubMed]
- McKinnell, I.W.; Ishibashi, J.; Le Grand, F.; Punch, V.G.; Addicks, G.C.; Greenblatt, J.F.; Dilworth, F.J.; Rudnicki, M.A. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat. Cell Biol. 2008, 10, 77–84. [Google Scholar] [CrossRef]
- Diao, Y.; Guo, X.; Li, Y.; Sun, K.; Lu, L.; Jiang, L.; Fu, X.; Zhu, H.; Sun, H.; Wang, H.; et al. Pax3/7BP is a Pax7- and Pax3-binding protein that regulates the proliferation of muscle precursor cells by an epigenetic mechanism. Cell Stem Cell 2012, 11, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, Y.; Wang, Y.X.; McKinnell, I.W.; Bedford, M.T.; Rudnicki, M.A. Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell 2012, 11, 333–345. [Google Scholar] [CrossRef]
- Soleimani, V.D.; Punch, V.G.; Kawabe, Y.; Jones, A.E.; Palidwor, G.A.; Porter, C.J.; Cross, J.W.; Carvajal, J.J.; Kockx, C.E.; van IJcken, W.F.; et al. Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev. Cell 2012, 22, 1208–1220. [Google Scholar] [CrossRef]
- Rampalli, S.; Li, L.; Mak, E.; Ge, K.; Brand, M.; Tapscott, S.J.; Dilworth, F.J. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat. Struct. Mol. Biol. 2007, 14, 1150–1156. [Google Scholar] [CrossRef]
- Sebastian, S.; Sreenivas, P.; Sambasivan, R.; Cheedipudi, S.; Kandalla, P.; Pavlath, G.K.; Dhawan, J. MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin A2 expression, and promotes myogenic differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 4719–4724. [Google Scholar] [CrossRef]
- Matteini, F.; Andresini, O.; Petrai, S.; Battistelli, C.; Rossi, M.N.; Maione, R. Poly(ADP-ribose) Polymerase 1 (PARP1) restrains MyoD-dependent gene expression during muscle differentiation. Sci. Rep. 2020, 10, 15086. [Google Scholar] [CrossRef] [PubMed]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef]
- Yi, X.; Tao, Y.; Lin, X.; Dai, Y.; Yang, T.; Yue, X.; Jiang, X.; Li, X.; Jiang, D.S.; Andrade, K.C.; et al. Histone methyltransferase Setd2 is critical for the proliferation and differentiation of myoblasts. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 697–707. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, L.; Robin, P.; Mathieu, J.R.; Souidi, M.; Hinaux, H.; Rougeulle, C.; Harel-Bellan, A.; Ameyar-Zazoua, M.; Ait-Si-Ali, S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol. Cell 2010, 37, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef]
- Zhang, R.H.; Judson, R.N.; Liu, D.Y.; Kast, J.; Rossi, F.M. The lysine methyltransferase Ehmt2/G9a is dispensable for skeletal muscle development and regeneration. Skelet. Muscle 2016, 6, 22. [Google Scholar] [CrossRef]
- Ling, B.M.; Gopinadhan, S.; Kok, W.K.; Shankar, S.R.; Gopal, P.; Bharathy, N.; Wang, Y.; Taneja, R. G9a mediates Sharp-1-dependent inhibition of skeletal muscle differentiation. Mol. Biol. Cell 2012, 23, 4778–4785. [Google Scholar] [CrossRef]
- Verrier, L.; Escaffit, F.; Chailleux, C.; Trouche, D.; Vandromme, M. A new isoform of the histone demethylase JMJD2A/KDM4A is required for skeletal muscle differentiation. PLoS Genet. 2011, 7, e1001390. [Google Scholar] [CrossRef]
- Choi, J.H.; Song, Y.J.; Lee, H. The histone demethylase KDM4B interacts with MyoD to regulate myogenic differentiation in C2C12 myoblast cells. Biochem. Biophys. Res. Commun. 2015, 456, 872–878. [Google Scholar] [CrossRef]
- Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Borchers, C.; Tempst, P.; Zhang, Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 2001, 8, 1207–1217. [Google Scholar] [CrossRef]
- Asp, P.; Blum, R.; Vethantham, V.; Parisi, F.; Micsinai, M.; Cheng, J.; Bowman, C.; Kluger, Y.; Dynlacht, B.D. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc. Natl. Acad. Sci. USA 2011, 108, E149–E158. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, F.J.; Blais, A. Epigenetic regulation of satellite cell activation during muscle regeneration. Stem Cell Res. Ther. 2011, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, K.; Zare, H.; Wang, A.H.; Sartorelli, V. Polycomb protein Ezh1 promotes RNA polymerase II elongation. Mol. Cell 2012, 45, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Okada, S.; Konno, D.; Odawara, J.; Yoshimi, T.; Yoshimura, S.; Kumamaru, H.; Saiwai, H.; Tsubota, T.; Kurumizaka, H.; et al. Chd2 interacts with H3.3 to determine myogenic cell fate. EMBO J. 2012, 31, 2994–3007. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Maehara, K.; Sato, Y.; Konno, D.; Tachibana, T.; Kimura, H.; Ohkawa, Y. Incorporation of histone H3.1 suppresses the lineage potential of skeletal muscle. Nucleic Acids Res. 2015, 43, 775–786. [Google Scholar] [CrossRef]
- Yang, J.H.; Song, Y.; Seol, J.H.; Park, J.Y.; Yang, Y.J.; Han, J.W.; Youn, H.D.; Cho, E.J. Myogenic transcriptional activation of MyoD mediated by replication-independent histone deposition. Proc. Natl. Acad. Sci. USA 2011, 108, 85–90. [Google Scholar] [CrossRef]
- Bae, S.; Lesch, B.J. H3K4me1 Distribution Predicts Transcription State and Poising at Promoters. Front. Cell Dev. Biol. 2020, 8, 289. [Google Scholar] [CrossRef]
- Ringrose, L.; Paro, R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu. Rev. Genet. 2004, 38, 413–443. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell 2010, 39, 736–749. [Google Scholar] [CrossRef]
- Minotti, R.; Andersson, A.; Hottiger, M.O. ARTD1 Suppresses Interleukin 6 Expression by Repressing MLL1-Dependent Histone H3 Trimethylation. Mol. Cell Biol. 2015, 35, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, D.R.; Marmorstein, R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FEBS J. 2013, 280, 5570–5581. [Google Scholar] [CrossRef]
- Elmallah, M.I.Y.; Micheau, O. Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy. Cancers 2019, 11, 850. [Google Scholar] [CrossRef] [PubMed]
- Furdas, S.D.; Kannan, S.; Sippl, W.; Jung, M. Small molecule inhibitors of histone acetyltransferases as epigenetic tools and drug candidates. Arch. Pharm. 2012, 345, 7–21. [Google Scholar] [CrossRef]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef]
- Doi, M.; Hirayama, J.; Sassone-Corsi, P. Circadian regulator CLOCK is a histone acetyltransferase. Cell 2006, 125, 497–508. [Google Scholar] [CrossRef]
- Addicks, G.C.; Zhang, H.; Ryu, D.; Vasam, G.; Green, A.E.; Marshall, P.L.; Patel, S.; Kang, B.E.; Kim, D.; Katsyuba, E.; et al. GCN5 maintains muscle integrity by acetylating YY1 to promote dystrophin expression. J. Cell Biol. 2022, 221, e202104022. [Google Scholar] [CrossRef]
- Santi, S.; Cenni, V.; Capanni, C.; Lattanzi, G.; Mattioli, E. PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation. Cells 2020, 9, 1735. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Jang, S.M.; Kim, C.H.; An, J.H.; Kang, E.J.; Choi, K.H. Tip60 regulates myoblast differentiation by enhancing the transcriptional activity of MyoD via their physical interactions. FEBS J. 2011, 278, 4394–4404. [Google Scholar] [CrossRef] [PubMed]
- Eckner, R.; Yao, T.P.; Oldread, E.; Livingston, D.M. Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes Dev. 1996, 10, 2478–2490. [Google Scholar] [CrossRef]
- Roth, J.F.; Shikama, N.; Henzen, C.; Desbaillets, I.; Lutz, W.; Marino, S.; Wittwer, J.; Schorle, H.; Gassmann, M.; Eckner, R. Differential role of p300 and CBP acetyltransferase during myogenesis: p300 acts upstream of MyoD and Myf5. EMBO J. 2003, 22, 5186–5196. [Google Scholar] [CrossRef] [PubMed]
- Mal, A.; Sturniolo, M.; Schiltz, R.L.; Ghosh, M.K.; Harter, M.L. A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: Inhibition of the myogenic program. EMBO J. 2001, 20, 1739–1753. [Google Scholar] [CrossRef]
- Bossone, K.A.; Ellis, J.A.; Holaska, J.M. Histone acetyltransferase inhibition rescues differentiation of emerin-deficient myogenic progenitors. Muscle Nerve 2020, 62, 128–136. [Google Scholar] [CrossRef]
- Qian, Z.; Ye, J.; Li, J.; Che, Y.; Yu, W.; Xu, P.; Lin, J.; Ye, F.; Xu, X.; Su, Z.; et al. Decrotonylation of AKT1 promotes AKT1 phosphorylation and activation during myogenic differentiation. J. Adv. Res. 2023, 50, 117–133. [Google Scholar] [CrossRef]
- Marroncelli, N.; Bianchi, M.; Bertin, M.; Consalvi, S.; Saccone, V.; De Bardi, M.; Puri, P.L.; Palacios, D.; Adamo, S.; Moresi, V. HDAC4 regulates satellite cell proliferation and differentiation by targeting P21 and Sharp1 genes. Sci. Rep. 2018, 8, 3448. [Google Scholar] [CrossRef]
- Renzini, A.; Marroncelli, N.; Noviello, C.; Moresi, V.; Adamo, S. HDAC4 Regulates Skeletal Muscle Regeneration via Soluble Factors. Front. Physiol. 2018, 9, 1387. [Google Scholar] [CrossRef]
- Haberland, M.; Arnold, M.A.; McAnally, J.; Phan, D.; Kim, Y.; Olson, E.N. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol. Cell Biol. 2007, 27, 518–525. [Google Scholar] [CrossRef]
- Myers, M.J.; Shepherd, D.L.; Durr, A.J.; Stanton, D.S.; Mohamed, J.S.; Hollander, J.M.; Alway, S.E. The role of SIRT1 in skeletal muscle function and repair of older mice. J. Cachexia Sarcopenia Muscle 2019, 10, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Lee, M.M.; Park, S.; Jeong, K.S. Sirt2 positively regulates muscle regeneration after Notexin-induced muscle injury. Exp. Mol. Pathol. 2022, 127, 104798. [Google Scholar] [CrossRef]
- Lin, L.; Chen, K.; Abdel Khalek, W.; Ward, J.L., 3rd; Yang, H.; Chabi, B.; Wrutniak-Cabello, C.; Tong, Q. Regulation of skeletal muscle oxidative capacity and muscle mass by SIRT3. PLoS ONE 2014, 9, e85636. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.K.; An, T.H.; Son, M.J.; Lee, D.S.; Kang, H.S.; Lee, E.W.; Han, B.S.; Kim, W.K.; Bae, K.H.; Oh, K.J.; et al. HDAC11 Inhibits Myoblast Differentiation through Repression of MyoD-Dependent Transcription. Mol. Cells 2017, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Alvarez, Y.; Hurtado, E.; Munoz, M.; Garcia-Tunon, I.; Rech, G.E.; Pluvinet, R.; Sumoy, L.; Pendas, A.M.; Peinado, M.A.; Suelves, M. Loss of HDAC11 accelerates skeletal muscle regeneration in mice. FEBS J. 2021, 288, 1201–1223. [Google Scholar] [CrossRef]
- Ochiai, N.; Nishizuka, M.; Osada, S.; Imagawa, M. Fad24, a Positive Regulator of Adipogenesis, Is Required for S Phase Re-entry of C2C12 Myoblasts Arrested in G0 Phase and Involved in p27(Kip1) Expression at the Protein Level. Biol. Pharm. Bull. 2016, 39, 807–814. [Google Scholar] [CrossRef]
- LaBarge, S.A.; Migdal, C.W.; Buckner, E.H.; Okuno, H.; Gertsman, I.; Stocks, B.; Barshop, B.A.; Nalbandian, S.R.; Philp, A.; McCurdy, C.E.; et al. p300 is not required for metabolic adaptation to endurance exercise training. FASEB J. 2016, 30, 1623–1633. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Y.; Hamed, M.; Lacroix, N.; Li, Q. Molecular Basis for the Regulation of Transcriptional Coactivator p300 in Myogenic Differentiation. Sci. Rep. 2015, 5, 13727. [Google Scholar] [CrossRef]
- Joung, H.; Kwon, S.; Kim, K.H.; Lee, Y.G.; Shin, S.; Kwon, D.H.; Lee, Y.U.; Kook, T.; Choe, N.; Kim, J.C.; et al. Sumoylation of histone deacetylase 1 regulates MyoD signaling during myogenesis. Exp. Mol. Med. 2018, 50, e427. [Google Scholar] [CrossRef]
- Ferrari, L.; Bragato, C.; Brioschi, L.; Spreafico, M.; Esposito, S.; Pezzotta, A.; Pizzetti, F.; Moreno-Fortuny, A.; Bellipanni, G.; Giordano, A.; et al. HDAC8 regulates canonical Wnt pathway to promote differentiation in skeletal muscles. J. Cell Physiol. 2019, 234, 6067–6076. [Google Scholar] [CrossRef]
- Habibian, J.S.; Bolino, M.J.; Ferguson, B.S. HDAC8 regulates protein kinase D phosphorylation in skeletal myoblasts in response to stress signaling. Biochem. Biophys. Res. Commun. 2023, 650, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 2000, 6, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Miska, E.A.; Karlsson, C.; Langley, E.; Nielsen, S.J.; Pines, J.; Kouzarides, T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999, 18, 5099–5107. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000, 408, 106–111. [Google Scholar] [CrossRef]
- Zhao, J.; Shen, X.; Cao, X.; He, H.; Han, S.; Chen, Y.; Cui, C.; Wei, Y.; Wang, Y.; Li, D.; et al. HDAC4 Regulates the Proliferation, Differentiation and Apoptosis of Chicken Skeletal Muscle Satellite Cells. Animals 2020, 10, 84. [Google Scholar] [CrossRef]
- Han, Z.; Chang, C.; Zhu, W.; Zhang, Y.; Zheng, J.; Kang, X.; Jin, G.; Gong, Z. Role of SIRT2 in regulating the dexamethasone-activated autophagy pathway in skeletal muscle atrophy. Biochem. Cell Biol. 2021, 99, 562–569. [Google Scholar] [CrossRef]
- Song, M.Y.; Han, C.Y.; Moon, Y.J.; Lee, J.H.; Bae, E.J.; Park, B.H. Sirt6 reprograms myofibers to oxidative type through CREB-dependent Sox6 suppression. Nat. Commun. 2022, 13, 1808. [Google Scholar] [CrossRef]
- Zhang, R.; Pan, Y.; Feng, W.; Zhao, Y.; Yang, Y.; Wang, L.; Zhang, Y.; Cheng, J.; Jiang, Q.; Zheng, Z.; et al. HDAC11 Regulates the Proliferation of Bovine Muscle Stem Cells through the Notch Signaling Pathway and Inhibits Muscle Regeneration. J. Agric. Food Chem. 2022, 70, 9166–9178. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Xie, Y.; Hu, H.; Liu, M.; Zhou, T.; Cheng, X.; Huang, W.; Cao, L. The role and mechanism of histone lactylation in health and diseases. Front. Genet. 2022, 13, 949252. [Google Scholar] [CrossRef]
- Ohno, Y.; Ando, K.; Ito, T.; Suda, Y.; Matsui, Y.; Oyama, A.; Kaneko, H.; Yokoyama, S.; Egawa, T.; Goto, K. Lactate Stimulates a Potential for Hypertrophy and Regeneration of Mouse Skeletal Muscle. Nutrients 2019, 11, 869. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Shibasaki, A.; Naka, A.; Saito, H.; Iida, K. Lactate Promotes Myoblast Differentiation and Myotube Hypertrophy via a Pathway Involving MyoD In Vitro and Enhances Muscle Regeneration In Vivo. Int. J. Mol. Sci. 2018, 19, 3649. [Google Scholar] [CrossRef]
- Willkomm, L.; Schubert, S.; Jung, R.; Elsen, M.; Borde, J.; Gehlert, S.; Suhr, F.; Bloch, W. Lactate regulates myogenesis in C2C12 myoblasts in vitro. Stem Cell Res. 2014, 12, 742–753. [Google Scholar] [CrossRef]
- Galle, E.; Wong, C.W.; Ghosh, A.; Desgeorges, T.; Melrose, K.; Hinte, L.C.; Castellano-Castillo, D.; Engl, M.; de Sousa, J.A.; Ruiz-Ojeda, F.J.; et al. H3K18 lactylation marks tissue-specific active enhancers. Genome Biol. 2022, 23, 207. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wu, G.; Liu, K.; Chen, Q.; Tao, J.; Liu, H.; Shen, M. Lactate promotes myogenesis via activating H3K9 lactylation-dependent up-regulation of Neu2 expression. J. Cachexia Sarcopenia Muscle 2023, 14, 2851–2865. [Google Scholar] [CrossRef] [PubMed]
- Desgeorges, T.; Galle, E.; Zhang, J.; von Meyenn, F.; De Bock, K. Histone lactylation in macrophages is predictive for gene expression changes during ischemia induced-muscle regeneration. Mol. Metab. 2024, 83, 101923. [Google Scholar] [CrossRef]
- Yoshihara, T.; Machida, S.; Tsuzuki, T.; Kakigi, R.; Chang, S.W.; Sugiura, T.; Naito, H. Age-related changes in histone modification in rat gastrocnemius muscle. Exp. Gerontol. 2019, 125, 110658. [Google Scholar] [CrossRef]
- Kawano, F.; Nimura, K.; Ishino, S.; Nakai, N.; Nakata, K.; Ohira, Y. Differences in histone modifications between slow- and fast-twitch muscle of adult rats and following overload, denervation, or valproic acid administration. J. Appl. Physiol. 2015, 119, 1042–1052. [Google Scholar] [CrossRef]
- Ryder, D.J.; Judge, S.M.; Beharry, A.W.; Farnsworth, C.L.; Silva, J.C.; Judge, A.R. Identification of the Acetylation and Ubiquitin-Modified Proteome during the Progression of Skeletal Muscle Atrophy. PLoS ONE 2015, 10, e0136247. [Google Scholar] [CrossRef]
- Liang, W.; Xu, F.; Li, L.; Peng, C.; Sun, H.; Qiu, J.; Sun, J. Epigenetic control of skeletal muscle atrophy. Cell Mol. Biol. Lett. 2024, 29, 99. [Google Scholar] [CrossRef]
- Acharyya, S.; Sharma, S.M.; Cheng, A.S.; Ladner, K.J.; He, W.; Kline, W.; Wang, H.; Ostrowski, M.C.; Huang, T.H.; Guttridge, D.C. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: Implications in duchenne muscular dystrophy. PLoS ONE 2010, 5, e12479. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.; Carathers, M.; Li, Z.W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef]
- Lu, X.; Liang, B.; Li, S.; Chen, Z.; Chang, W. Modulation of HOXA9 after skeletal muscle denervation and reinnervation. Am. J. Physiol. Cell Physiol. 2020, 318, C1154–C1165. [Google Scholar] [CrossRef] [PubMed]
- Proserpio, V.; Fittipaldi, R.; Ryall, J.G.; Sartorelli, V.; Caretti, G. The methyltransferase SMYD3 mediates the recruitment of transcriptional cofactors at the myostatin and c-Met genes and regulates skeletal muscle atrophy. Genes Dev. 2013, 27, 1299–1312. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Cai, W.; Wang, C.H.; Cederquist, C.T.; Damasio, M.; Homan, E.P.; Batista, T.; Ramirez, A.K.; Gupta, M.K.; Steger, M.; et al. FoxK1 and FoxK2 in insulin regulation of cellular and mitochondrial metabolism. Nat. Commun. 2019, 10, 1582. [Google Scholar] [CrossRef]
- Senf, S.M.; Sandesara, P.B.; Reed, S.A.; Judge, A.R. p300 Acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. Am. J. Physiol. Cell Physiol. 2011, 300, C1490–C1501. [Google Scholar] [CrossRef] [PubMed]
- Bertaggia, E.; Coletto, L.; Sandri, M. Posttranslational modifications control FoxO3 activity during denervation. Am. J. Physiol. Cell Physiol. 2012, 302, C587–C596. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Chamberlain, W.; Gonnella, P.; Alamdari, N.; Aversa, Z.; Hasselgren, P.O. Multiple muscle wasting-related transcription factors are acetylated in dexamethasone-treated muscle cells. Biochem. Cell Biol. 2012, 90, 200–208. [Google Scholar] [CrossRef]
- Sin, T.K.; Zhu, J.Z.; Zhang, G.; Li, Y.P. p300 Mediates Muscle Wasting in Lewis Lung Carcinoma. Cancer Res. 2019, 79, 1331–1342. [Google Scholar] [CrossRef]
- Zhang, G.; Jin, B.; Li, Y.P. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011, 30, 4323–4335. [Google Scholar] [CrossRef]
- Fan, Z.; Wu, J.; Chen, Q.N.; Lyu, A.K.; Chen, J.L.; Sun, Y.; Lyu, Q.; Zhao, Y.X.; Guo, A.; Liao, Z.Y.; et al. Type 2 diabetes-induced overactivation of P300 contributes to skeletal muscle atrophy by inhibiting autophagic flux. Life Sci. 2020, 258, 118243. [Google Scholar] [CrossRef] [PubMed]
- Sin, T.K.; Zhang, G.; Zhang, Z.; Zhu, J.Z.; Zuo, Y.; Frost, J.A.; Li, M.; Li, Y.P. Cancer-Induced Muscle Wasting Requires p38beta MAPK Activation of p300. Cancer Res. 2021, 81, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Beharry, A.W.; Sandesara, P.B.; Roberts, B.M.; Ferreira, L.F.; Senf, S.M.; Judge, A.R. HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J. Cell Sci. 2014, 127, 1441–1453. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Martone, J.; Girardi, E.; Cesana, M.; Incitti, T.; Morlando, M.; Nicoletti, C.; Santini, T.; Sthandier, O.; Barberi, L.; et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 2010, 12, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef]
- Spreafico, M.; Cafora, M.; Bragato, C.; Capitanio, D.; Marasca, F.; Bodega, B.; De Palma, C.; Mora, M.; Gelfi, C.; Marozzi, A.; et al. Targeting HDAC8 to ameliorate skeletal muscle differentiation in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105750. [Google Scholar] [CrossRef]
- Berdeaux, R.; Goebel, N.; Banaszynski, L.; Takemori, H.; Wandless, T.; Shelton, G.D.; Montminy, M. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat. Med. 2007, 13, 597–603. [Google Scholar] [CrossRef]
- Cohen, T.J.; Waddell, D.S.; Barrientos, T.; Lu, Z.; Feng, G.; Cox, G.A.; Bodine, S.C.; Yao, T.P. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. J. Biol. Chem. 2007, 282, 33752–33759. [Google Scholar] [CrossRef]
- Pigna, E.; Simonazzi, E.; Sanna, K.; Bernadzki, K.M.; Proszynski, T.; Heil, C.; Palacios, D.; Adamo, S.; Moresi, V. Histone deacetylase 4 protects from denervation and skeletal muscle atrophy in a murine model of amyotrophic lateral sclerosis. EBioMedicine 2019, 40, 717–732. [Google Scholar] [CrossRef]
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.J.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A.; et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010, 143, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhou, Y.; Li, L.; Zhao, J.; Qin, W.; Jin, Z.; Liu, W. Increase in HDAC9 suppresses myoblast differentiation via epigenetic regulation of autophagy in hypoxia. Cell Death Dis. 2019, 10, 552. [Google Scholar] [CrossRef]
- Osseni, A.; Ravel-Chapuis, A.; Belotti, E.; Scionti, I.; Gangloff, Y.G.; Moncollin, V.; Mazelin, L.; Mounier, R.; Leblanc, P.; Jasmin, B.J.; et al. Pharmacological inhibition of HDAC6 improves muscle phenotypes in dystrophin-deficient mice by downregulating TGF-beta via Smad3 acetylation. Nat. Commun. 2022, 13, 7108. [Google Scholar] [CrossRef]
- Surinlert, P.; Thitiphatphuvanon, T.; Khimmaktong, W.; Pholpramoo, C.; Tipbunjong, C. Hyperglycemia induced C2C12 myoblast cell cycle arrest and skeletal muscle atrophy by modulating sirtuins gene expression in rats. Pol. J. Vet. Sci. 2021, 24, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, S.Y.; Lim, Y. Lespedeza bicolor extract supplementation reduced hyperglycemia-induced skeletal muscle damage by regulation of AMPK/SIRT/PGC1alpha-related energy metabolism in type 2 diabetic mice. Nutr. Res. 2023, 110, 1–13. [Google Scholar] [CrossRef]
- Lee, D.; Goldberg, A.L. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J. Biol. Chem. 2013, 288, 30515–30526. [Google Scholar] [CrossRef] [PubMed]
- Dugdale, H.F.; Hughes, D.C.; Allan, R.; Deane, C.S.; Coxon, C.R.; Morton, J.P.; Stewart, C.E.; Sharples, A.P. The role of resveratrol on skeletal muscle cell differentiation and myotube hypertrophy during glucose restriction. Mol. Cell Biochem. 2018, 444, 109–123. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Igarashi, M.; Nasamu, A.S.; Knezevic, J.; Guarente, L. Muscle-specific SIRT1 gain-of-function increases slow-twitch fibers and ameliorates pathophysiology in a mouse model of duchenne muscular dystrophy. PLoS Genet. 2014, 10, e1004490. [Google Scholar] [CrossRef]
- Simionescu-Bankston, A.; Kumar, A. Noncoding RNAs in the regulation of skeletal muscle biology in health and disease. J. Mol. Med. 2016, 94, 853–866. [Google Scholar] [CrossRef]
- Lee, S.J.; Huynh, T.V.; Lee, Y.S.; Sebald, S.M.; Wilcox-Adelman, S.A.; Iwamori, N.; Lepper, C.; Matzuk, M.M.; Fan, C.M. Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathway. Proc. Natl. Acad. Sci. USA 2012, 109, E2353–E2360. [Google Scholar] [CrossRef]
- Hitachi, K.; Tsuchida, K. Role of microRNAs in skeletal muscle hypertrophy. Front. Physiol. 2013, 4, 408. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; Fairlie, E.; Garnham, A.P.; Hargreaves, M. Exercise-induced histone modifications in human skeletal muscle. J. Physiol. 2009, 587, 5951–5958. [Google Scholar] [CrossRef] [PubMed]
- Thalacker-Mercer, A.; Stec, M.; Cui, X.; Cross, J.; Windham, S.; Bamman, M. Cluster analysis reveals differential transcript profiles associated with resistance training-induced human skeletal muscle hypertrophy. Physiol. Genomics 2013, 45, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef]
- Chabert, C.; Khochbin, S.; Rousseaux, S.; Furze, R.; Smithers, N.; Prinjha, R.; Schlattner, U.; Pison, C.; Dubouchaud, H. Muscle hypertrophy in hypoxia with inflammation is controlled by bromodomain and extra-terminal domain proteins. Sci. Rep. 2017, 7, 12133. [Google Scholar] [CrossRef]
- Moretti, I.; Ciciliot, S.; Dyar, K.A.; Abraham, R.; Murgia, M.; Agatea, L.; Akimoto, T.; Bicciato, S.; Forcato, M.; Pierre, P.; et al. MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat. Commun. 2016, 7, 12397. [Google Scholar] [CrossRef]
- Winbanks, C.E.; Beyer, C.; Hagg, A.; Qian, H.; Sepulveda, P.V.; Gregorevic, P. miR-206 represses hypertrophy of myogenic cells but not muscle fibers via inhibition of HDAC4. PLoS ONE 2013, 8, e73589. [Google Scholar] [CrossRef]
- Koltai, E.; Bori, Z.; Chabert, C.; Dubouchaud, H.; Naito, H.; Machida, S.; Davies, K.J.; Murlasits, Z.; Fry, A.C.; Boldogh, I.; et al. SIRT1 may play a crucial role in overload-induced hypertrophy of skeletal muscle. J. Physiol. 2017, 595, 3361–3376. [Google Scholar] [CrossRef]
- Khurana, T.S.; Hoffman, E.P.; Kunkel, L.M. Identification of a chromosome 6-encoded dystrophin-related protein. J. Biol. Chem. 1990, 265, 16717–16720. [Google Scholar] [CrossRef]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.J.; Kunkel, L.M. The Pathogenesis and Therapy of Muscular Dystrophies. Annu. Rev. Genomics Hum. Genet. 2015, 16, 281–308. [Google Scholar] [CrossRef]
- Colussi, C.; Gurtner, A.; Rosati, J.; Illi, B.; Ragone, G.; Piaggio, G.; Moggio, M.; Lamperti, C.; D’Angelo, G.; Clementi, E.; et al. Nitric oxide deficiency determines global chromatin changes in Duchenne muscular dystrophy. FASEB J. 2009, 23, 2131–2141. [Google Scholar] [CrossRef]
- Consalvi, S.; Saccone, V.; Mozzetta, C. Histone deacetylase inhibitors: A potential epigenetic treatment for Duchenne muscular dystrophy. Epigenomics 2014, 6, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Sandona, M.; Cavioli, G.; Renzini, A.; Cedola, A.; Gigli, G.; Coletti, D.; McKinsey, T.A.; Moresi, V.; Saccone, V. Histone Deacetylases: Molecular Mechanisms and Therapeutic Implications for Muscular Dystrophies. Int. J. Mol. Sci. 2023, 24, 4306. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, A.M.; Guo, X.; Bartkuhn, M.; Gunther, S.; Kunne, C.; Smolka, C.; Atzberger, A.; Gartner, U.; Mamchaoui, K.; Bober, E.; et al. Inactivation of Sirt6 ameliorates muscular dystrophy in mdx mice by releasing suppression of utrophin expression. Nat. Commun. 2022, 13, 4184. [Google Scholar] [CrossRef] [PubMed]
- Renzini, A.; Marroncelli, N.; Cavioli, G.; Di Francescantonio, S.; Forcina, L.; Lambridis, A.; Di Giorgio, E.; Valente, S.; Mai, A.; Brancolini, C.; et al. Cytoplasmic HDAC4 regulates the membrane repair mechanism in Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 1339–1359. [Google Scholar] [CrossRef]
- Givinostat (Duvyzat) for Duchenne muscular dystrophy. Med. Lett. Drugs Ther. 2024, 66, 204–205. [CrossRef]
- Tonini, M.M.; Passos-Bueno, M.R.; Cerqueira, A.; Matioli, S.R.; Pavanello, R.; Zatz, M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 2004, 14, 33–38. [Google Scholar] [CrossRef]
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camano, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef]
- Zeng, W.; de Greef, J.C.; Chen, Y.Y.; Chien, R.; Kong, X.; Gregson, H.C.; Winokur, S.T.; Pyle, A.; Robertson, K.D.; Schmiesing, J.A.; et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009, 5, e1000559. [Google Scholar] [CrossRef]
- Jiang, G.; Yang, F.; van Overveld, P.G.; Vedanarayanan, V.; van der Maarel, S.; Ehrlich, M. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum. Mol. Genet. 2003, 12, 2909–2921. [Google Scholar] [CrossRef]
- Haynes, P.; Bomsztyk, K.; Miller, D.G. Sporadic DUX4 expression in FSHD myocytes is associated with incomplete repression by the PRC2 complex and gain of H3K9 acetylation on the contracted D4Z4 allele. Epigenetics Chromatin 2018, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Strafella, C.; Caputo, V.; Bortolani, S.; Torchia, E.; Megalizzi, D.; Trastulli, G.; Monforte, M.; Colantoni, L.; Caltagirone, C.; Ricci, E.; et al. Whole exome sequencing highlights rare variants in CTCF, DNMT1, DNMT3A, EZH2 and SUV39H1 as associated with FSHD. Front. Genet. 2023, 14, 1235589. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016, 44, 5161–5173. [Google Scholar] [CrossRef]
- Bosnakovski, D.; da Silva, M.T.; Sunny, S.T.; Ener, E.T.; Toso, E.A.; Yuan, C.; Cui, Z.; Walters, M.A.; Jadhav, A.; Kyba, M. A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci. Adv. 2019, 5, eaaw7781. [Google Scholar] [CrossRef]
- Ma, Z.; Chen, L.; Wang, Y.; Zhang, S.; Zheng, J.; Luo, Y.; Wang, C.; Zeng, H.; Xue, L.; Tan, Z.; et al. Novel insights of EZH2-mediated epigenetic modifications in degenerative musculoskeletal diseases. Ageing Res. Rev. 2023, 90, 102034. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Kosinsky, R.L. Epigenetics of Aging and Aging-Associated Diseases. Int. J. Mol. Sci. 2021, 22, 401. [Google Scholar] [CrossRef]
- Molinari, S.; Imbriano, C.; Moresi, V.; Renzini, A.; Belluti, S.; Lozanoska-Ochser, B.; Gigli, G.; Cedola, A. Histone deacetylase functions and therapeutic implications for adult skeletal muscle metabolism. Front. Mol. Biosci. 2023, 10, 1130183. [Google Scholar] [CrossRef]
- Setiawan, T.; Sari, I.N.; Wijaya, Y.T.; Julianto, N.M.; Muhammad, J.A.; Lee, H.; Chae, J.H.; Kwon, H.Y. Cancer cachexia: Molecular mechanisms and treatment strategies. J. Hematol. Oncol. 2023, 16, 54. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, T.; Shao, W.; Lu, R.; Wang, L.; Liu, H.; Jiang, B.; Li, S.; Zhuo, H.; Wang, S.; et al. Amiloride ameliorates muscle wasting in cancer cachexia through inhibiting tumor-derived exosome release. Skelet. Muscle 2021, 11, 17. [Google Scholar] [CrossRef]
- Xu, K.; Wang, Y.; Wang, F.; Guo, Y.; Ren, Y.; Low, V.; Cho, S.; Liu, Q.; Qiu, Y.; Li, X.; et al. SIRT6 Ameliorates Cancer Cachexia-Associated Adipose Wasting by Suppressing TNFR2 Signalling in Mice. J. Cachexia Sarcopenia Muscle 2025, 16, e13734. [Google Scholar] [CrossRef]
- Liu, D.; Wei, B.; Liang, L.; Sheng, Y.; Sun, S.; Sun, X.; Li, M.; Li, H.; Yang, C.; Peng, Y.; et al. The Circadian Clock Component RORA Increases Immunosurveillance in Melanoma by Inhibiting PD-L1 Expression. Cancer Res. 2024, 84, 2265–2281. [Google Scholar] [CrossRef]
- Sincennes, M.C.; Brun, C.E.; Rudnicki, M.A. Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease. Stem Cells Transl. Med. 2016, 5, 282–290. [Google Scholar] [CrossRef]
- Bianchi, M.; Renzini, A.; Adamo, S.; Moresi, V. Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation. Int. J. Mol. Sci. 2017, 18, 840. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, X.; Yan, J.; Chen, M.; Zhu, M.; Tang, Y.; Liu, S.; Tang, Z. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yao, H.; Lin, S.; Zhu, X.; Shen, Z.; Lu, G.; Poon, W.S.; Xie, D.; Lin, M.C.; Kung, H.F. Transcriptional and epigenetic regulation of human microRNAs. Cancer Lett. 2013, 331, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, J.; Tretyn, A. MicroRNA-Mediated Regulation of Histone-Modifying Enzymes in Cancer: Mechanisms and Therapeutic Implications. Biomolecules 2023, 13, 1590. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, K.; Cziesielski, M.J.; Mariappan, K.G.; Cui, G.; Aranda, M. Histone modifications and DNA methylation act cooperatively in regulating symbiosis genes in the sea anemone Aiptasia. BMC Biol. 2022, 20, 265. [Google Scholar] [CrossRef]
- Dubois, V.; Laurent, M.; Boonen, S.; Vanderschueren, D.; Claessens, F. Androgens and skeletal muscle: Cellular and molecular action mechanisms underlying the anabolic actions. Cell Mol. Life Sci. 2012, 69, 1651–1667. [Google Scholar] [CrossRef]
- Nuccio, A.; Nogueira-Ferreira, R.; Moreira-Pais, A.; Attanzio, A.; Duarte, J.A.; Luparello, C.; Ferreira, R. The contribution of mitochondria to age-related skeletal muscle wasting: A sex-specific perspective. Life Sci. 2024, 336, 122324. [Google Scholar] [CrossRef]
- Pataky, M.W.; Dasari, S.; Michie, K.L.; Sevits, K.J.; Kumar, A.A.; Klaus, K.A.; Heppelmann, C.J.; Robinson, M.M.; Carter, R.E.; Lanza, I.R.; et al. Impact of biological sex and sex hormones on molecular signatures of skeletal muscle at rest and in response to distinct exercise training modes. Cell Metab. 2023, 35, 1996–2010. [Google Scholar] [CrossRef]
- Urban, R.J.; Bodenburg, Y.H.; Gilkison, C.; Foxworth, J.; Coggan, A.R.; Wolfe, R.R.; Ferrando, A. Testosterone administration to elderly men increases skeletal muscle strength and protein synthesis. Am. J. Physiol. 1995, 269, E820–E826. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; Guerra, M.; Di Rosa, V.; Compagnucci, C.; Sette, C. Combined treatment with the histone deacetylase inhibitor LBH589 and a splice-switch antisense oligonucleotide enhances SMN2 splicing and SMN expression in Spinal Muscular Atrophy cells. J. Neurochem. 2020, 153, 264–275. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Histone Methylation Properties | Histone Methylation Types | Regulator | Function | Mechanism | Ref. |
|---|---|---|---|---|---|
| Inhibitory markers | H3K9 methylation | Suv39H1 | Maintain myoblasts in proliferative stage | Interact with phosphorylated MyoD; deposit H3K9me2 | [50,51,52] |
| Inhibit myogenic differentiation | Interact with MyoD; deposit H3K9me3 | [52,53] | |||
| G9a | Inhibit myogenic differentiation | Interact with Msk1; deposit H3K9me2 | [54,55] | ||
| LSD1 | Promote myogenic differentiation | Interact with MEF2; remove H3K9me2 and H3K9me3 | [56] | ||
| KDM4A | Promote myogenic differentiation and muscle regeneration | Remove H3K9me3 | [57] | ||
| H3K27 methylation | EZH2 | Promote myoblast proliferation and inhibit differentiation | Interact with YY1; deposit H3K27me3 | [58,59] | |
| EZH1 | Promote myogenic differentiation | Interact with Pol II Complex; deposit H3K4me3 | [57] | ||
| Msk1 | Promote myogenic differentiation | Phosphorylize H3S28; remove H3K27me3 | [60,61] | ||
| UTX | Promote myogenic differentiation | Interact with Six4; remove H3K27me3 | [62] | ||
| H4K20 methylation | Suv4-20h1 | Maintain quiescent state of skeletal muscle stem cells | Promote fHC formation; deposit H4K20me2 | [63] | |
| Permissive markers | H3K4 methylation | Set7 | Promote myogenic differentiation | Interact with MyoD; deposit H3K4me1 | [64] |
| TrxG | Promote myoblast proliferation | Interact with methylated Pax7; deposit H3K4me3 | [65,66,67,68] | ||
| Promote myogenic differentiation | Interact with phosphorylated MEF2D | [69] | |||
| MLL5 | Promote myoblast proliferation and differentiation | Regulate LSD1 and SET7/9; deposit H3K4me2/3 | [70] | ||
| PARP1 | Inhibit myogenic differentiation | Interact with MyoD binding regions; remove H3K4me3 | [71] | ||
| H3K36 methylation | Setd2 | Promote myoblast proliferation and differentiation | Inhibit p21, MyoG, and MyHC | [72,73] |
| Histone Acetylation Properties | Histone Acetylation Types | Regulator | Function | Mechanism | Ref. |
|---|---|---|---|---|---|
| HATs | GNAT family | GCN5 | Preserve muscle integrity | Interact with and acetylate YY1 | [100] |
| PCAF | Promote myogenic differentiation | Interact with lamin A/C and acetylate HDAC2 | [101] | ||
| MYST family | Tip60 | Promote myogenic differentiation | Interact with MyoD | [102] | |
| P300/CBP family | P300 | Promote myogenic differentiation | Bind to bHLH or act upstream of Myf5 and MyoD | [103,104] | |
| HDACs | Class I | HDAC1 | Inhibit myogenic differentiation | Interact with MyoD | [105] |
| HDAC3 | Promote myogenic differentiation | Activate EMD and reduce H4K5ac | [106] | ||
| HDAC8 | Promote myogenic differentiation | Interact with EZH2 | [107] | ||
| Class II | HDAC4 | Promote myoblast proliferation, differentiation, and regeneration | Inhibit Cdkn1a and Sharp1 | [108,109] | |
| HDAC9 | Inhibit myogenic differentiation | Interact with MEF2 | [110] | ||
| Class III | SIRT1 | Improve muscle fatigue resistance after repair from muscle injury | Interact with p53 | [111] | |
| SIRT2 | Promote muscle regeneration | Enhance MRFs and CDKs, and inhibit atrogin1 | [112] | ||
| SIRT3 | Promote formation of oxidative muscle fiber | Activate AMPK and PPARδ | [113] | ||
| Class IV | HDAC11 | Inhibit myogenic differentiation and muscle regeneration | Inhibit MyoD activity | [114,115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Hu, L.; Liu, Z.; Wang, S. The Functions and Regulatory Mechanisms of Histone Modifications in Skeletal Muscle Development and Disease. Int. J. Mol. Sci. 2025, 26, 3644. https://doi.org/10.3390/ijms26083644
Huang Z, Hu L, Liu Z, Wang S. The Functions and Regulatory Mechanisms of Histone Modifications in Skeletal Muscle Development and Disease. International Journal of Molecular Sciences. 2025; 26(8):3644. https://doi.org/10.3390/ijms26083644
Chicago/Turabian StyleHuang, Zining, Linqing Hu, Zhiwei Liu, and Shanshan Wang. 2025. "The Functions and Regulatory Mechanisms of Histone Modifications in Skeletal Muscle Development and Disease" International Journal of Molecular Sciences 26, no. 8: 3644. https://doi.org/10.3390/ijms26083644
APA StyleHuang, Z., Hu, L., Liu, Z., & Wang, S. (2025). The Functions and Regulatory Mechanisms of Histone Modifications in Skeletal Muscle Development and Disease. International Journal of Molecular Sciences, 26(8), 3644. https://doi.org/10.3390/ijms26083644