

Targeting Siderophore Biosynthesis to Thwart Microbial Growth




Abstract

1. Introduction
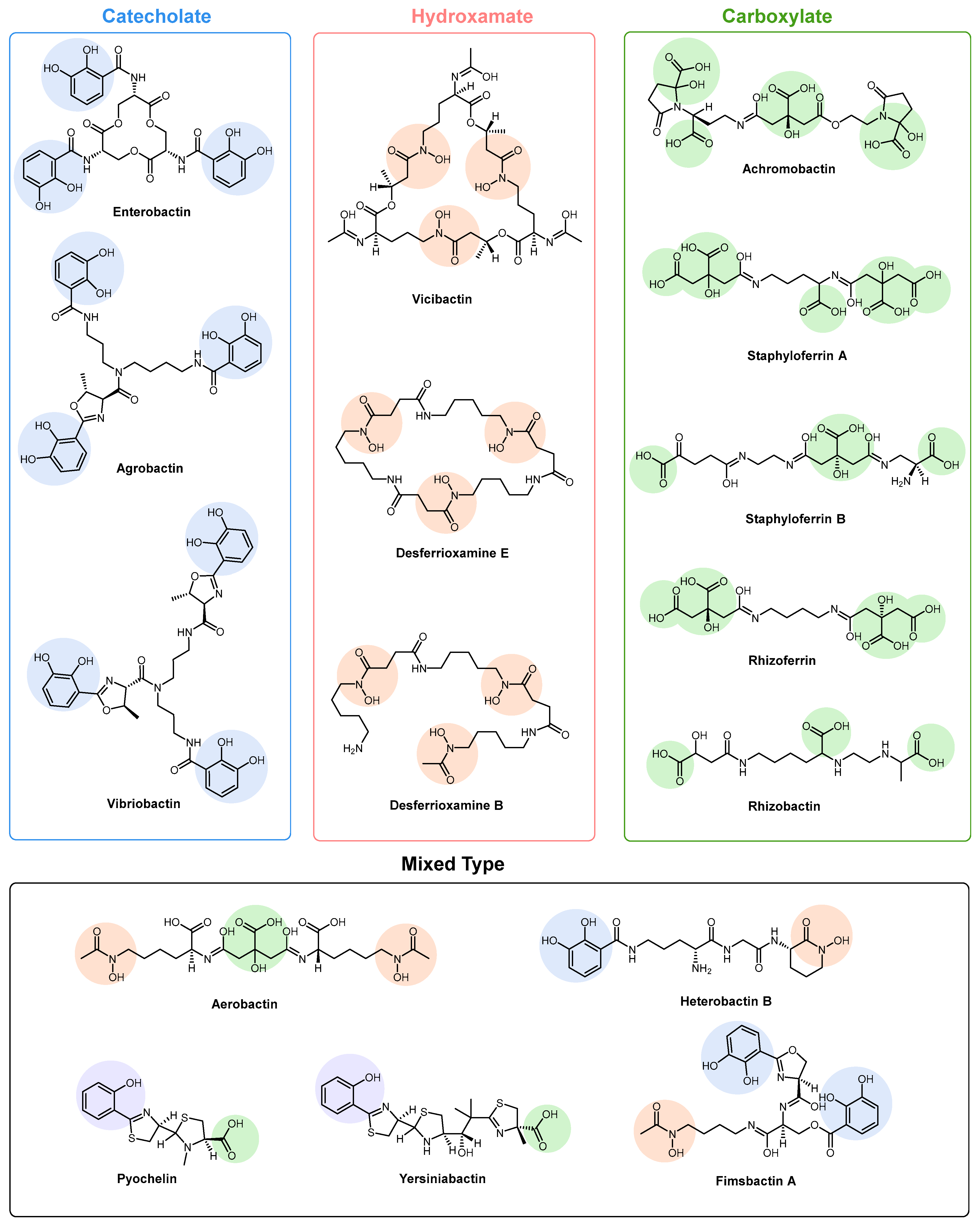
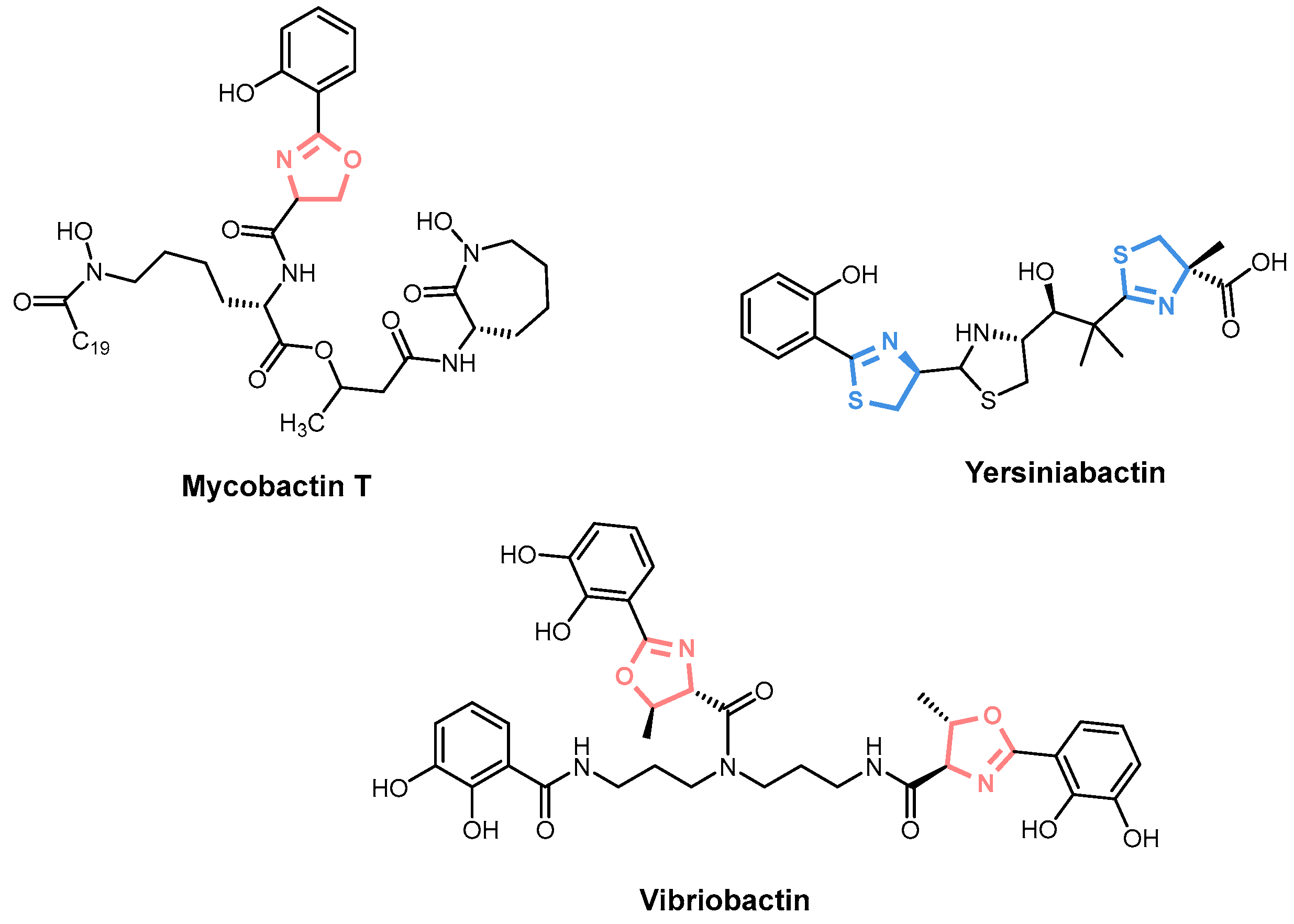
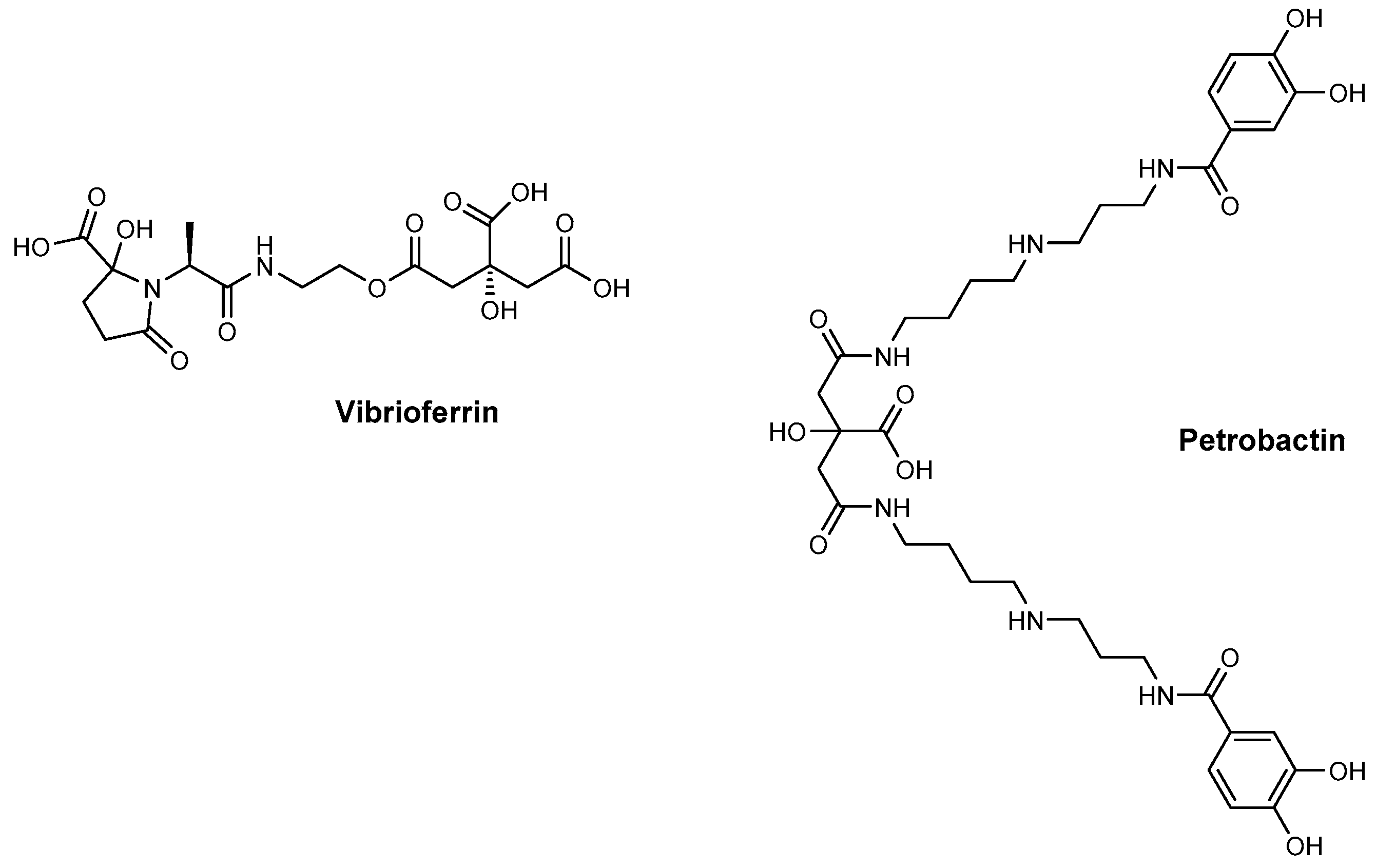
2. Siderophore Biosynthesis
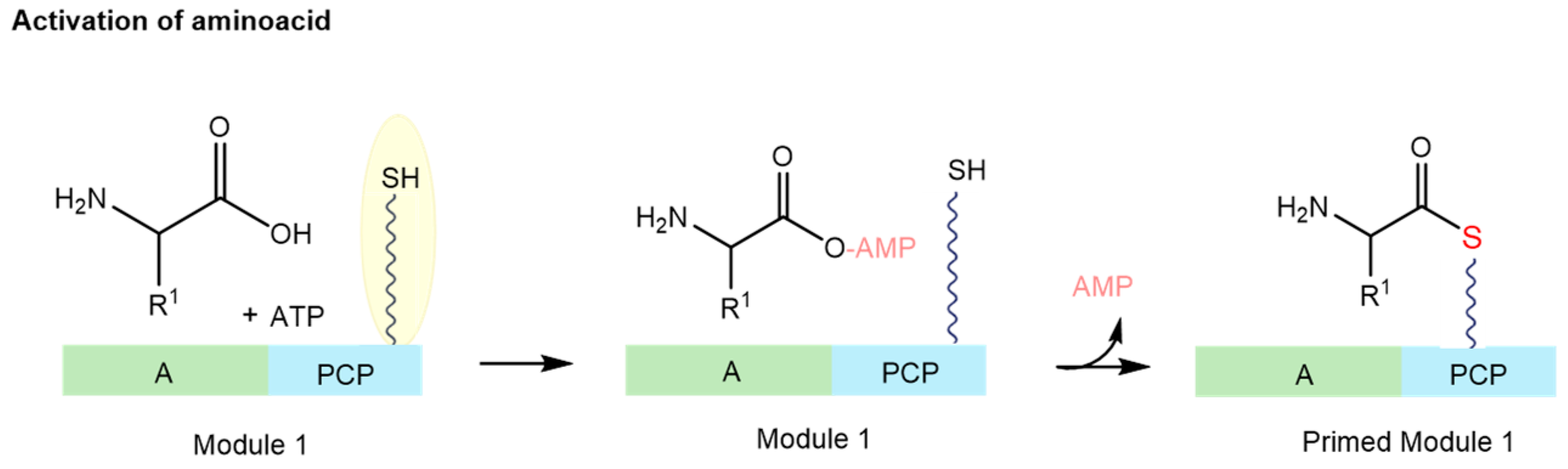
2.1. Nonribosomal Peptide Synthetase (NRPS) Biosynthesis
2.2. Polyketide Synthase (PKS) Biosynthesis
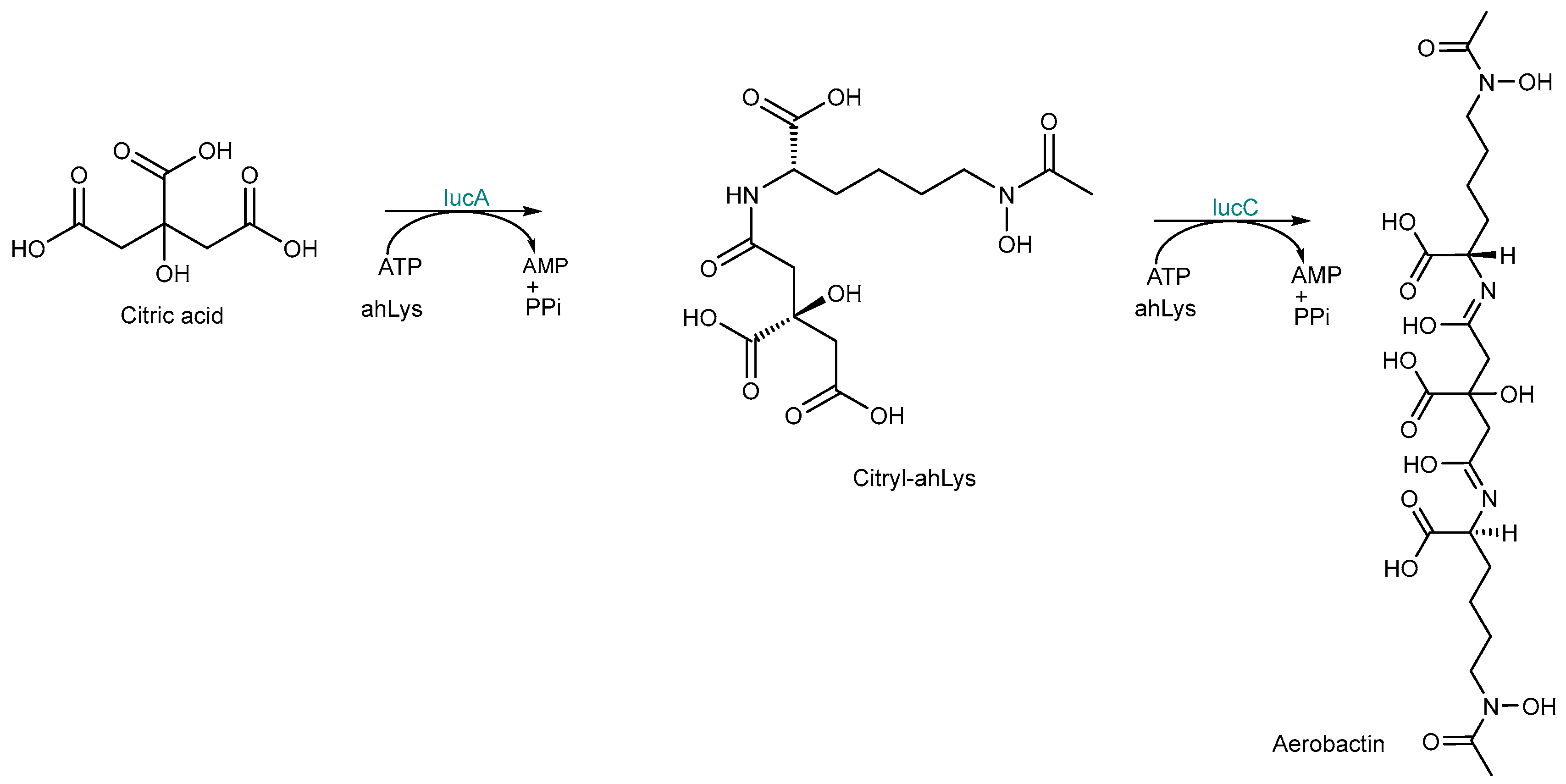
2.3. NRPS-Independent Siderophore (NIS) Synthetase Biosynthesis
3. Iron Acquisition
3.1. Siderophore Secretion
3.2. Cellular Transport Systems for Uptake of Siderophore-Delivered Iron in Bacteria and Fungi
4. Inhibition of Siderophore Biosynthesis
4.1. Inhibitors Targeting NRPS Biosynthesis
4.1.1. Phosphopantetheinyl Transferase Inhibitors
4.1.2. Inhibitors of the Adenylation Domain
4.1.3. An NRPS Enzyme Used as an Inhibitor
4.2. PKS Biosynthesis Inhibitors
4.3. Inhibitors Targeting NIS Biosynthesis
4.4. Inhibition of Siderophore Biosynthesis in Pathogenic Fungi
4.5. Nanoparticle Delivery Systems
4.6. CRISPR-Based Approaches
5. Challenges and Opportunities
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A | Adenylation |
| ABC | ATP-binding cassette |
| ACP | Acyl carrier proteins |
| AMP | Adenosine monophosphate |
| AMS | Adenosine monosulfamate |
| AT | Acyltransferase domain |
| ATP | Adenosine triphosphate |
| BCG | Bacillus Calmette-Guerin |
| C | Condensation |
| CM | Cytosolic membrane |
| CoA | Coenzime A |
| Cy | Cyclization |
| DHB | Dihydroxybenzoate |
| DNA | Deoxyribonucleic Acid |
| E | Epimerization |
| EC50 | Half maximal effective concentration |
| EMA | European Medicines Agency |
| FAAL | Fatty acyl-AMP ligase |
| FACL | Fatty acyl-CoA ligase |
| FAS | Fatty acid synthase |
| FDA | Food and Drug Administration |
| Fur | Ferric uptake regulator |
| IC50 | Half maximal inhibitory concentration |
| K | Ketosynthase domain |
| Ki | Inhibitor constant |
| MbtA | Mycobacterium tuberculosis salicylate adenylation enzyme |
| MbtI | Mycobacterium tuberculosis salicylate synthase |
| MFP | Membrane fusion protein |
| MFS | Major facilitator superfamily |
| Mt | Methylation |
| NADH | 1,4-Dihydronicotinamide adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NIS | NRPS-independent |
| NRPS | Nonribosomal peptide synthetase |
| NTM | Non-tuberculous mycobacteria |
| NTP | Nucleoside triphosphate |
| OM | Outer membrane |
| OMF | Outer membrane factor |
| Ox | Oxidation |
| P | Carrier domain |
| PBP | Periplasmic binding proteins |
| PCP | Peptidyl carrier protein |
| PKS | Polyketide synthase |
| PPTase | Phosphopantetheinyl transferases |
| RNA | Ribonucleic acid |
| RND | Gram-negative-specific resistance/nodulation/cell division |
| SAL-AMS | 5′-O-[N-(salicyl)-sulfamoyl] adenosine |
| SBP | Siderophore-binding protein |
| T | Thiolation |
| TBDT | TonB-dependent transporter |
| TE | Thioesterase |
| tRNA | Transfer ribonucleic acid |
References
- Rogers Van Katwyk, S.; Poirier, M.J.P.; Chandy, S.J.; Faure, K.; Fisher, C.; Lhermie, G.; Moodley, A.; Sarkar, S.; Sophie, M.; Strong, K.; et al. 1–10-100: Unifying goals to mobilize global action on antimicrobial resistance. Glob. Health 2024, 20, 66. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Jonas, O.B.; Irwin, A.; Berthe, F.C.J.; Le Gall, F.G.; Marquez, P.V. Drug-Resistant Infections: A Threat to Our Economic Future (Vol. 2): Final Report; HNP/Agriculture Global Antimicrobial Resistance Initiative; World Bank Group: Washington, DC, USA, 2017. (In English) [Google Scholar]
- European Centre for Disease Prevention and Control. Antimicrobial Consumption in the EU/EEA (ESAC-Net)—Annual Epidemiological Report 2023; ECDC: Stockholm, Sweden, 2024. [Google Scholar]
- Breijyeh, Z.; Karaman, R. Design and Synthesis of Novel Antimicrobial Agents. Antibiotics 2023, 12, 628. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report: 2022; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Schalk, I.J. Bacterial siderophores: Diversity, uptake pathways and applications. Nat. Rev. Microbiol. 2025, 23, 24–40. [Google Scholar] [CrossRef]
- Rodríguez, D.; González-Bello, C. Siderophores: Chemical tools for precise antibiotic delivery. Bioorg. Med. Chem. Lett. 2023, 87, 129282. [Google Scholar] [CrossRef]
- Gomes, A.F.R.; Almeida, M.C.; Sousa, E.; Resende, D.I.S.P. Siderophores and metallophores: Metal complexation weapons to fight environmental pollution. Sci. Total Environ. 2024, 932, 173044. [Google Scholar] [CrossRef]
- Khairnar, A.; Goyal, A.K. Iron Harvesters: Exploring Microbial Siderophores and Their Diverse Applications in Biomedicine. Biomed. Mat. Dev. 2025, 3, 277–287. [Google Scholar] [CrossRef]
- Schalk, I.J.; Mislin, G.L.A. Bacterial Iron Uptake Pathways: Gates for the Import of Bactericide Compounds. J. Med. Chem. 2017, 60, 4573–4576. [Google Scholar] [CrossRef]
- Almeida, M.C.; da Costa, P.M.; Sousa, E.; Resende, D.I.S.P. Emerging Target-Directed Approaches for the Treatment and Diagnosis of Microbial Infections. J. Med. Chem. 2023, 66, 32–70. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, A.; Srivastava, A. Xenosiderophores: Bridging the gap in microbial iron acquisition strategies. World J. Microbiol. Biotechnol. 2025, 41, 69. [Google Scholar] [CrossRef]
- Silvestri, L.; Pettinato, M.; Furiosi, V.; Bavuso Volpe, L.; Nai, A.; Pagani, A. Managing the Dual Nature of Iron to Preserve Health. Int. J. Mol. Sci. 2023, 24, 3995. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Teh, M.R.; Armitage, A.E.; Drakesmith, H. Why cells need iron: A compendium of iron utilisation. Trends Endocrinol. Metab. 2024, 35, 1026–1049. [Google Scholar] [CrossRef]
- Passari, A.K.; Ruiz-Villafán, B.; Cruz-Bautista, R.; Díaz-Domínguez, V.; Rodríguez-Sanoja, R.; Sanchez, S. Opportunities and challenges of microbial siderophores in the medical field. Appl. Microbiol. Biotechnol. 2023, 107, 6751–6759. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.; Lima, T.; Correia, A.; Silva, A.M.; Soares, C.; Morais, S.; Weißelberg, S.; Vilanova, M.; Rohde, H.; Cerca, N. Siderophore-Mediated Iron Acquisition Plays a Critical Role in Biofilm Formation and Survival of Staphylococcus epidermidis Within the Host. Front. Med. 2021, 8, 799227. [Google Scholar] [CrossRef]
- Chen, J.; Guo, Y.; Lu, Y.; Wang, B.; Sun, J.; Zhang, H.; Wang, H. Chemistry and Biology of Siderophores from Marine Microbes. Mar. Drugs 2019, 17, 562. [Google Scholar] [CrossRef]
- Khasheii, B.; Mahmoodi, P.; Mohammadzadeh, A. Siderophores: Importance in bacterial pathogenesis and applications in medicine and industry. Microbiol. Res. 2021, 250, 126790. [Google Scholar] [CrossRef]
- LeBlanc, A.R.; Wuest, W.M. Siderophores: A Case Study in Translational Chemical Biology. Biochemistry 2024, 63, 1877–1891. [Google Scholar] [CrossRef]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef]
- Roemhild, K.; von Maltzahn, F.; Weiskirchen, R.; Knüchel, R.; von Stillfried, S.; Lammers, T. Iron metabolism: Pathophysiology and pharmacology. Trends Pharmacol. Sci. 2021, 42, 640–656. [Google Scholar] [CrossRef]
- Timofeeva, A.M.; Galyamova, M.R.; Sedykh, S.E. Bacterial Siderophores: Classification, Biosynthesis, Perspectives of Use in Agriculture. Plants 2022, 11, 3065. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, L.; Wang, X.; Shah, D.; Song, X.; Kumar, V.; Shakoor, A.; Tripathi, K.; Ramteke, P.W.; Rani, R. Biosynthesis Pathways, Transport Mechanisms and Biotechnological Applications of Fungal Siderophores. J. Fungi 2021, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Puja, H.; Mislin, G.L.A.; Rigouin, C. Engineering Siderophore Biosynthesis and Regulation Pathways to Increase Diversity and Availability. Biomolecules 2023, 13, 959. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kronstad, J.W.; Jung, W.H. Siderophore Biosynthesis and Transport Systems in Model and Pathogenic Fungi. J. Microbiol. Biotechnol. 2024, 34, 1551–1562. [Google Scholar] [CrossRef]
- Shankar, G.; Akhter, Y. Stealing Survival: Iron Acquisition Strategies of Mycobacterium tuberculosis. Biochimie 2024, 227, 37–60. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Cazzaniga, G.; Gelain, A.; Tresoldi, A.; Cocorullo, M.; Roversi, M.; Chiarelli, L.R.; Tomaiuolo, M.; Delre, P.; et al. Targeting Siderophore-Mediated Iron Uptake in M. abscessus: A New Strategy to Limit the Virulence of Non-Tuberculous Mycobacteria. Pharmaceutics 2023, 15, 502. [Google Scholar] [CrossRef]
- Swayambhu, G.; Bruno, M.; Gulick, A.M.; Pfeifer, B.A. Siderophore natural products as pharmaceutical agents. Curr. Opin. Biotechnol. 2021, 69, 242–251. [Google Scholar] [CrossRef]
- Hider, R.C.; Kong, X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27, 637–657. [Google Scholar] [CrossRef]
- Gomes, A.F.R.; Sousa, E.; Resende, D.I.S.P. A Practical Toolkit for the Detection, Isolation, Quantification, and Characterization of Siderophores and Metallophores in Microorganisms. ACS Omega 2024, 9, 26863–26877. [Google Scholar] [CrossRef]
- Barry, S.M.; Challis, G.L. Recent advances in siderophore biosynthesis. Curr. Opin. Chem. Biol. 2009, 13, 205–215. [Google Scholar] [CrossRef]
- Lamb, A.L. Breaking a pathogen’s iron will: Inhibiting siderophore production as an antimicrobial strategy. Biochim. Biophys. Acta 2015, 1854, 1054–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Peng, Z.; Huang, Z.; Fang, J.; Li, X.; Qiu, X. Functional Diversity and Engineering of the Adenylation Domains in Nonribosomal Peptide Synthetases. Mar. Drugs 2024, 22, 349. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Peng, Z.; Zhang, M.; Li, X.; Qiu, X. Structure, Function and Engineering of the Nonribosomal Peptide Synthetase Condensation Domain. Int. J. Mol. Sci. 2024, 25, 11774. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Harder, T.; Ostrowski, A.D.; Carrano, C.J. Photoactive siderophores: Structure, function and biology. J. Inorg. Biochem. 2021, 221, 111457. [Google Scholar] [CrossRef]
- Miyanaga, A.; Kudo, F.; Eguchi, T. Recent advances in the structural analysis of adenylation domains in natural product biosynthesis. Curr. Opin. Chem. Biol. 2022, 71, 102212. [Google Scholar] [CrossRef]
- Xie, B.; Wei, X.; Wan, C.; Zhao, W.; Song, R.; Xin, S.; Song, K. Exploring the Biological Pathways of Siderophores and Their Multidisciplinary Applications: A Comprehensive Review. Molecules 2024, 29, 2318. [Google Scholar] [CrossRef]
- Miethke, M.; Marahiel, M.A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef]
- Carroll, C.S.; and Moore, M.M. Ironing out siderophore biosynthesis: A review of non-ribosomal peptide synthetase (NRPS)-independent siderophore synthetases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 356–381. [Google Scholar] [CrossRef]
- Bailey, D.C.; Alexander, E.; Rice, M.R.; Drake, E.J.; Mydy, L.S.; Aldrich, C.C.; Gulick, A.M. Structural and functional delineation of aerobactin biosynthesis in hypervirulent Klebsiella pneumoniae. J. Biol. Chem. 2018, 293, 7841–7852. [Google Scholar] [CrossRef]
- Sargun, A.; Gerner, R.R.; Raffatellu, M.; Nolan, E.M. Harnessing Iron Acquisition Machinery to Target Enterobacteriaceae. J. Infect. Dis. 2020, 223, S307–S313. [Google Scholar] [CrossRef]
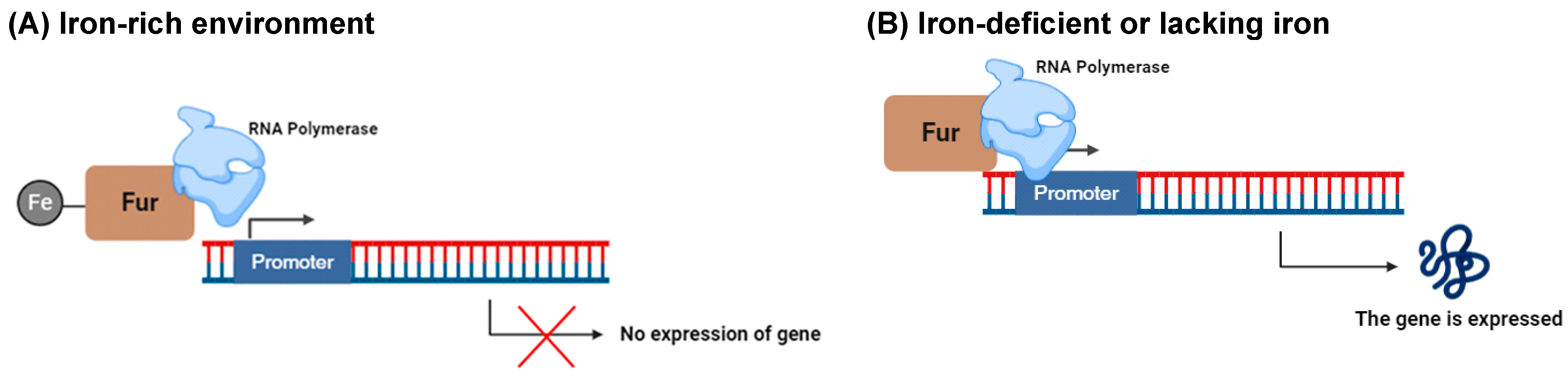
- Santos, R.E.R.d.S.; Batista, B.B.; Neto, J.F.d.S. Ferric Uptake Regulator Fur Coordinates Siderophore Production and Defense against Iron Toxicity and Oxidative Stress and Contributes to Virulence in Chromobacterium violaceum. Appl. Environ. Microbiol. 2020, 86, e01620-20. [Google Scholar] [CrossRef] [PubMed]
- Bin, X.; Pawelek, P.D. Evidence of isochorismate channeling between the Escherichia coli enterobactin biosynthetic enzymes EntC and EntB. Protein Sci. 2024, 33, e5122. [Google Scholar] [CrossRef] [PubMed]
- Hajiagha, M.N.; Kafil, H.S. Efflux pumps and microbial biofilm formation. Infect. Genet. Evol. 2023, 112, 105459. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, L.G.; Dey, D.; Shafer, W.M.; Conn, G.L. Structural and functional diversity of Resistance-Nodulation-Division (RND) efflux pump transporters with implications for antimicrobial resistance. Microbiol. Mol. Biol. Rev. 2024, 88, e00089-23. [Google Scholar] [CrossRef]
- Kronstad, J.W.; Caza, M. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front. Cell Infect. Microbiol. 2013, 4, 80. [Google Scholar] [CrossRef]
- Boyer, E.; Dessolin, J.; Lustig, M.; Decossas, M.; Phan, G.; Cece, Q.; Durand, G.; Dubois, V.; Sansen, J.; Taveau, J.-C.; et al. Molecular Determinants for OMF Selectivity in Tripartite RND Multidrug Efflux Systems. Antibiotics 2022, 11, 126. [Google Scholar] [CrossRef]
- Thomas, C.; Tampé, R. Structural and Mechanistic Principles of ABC Transporters. Annu. Rev. Biochem. 2020, 89, 605–636. [Google Scholar] [CrossRef]
- Klebba, P.E.; Newton, S.M.C.; Six, D.A.; Kumar, A.; Yang, T.; Nairn, B.L.; Munger, C.; Chakravorty, S. Iron Acquisition Systems of Gram-negative Bacterial Pathogens Define TonB-Dependent Pathways to Novel Antibiotics. Chem. Rev. 2021, 121, 5193–5239. [Google Scholar] [CrossRef]
- Braun, V.; Ratliff, A.C.; Celia, H.; Buchanan, S.K. Energization of Outer Membrane Transport by the ExbB ExbD Molecular Motor. J. Bacteriol. 2023, 205, e00035-23. [Google Scholar] [CrossRef]
- Braun, V. Substrate Uptake by TonB-Dependent Outer Membrane Transporters. Mol. Microbiol. 2024, 122, 929–947. [Google Scholar] [CrossRef]
- Uppalapati, S.R.; Sett, A.; Pathania, R. The Outer Membrane Proteins OmpA, CarO, and OprD of Acinetobacter baumannii Confer a Two-Pronged Defense in Facilitating Its Success as a Potent Human Pathogen. Front. Microbiol. 2020, 11, 589234. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.; Crump, M.P. The structural role of the carrier protein—Active controller or passive carrier. Nat. Prod. Rep. 2012, 29, 1111–1137. [Google Scholar] [CrossRef] [PubMed]
- Mercer, A.C.; Burkart, M.D. The ubiquitous carrier protein—A window to metabolite biosynthesis. Nat. Prod. Rep. 2007, 24, 750–773. [Google Scholar] [CrossRef] [PubMed]
- Kinatukara, P.; Patel, K.D.; Haque, A.S.; Singh, R.; Gokhale, R.S.; Sankaranarayananan, R. Structural insights into the regulation of NADPH binding to reductase domains of nonribosomal peptide synthetases: A concerted loop movement model. J. Struct. Biol. 2016, 194, 368–374. [Google Scholar] [CrossRef]
- Walsh, C.T.; Chen, H.; Keating, T.A.; Hubbard, B.K.; Losey, H.C.; Luo, L.; Marshall, C.G.; Miller, D.A.; Patel, H.M. Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines. Curr. Opin. Chem. Biol. 2001, 5, 525–534. [Google Scholar] [CrossRef]
- Linne, U.; Doekel, S.; Marahiel, M.A. Portability of epimerization domain and role of peptidyl carrier protein on epimerization activity in nonribosomal peptide synthetases. Biochemistry 2001, 40, 15824–15834. [Google Scholar] [CrossRef]
- Cho, Y.I.; Armstrong, C.L.; Sulpizio, A.; Acheampong, K.K.; Banks, K.N.; Bardhan, O.; Churchill, S.J.; Connolly-Sporing, A.E.; Crawford, C.E.W.; Cruz Parrilla, P.L.; et al. Engineered Chimeras Unveil Swappable Modular Features of Fatty Acid and Polyketide Synthase Acyl Carrier Proteins. Biochemistry 2022, 61, 217–227. [Google Scholar] [CrossRef]
- Walsh, C.T.; Gehring, A.M.; Weinreb, P.H.; Quadri, L.E.; Flugel, R.S. Post-translational modification of polyketide and nonribosomal peptide synthases. Curr. Opin. Chem. Biol. 1997, 1, 309–315. [Google Scholar] [CrossRef]
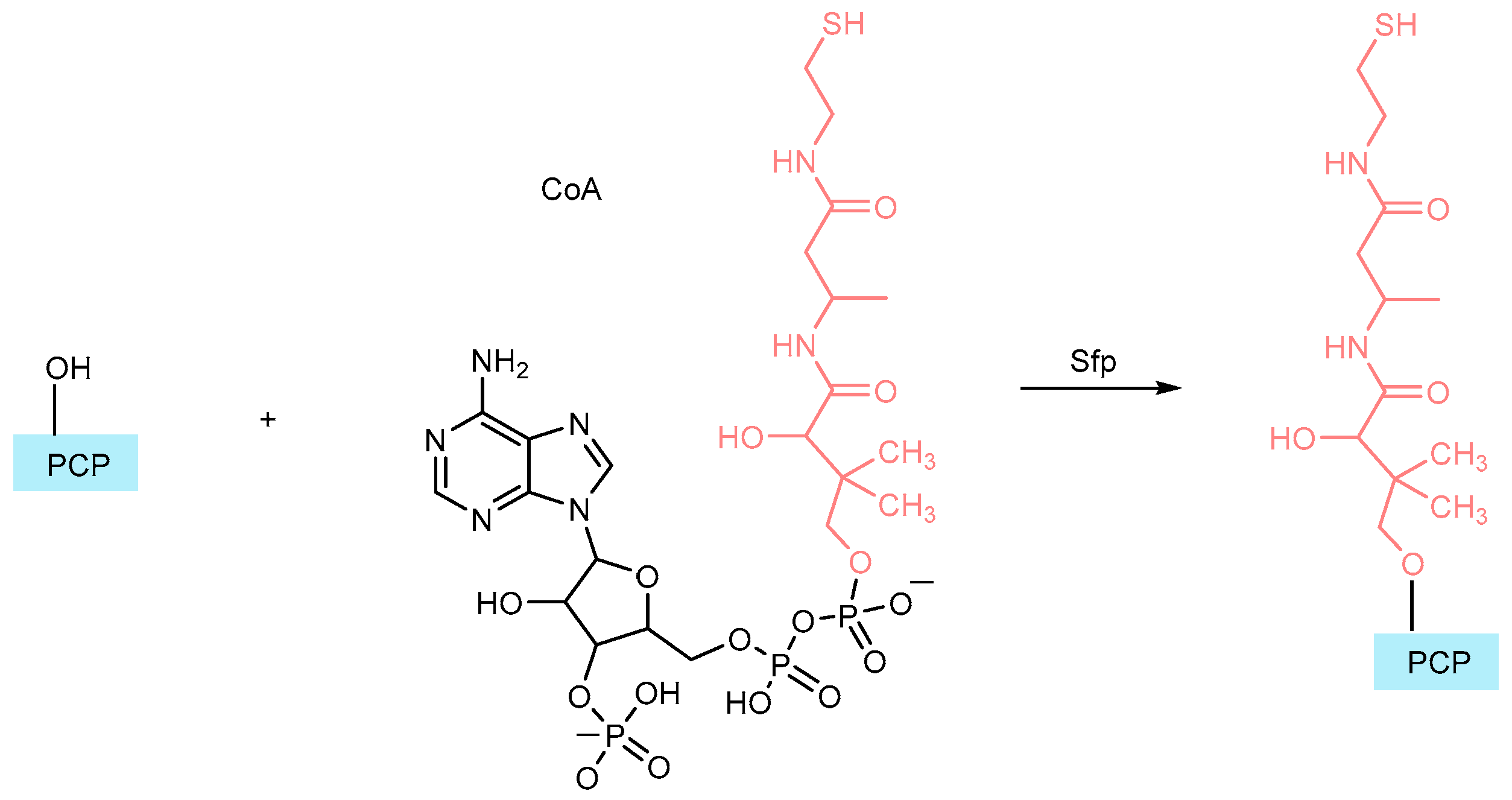
- Lambalot, R.H.; Gehring, A.M.; Flugel, R.S.; Zuber, P.; LaCelle, M.; Marahiel, M.A.; Reid, R.; Khosla, C.; Walsh, C.T. A new enzyme superfamily—The phosphopantetheinyl transferases. Chem. Biol. 1996, 3, 923–936. [Google Scholar] [CrossRef]
- Chalut, C.; Botella, L.; de Sousa-D’Auria, C.; Houssin, C.; Guilhot, C. The nonredundant roles of two 4′-phosphopantetheinyl transferases in vital processes of Mycobacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 8511–8516. [Google Scholar] [CrossRef]
- Brown, A.S.; Owen, J.G.; Jung, J.; Baker, E.N.; Ackerley, D.F. Inhibition of Indigoidine Synthesis as a High-Throughput Colourimetric Screen for Antibiotics Targeting the Essential Mycobacterium tuberculosis Phosphopantetheinyl Transferase PptT. Pharmaceutics 2021, 13, 1066. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Straight, P.D.; McLoughlin, S.M.; Zhou, Z.; Lin, A.J.; Golan, D.E.; Kelleher, N.L.; Kolter, R.; Walsh, C.T. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc. Natl. Acad. Sci. USA 2005, 102, 15815–15820. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.L.; Rai, G.; Yasgar, A.; Attene-Ramos, M.S.; Burkart, M.D.; Simeonov, A.; Jadhav, A.; Maloney, D.J. Discovery of ML 267 as a Novel Inhibitor of Pathogenic Sfp phosphopantetheinyl transferase (PPTase). In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Konno, S.; Ishikawa, F.; Suzuki, T.; Dohmae, N.; Kakeya, H.; Tanabe, G. A Chemoproteomics Approach to Investigate Phosphopantetheine Transferase Activity at the Cellular Level. ChemBioChem 2017, 18, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.L.; Rai, G.; Yasgar, A.; Daniel, T.; Baker, H.L.; Attene-Ramos, M.; Kosa, N.M.; Leister, W.; Burkart, M.D.; Jadhav, A.; et al. 4-(3-Chloro-5-(trifluoromethyl)pyridin-2-yl)-N-(4-methoxypyridin-2-yl)piperazine-1-carbothioamide (ML267), a Potent Inhibitor of Bacterial Phosphopantetheinyl Transferase That Attenuates Secondary Metabolism and Thwarts Bacterial Growth. J. Med. Chem. 2014, 57, 1063–1078. [Google Scholar] [CrossRef]
- Vickery, C.R.; Kosa, N.M.; Casavant, E.P.; Duan, S.; Noel, J.P.; Burkart, M.D. Structure, biochemistry, and inhibition of essential 4′-phosphopantetheinyl transferases from two species of Mycobacteria. ACS Chem. Biol. 2014, 9, 1939–1944. [Google Scholar] [CrossRef]
- Ferreras, J.A.; Ryu, J.S.; Di Lello, F.; Tan, D.S.; Quadri, L.E. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005, 1, 29–32. [Google Scholar] [CrossRef]
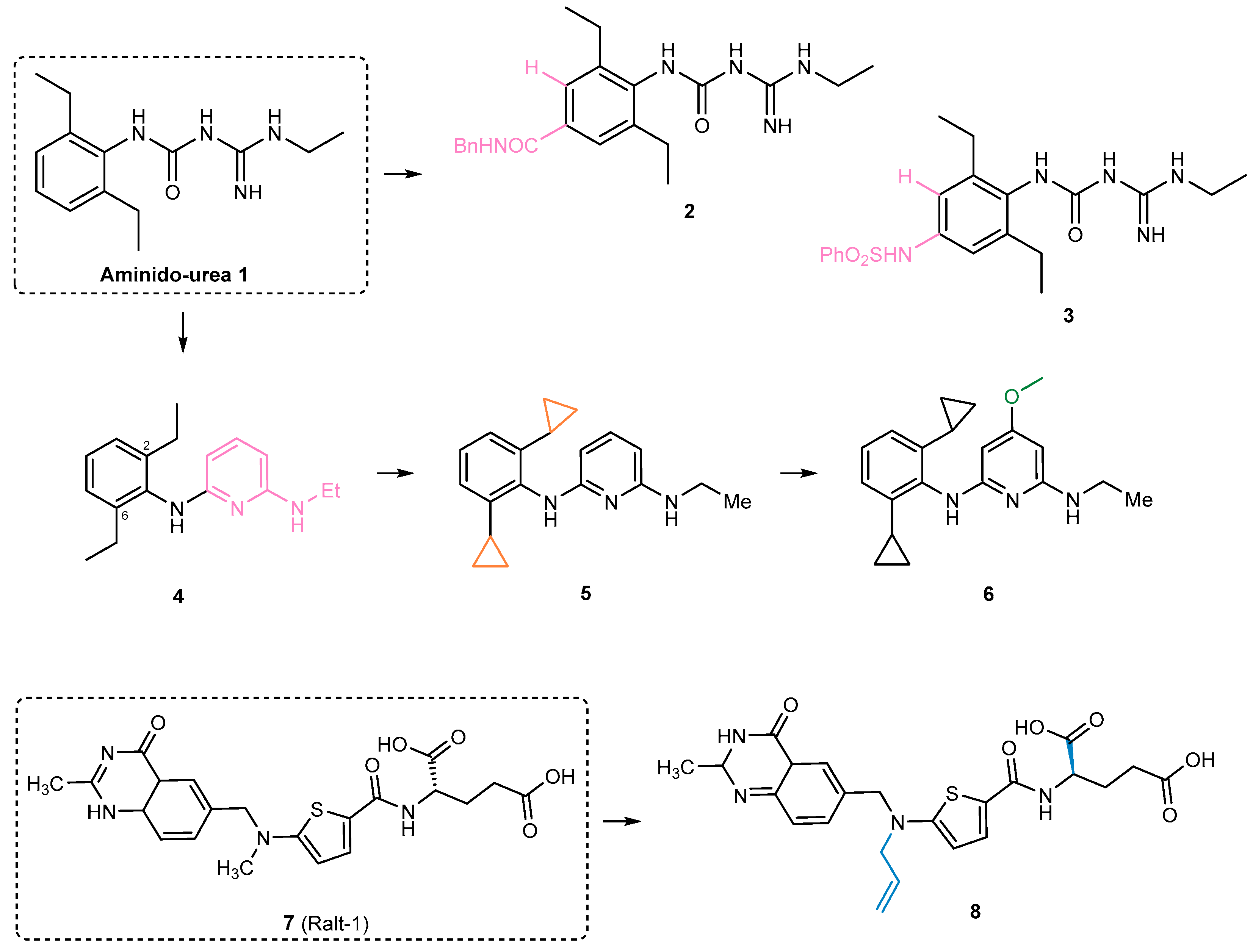
- Carivenc, C.; Maveyraud, L.; Blanger, C.; Ballereau, S.; Roy-Camille, C.; Nguyen, M.C.; Génisson, Y.; Guilhot, C.; Chalut, C.; Pedelacq, J.-D.; et al. Phosphopantetheinyl transferase binding and inhibition by amidino-urea and hydroxypyrimidinethione compounds. Sci. Rep. 2021, 11, 18042. [Google Scholar] [CrossRef]
- Nguyen, M.C.; Saurel, O.; Carivenc, C.; Gavalda, S.; Saitta, S.; Tran, M.P.; Milon, A.; Chalut, C.; Guilhot, C.; Mourey, L.; et al. Conformational flexibility of coenzyme A and its impact on the post-translational modification of acyl carrier proteins by 4′-phosphopantetheinyl transferases. FEBS J. 2020, 287, 4729–4746. [Google Scholar] [CrossRef]
- Ottavi, S.; Li, K.; Cacioppo, J.G.; Perkowski, A.J.; Ramesh, R.; Gold, B.S.; Ling, Y.; Roberts, J.; Singh, A.; Zhang, D.; et al. Mycobacterium tuberculosis PptT Inhibitors Based on Heterocyclic Replacements of Amidinoureas. ACS Med. Chem. Lett. 2023, 14, 970–976. [Google Scholar] [CrossRef]
- Ottavi, S.; Scarry, S.M.; Mosior, J.; Ling, Y.; Roberts, J.; Singh, A.; Zhang, D.; Goullieux, L.; Roubert, C.; Bacqué, E.; et al. In Vitro and In Vivo Inhibition of the Mycobacterium tuberculosis Phosphopantetheinyl Transferase PptT by Amidinoureas. J. Med. Chem. 2022, 65, 1996–2022. [Google Scholar] [CrossRef]
- Leblanc, C.; Prudhomme, T.; Tabouret, G.; Ray, A.; Burbaud, S.; Cabantous, S.; Mourey, L.; Guilhot, C.; Chalut, C. 4′-Phosphopantetheinyl transferase PptT, a new drug target required for Mycobacterium tuberculosis growth and persistence in vivo. PLoS Pathog. 2012, 8, e1003097. [Google Scholar] [CrossRef]
- Ballinger, E.; Mosior, J.; Hartman, T.; Burns-Huang, K.; Gold, B.; Morris, R.; Goullieux, L.; Blanc, I.; Vaubourgeix, J.; Lagrange, S.; et al. Opposing reactions in coenzyme A metabolism sensitize Mycobacterium tuberculosis to enzyme inhibition. Science 2019, 363, eaau8959. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cunningham, D.; Maroun, J.; Cervantes, A.; Glimelius, B. Raltitrexed: Current clinical status and future directions. Ann. Oncol. 2002, 13, 513–522. [Google Scholar] [CrossRef]
- Singh, A.; Ottavi, S.; Krieger, I.; Planck, K.; Perkowski, A.; Kaneko, T.; Davis, A.M.; Suh, C.; Zhang, D.; Goullieux, L.; et al. Redirecting raltitrexed from cancer cell thymidylate synthase to Mycobacterium tuberculosis phosphopantetheinyl transferase. Sci. Adv. 2024, 10, eadj6406. [Google Scholar] [CrossRef]
- Rohilla, A.; Khare, G.; Tyagi, A.K. A combination of docking and cheminformatics approaches for the identification of inhibitors against 4′ phosphopantetheinyl transferase of Mycobacterium tuberculosis. RSC Adv. 2018, 8, 328–341. [Google Scholar] [CrossRef]
- Quadri, L.E. Assembly of aryl-capped siderophores by modular peptide synthetases and polyketide synthases. Mol. Microbiol. 2000, 37, 1–12. [Google Scholar] [CrossRef]
- Nelson, K.M.; Viswanathan, K.; Dawadi, S.; Duckworth, B.P.; Boshoff, H.I.; Barry III, C.E.; Aldrich, C.C. Synthesis and pharmacokinetic evaluation of siderophore biosynthesis inhibitors for Mycobacterium tuberculosis. J. Med. Chem. 2015, 58, 5459–5475. [Google Scholar] [CrossRef]
- Miethke, M.; Bisseret, P.; Beckering, C.L.; Vignard, D.; Eustache, J.; Marahiel, M.A. Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 2006, 273, 409–419. [Google Scholar] [CrossRef]
- Dawadi, S.; Boshoff, H.I.M.; Park, S.W.; Schnappinger, D.; Aldrich, C.C. Conformationally Constrained Cinnolinone Nucleoside Analogues as Siderophore Biosynthesis Inhibitors for Tuberculosis. ACS Med. Chem. Lett. 2018, 9, 386–391. [Google Scholar] [CrossRef]
- Lun, S.; Guo, H.; Adamson, J.; Cisar, J.S.; Davis, T.D.; Chavadi, S.S.; Warren, J.D.; Quadri, L.E.N.; Tan, D.S.; Bishai, W.R. Pharmacokinetic and In Vivo Efficacy Studies of the Mycobactin Biosynthesis Inhibitor Salicyl-AMS in Mice. Antimicrob. Agents Chemother. 2013, 57, 5138–5140. [Google Scholar] [CrossRef]
- Lux, M.C.; Standke, L.C.; Tan, D.S. Targeting adenylate-forming enzymes with designed sulfonyladenosine inhibitors. J. Antibiot. 2019, 72, 325–349. [Google Scholar] [CrossRef]
- Dawadi, S.; Viswanathan, K.; Boshoff, H.I.; Barry, C.E., III; Aldrich, C.C. Investigation and Conformational Analysis of Fluorinated Nucleoside Antibiotics Targeting Siderophore Biosynthesis. J. Org. Chem. 2015, 80, 4835–4850. [Google Scholar] [CrossRef]
- Finking, R.; Neumüller, A.; Solsbacher, J.; Konz, D.; Kretzschmar, G.; Schweitzer, M.; Krumm, T.; Marahiel, M.A. Aminoacyl adenylate substrate analogues for the inhibition of adenylation domains of nonribosomal peptide synthetases. ChemBioChem 2003, 4, 903–906. [Google Scholar] [CrossRef]
- Ishikawa, F.; Konno, S.; Kakeya, H.; Tanabe, G. Development of a chemical scaffold for inhibiting nonribosomal peptide synthetases in live bacterial cells. Beilstein J. Org. Chem. 2024, 20, 445–451. [Google Scholar] [CrossRef]
- Neres, J.; Labello, N.P.; Somu, R.V.; Boshoff, H.I.; Wilson, D.J.; Vannada, J.; Chen, L.; Barry, C.E., III; Bennett, E.M.; Aldrich, C.C. Inhibition of Siderophore Biosynthesis in Mycobacterium tuberculosis with Nucleoside Bisubstrate Analogues: Structure−Activity Relationships of the Nucleobase Domain of 5′-O-[N-(Salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008, 51, 5349–5370. [Google Scholar] [CrossRef]
- Davis, T.D.; Mohandas, P.; Chiriac, M.I.; Bythrow, G.V.; Quadri, L.E.; Tan, D.S. Design, synthesis, and biological evaluation of α-hydroxyacyl-AMS inhibitors of amino acid adenylation enzymes. Bioorg. Med. Chem. Lett. 2016, 26, 5340–5345. [Google Scholar] [CrossRef]
- Cisar, J.S.; Ferreras, J.A.; Soni, R.K.; Quadri, L.E.; Tan, D.S. Exploiting ligand conformation in selective inhibition of non-ribosomal peptide synthetase amino acid adenylation with designed macrocyclic small molecules. J. Am. Chem. Soc. 2007, 129, 7752–7753. [Google Scholar] [CrossRef]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry, C.E. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef]
- Mori, M.; Cocorullo, M.; Tresoldi, A.; Cazzaniga, G.; Gelain, A.; Stelitano, G.; Chiarelli, L.R.; Tomaiuolo, M.; Delre, P.; Mangiatordi, G.F.; et al. Structural basis for specific inhibition of salicylate synthase from Mycobacterium abscessus. Eur. J. Med. Chem. 2024, 265, 116073. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Gelain, A.; Pini, E.; Chiarelli, L.R.; Sammartino, J.C.; Poli, G.; Tuccinardi, T.; Beretta, G.; Porta, A.; et al. Shedding X-ray Light on the Role of Magnesium in the Activity of Mycobacterium tuberculosis Salicylate Synthase (MbtI) for Drug Design. J. Med. Chem. 2020, 63, 7066–7080. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Stelitano, G.; Griego, A.; Chiarelli, L.R.; Cazzaniga, G.; Gelain, A.; Pini, E.; Camera, M.; Canzano, P.; Fumagalli, A.; et al. Synthesis and Assessment of the In Vitro and Ex Vivo Activity of Salicylate Synthase (Mbti) Inhibitors as New Candidates for the Treatment of Mycobacterial Infections. Pharmaceuticals 2022, 15, 992. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.L.; Wilson, D.J.; Aldrich, C.C.; Blanchard, J.S. Kinetic and inhibition studies of dihydroxybenzoate-AMP ligase from Escherichia coli. Biochemistry 2010, 49, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.P.; Lomino, J.V.; Wolfenden, R. Nanomolar inhibition of the enterobactin biosynthesis enzyme, EntE: Synthesis, substituent effects, and additivity. Bioorg. Med. Chem. Lett. 2006, 16, 3802–3805. [Google Scholar] [CrossRef]
- Sundlov, J.A.; Shi, C.; Wilson, D.J.; Aldrich, C.C.; Gulick, A.M. Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem. Biol. 2012, 19, 188–198. [Google Scholar] [CrossRef]
- Mitchell, C.A.; Shi, C.; Aldrich, C.C.; Gulick, A.M. Structure of PA1221, a Nonribosomal Peptide Synthetase Containing Adenylation and Peptidyl Carrier Protein Domains. Biochemistry 2012, 51, 3252–3263. [Google Scholar] [CrossRef]
- Balhara, M.; Ruhil, S.; Kumar, M.; Dhankhar, S.; Chhillar, A. An anti-Aspergillus protein from Escherichia coli DH 5α: Putative inhibitor of siderophore biosynthesis in Aspergillus fumigatus. Mycoses 2014, 57, 153–162. [Google Scholar] [CrossRef]
- Cummings, M.; Breitling, R.; Takano, E. Steps towards the synthetic biology of polyketide biosynthesis. FEMS Microb. Lett. 2014, 351, 116–125. [Google Scholar] [CrossRef]
- Meier, J.L.; Burkart, M.D. The chemical biology of modular biosynthetic enzymes. Chem. Soc. Rev. 2009, 38, 2012–2045. [Google Scholar] [CrossRef]
- Williams, G.J. Engineering polyketide synthases and nonribosomal peptide synthetases. Curr. Opin. Struct Biol. 2013, 23, 603–612. [Google Scholar] [CrossRef]
- Arora, P.; Goyal, A.; Natarajan, V.T.; Rajakumara, E.; Verma, P.; Gupta, R.; Yousuf, M.; Trivedi, O.A.; Mohanty, D.; Tyagi, A.; et al. Mechanistic and functional insights into fatty acid activation in Mycobacterium tuberculosis. Nat. Chem. Biol. 2009, 5, 166–173. [Google Scholar] [CrossRef]
- McQuade, T.J.; Shallop, A.D.; Sheoran, A.; DelProposto, J.E.; Tsodikov, O.V.; Garneau-Tsodikova, S. A nonradioactive high-throughput assay for screening and characterization of adenylation domains for nonribosomal peptide combinatorial biosynthesis. Anal. Biochem. 2009, 386, 244–250. [Google Scholar] [CrossRef]
- Tripathi, A.; Schofield, M.M.; Chlipala, G.E.; Schultz, P.J.; Yim, I.; Newmister, S.A.; Nusca, T.D.; Scaglione, J.B.; Hanna, P.C.; Tamayo-Castillo, G. Baulamycins A and B, broad-spectrum antibiotics identified as inhibitors of siderophore biosynthesis in Staphylococcus aureus and Bacillus anthracis. J. Am. Chem. Soc. 2014, 136, 1579–1586. [Google Scholar] [CrossRef]
- Schrettl, M.; Bignell, E.; Kragl, C.; Joechl, C.; Rogers, T.; Arst Jr, H.N.; Haynes, K.; Haas, H. Siderophore biosynthesis but not reductive iron assimilation is essential for Aspergillus fumigatus virulence. J. Exp. Med. 2004, 200, 1213–1219. [Google Scholar] [CrossRef]
- Jirí Houšt, J.S.; Havlícek, V. Antifungal Drugs. Metabolites 2020, 10, 106. [Google Scholar] [CrossRef]
- Martín del Campo, J.S.; Vogelaar, N.; Tolani, K.; Kizjakina, K.; Harich, K.; Sobrado, P. Inhibition of the flavin-dependent monooxygenase siderophore A (SidA) blocks siderophore biosynthesis and Aspergillus fumigatus growth. ACS Chem. Biol. 2016, 11, 3035–3042. [Google Scholar] [CrossRef]
- Haas, H. Fungal siderophore metabolism with a focus on Aspergillus fumigatus. Nat. Prod. Rep. 2014, 31, 1266–1276. [Google Scholar] [CrossRef]
- Ha, S.; Kim, J.; Seo, H.W.; Kim, L.; Yi, Y.-S.; Seo, S.E.; Kim, K.H.; Kim, S.; An, J.E.; Kim, G.-J.; et al. Siderophore-Functionalized Nanodrug for Treating Antibiotic-Resistant Bacteria. ACS Nano 2025, 19, 5131–5145. [Google Scholar] [CrossRef]
- Kharga, K.; Shubhang, J.; Tanvi, V.; and Kumar, L. Current developments and prospects of the antibiotic delivery systems. Crit. Rev. Microbiol. 2025, 51, 44–83. [Google Scholar] [CrossRef]
- Kotb, E.; Ahmed, A.A.; Saleh, T.A.; Ajeebi, A.M.; Al-Gharsan, M.S.; Aldahmash, N.F. Pseudobactins bounded iron nanoparticles for control of an antibiotic-resistant Pseudomonas aeruginosa ryn32. Biotechnol. Prog. 2020, 36, e2907. [Google Scholar] [CrossRef]
- Jeong, G.-J.; Khan, F.; Khan, S.; Tabassum, N.; Mehta, S.; Kim, Y.-M. Pseudomonas aeruginosa virulence attenuation by inhibiting siderophore functions. Appl. Microbiol. Biotechnol. 2023, 107, 1019–1038. [Google Scholar] [CrossRef]
- Khan, F.; Kang, M.-G.; Jo, D.-M.; Chandika, P.; Jung, W.-K.; Kang, H.W.; Kim, Y.-M. Phloroglucinol-Gold and -Zinc Oxide Nanoparticles: Antibiofilm and Antivirulence Activities towards Pseudomonas aeruginosa PAO1. Mar. Drugs 2021, 19, 601. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Li, Z.; Song, C.; Kuipers, O.P. Exploring plant-microbe interactions of the rhizobacteria Bacillus subtilis and Bacillus mycoides by use of the CRISPR-Cas9 system. Environ. Microbiol. 2018, 20, 4245–4260. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.G.; Gu, B.; Cha, Y.; Ha, J.; Lee, Y.; Kim, G.; Cho, B.-K.; Oh, M.-K. Engineered CRISPR-Cas9 for Streptomyces sp. genome editing to improve specialized metabolite production. Nat. Commun. 2025, 16, 874. [Google Scholar] [CrossRef]






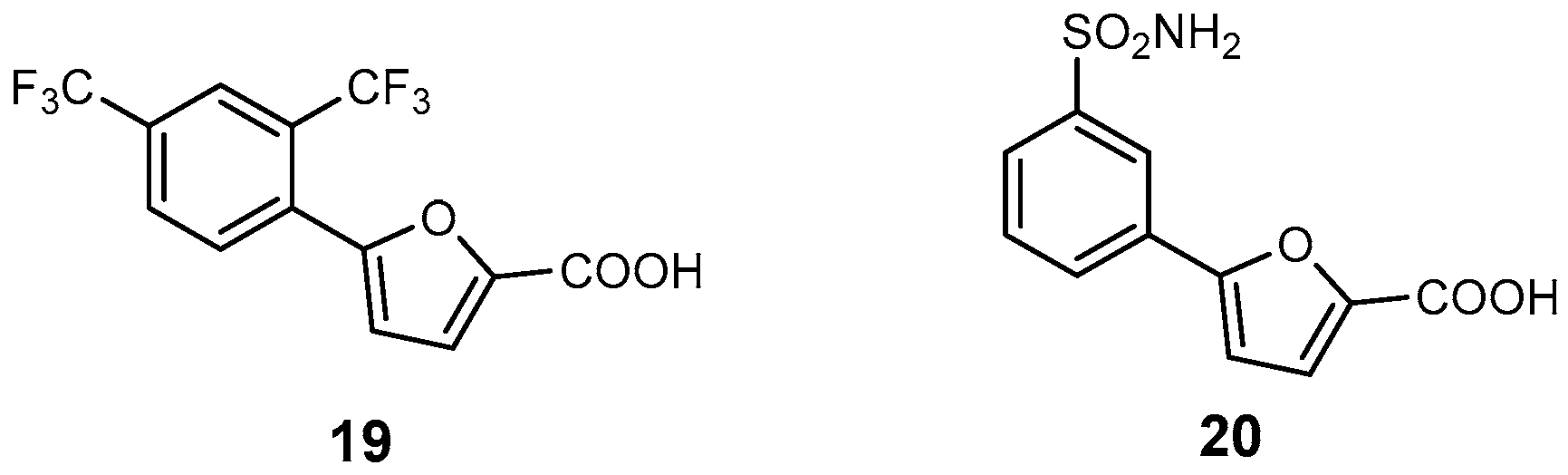




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Enzyme | Enzyme Inhibition (IC50, μM) | Target Species | Preclinical Assays | Reference |
|---|---|---|---|---|---|
| Inhibitors targeting NRPS biosynthesis | |||||
| Phosphopantetheinyl transferase inhibitors | |||||
| ML267 | Sfp AcpS | 0.29 8.1 | B. subtilis | In vitro inhibition In vitro ADME profile In vivo PK profile Mechanism of action | [67] |
| Calmidazolium chloride | VibB MAS | 2.0 4.9 | V. cholerae M. tuberculosis | In vitro inhibition Mechanism of action | [69] |
| Sanguinarine chloride | VibB MAS | 22 4.9 | V. cholerae M. tuberculosis | In vitro inhibition Mechanism of action | [69] |
| 1 | PptAb PcpS PptT | N.A. N.A. 2.3 | M. abscessus P. aeruginosa M. tuberculosis | In vitro inhibition | [71] [71] [74] |
| 2 | PptT | 2.5 | M. tuberculosis | In vitro inhibition | [74] |
| 3 | PptT | 3.8 | M. tuberculosis | In vitro inhibition | [74] |
| 4 | PptT | 0.71 | M. tuberculosis | In vitro inhibition Cardiotoxicity studies | [73] |
| 5 | PptT | 8.4 | M. tuberculosis | In vitro inhibition | [73] |
| 6 | PptT | 0.72 | M. tuberculosis | In vitro inhibition Cardiotoxicity studies | [73] |
| 7 | PptT | 0.065 | M. tuberculosis | In vitro inhibition Cytotoxicity studies | [78] |
| 8 | PptT | 0.018 | M. tuberculosis | In vitro inhibition | [78] |
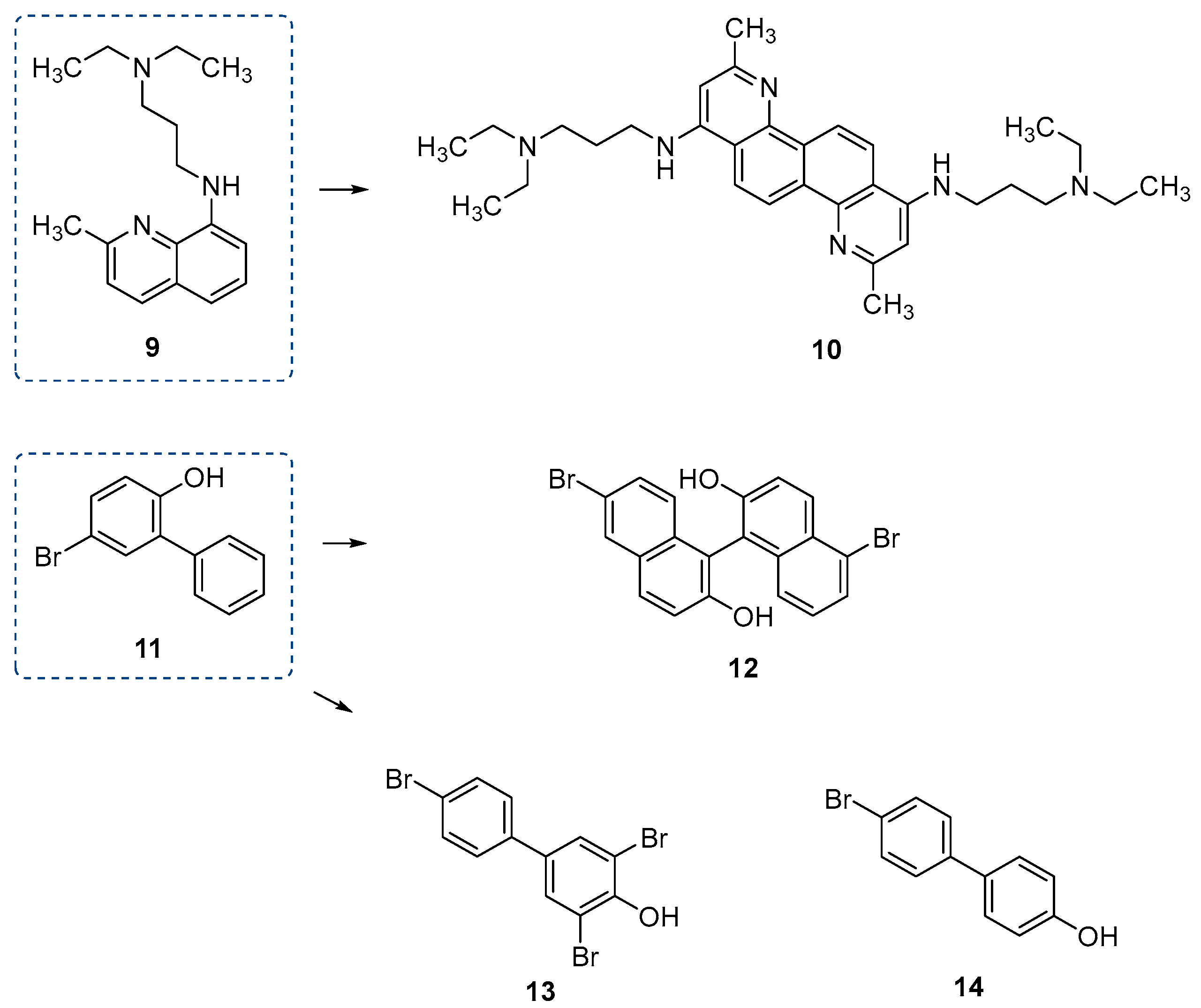
| 9 | PptT | 8.29 | M. tuberculosis | In vitro inhibition | [79] |
| 10 | PptT | 0.49 | M. tuberculosis | In vitro inhibition | [79] |
| 11 | PptT | 173.16 | M. tuberculosis | In vitro inhibition | [79] |
| 12 | PptT | 9.28 | M. tuberculosis | In vitro inhibition | [79] |
| 13 | PptT | 12.29 | M. tuberculosis | In vitro inhibition | [79] |
| 14 | PptT | 28.58 | M. tuberculosis | In vitro inhibition | [79] |
| Inhibitors of the adenylation domain | |||||
| Salicyl-AMS | MbtA YbtE PchD DhbE | 10.7 14.7 12.5 N.A. | M. tuberculosis Y. pestis P. aeruginosa B. subtilis | In vitro inhibition In vivo efficacy and PK studies | [70] [70] [70] [82] |
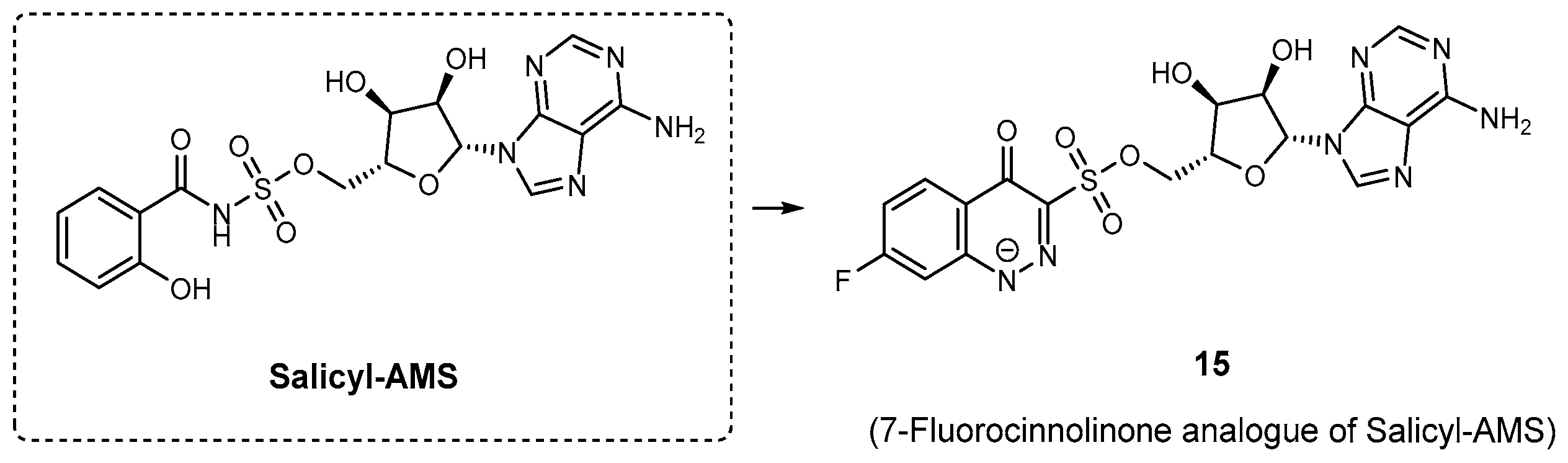
| 15 | MbtA | N.A. | M. tuberculosis | In vitro inhibition | [83] |
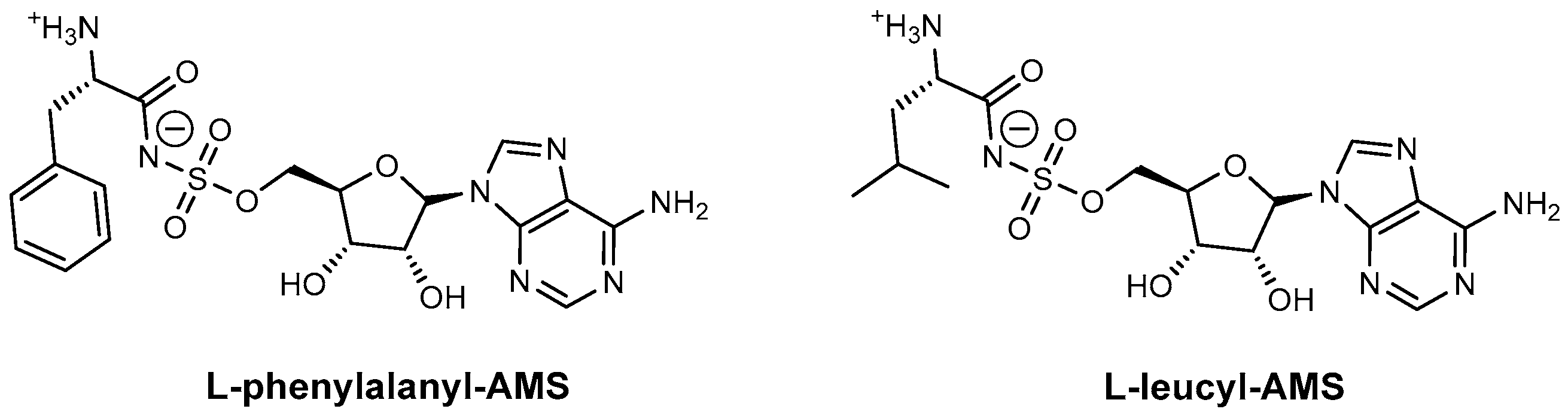
| L-Phenylalanyl-AMS | GrsA SrfA-C | N.A. | B. brevis B. subtilis | In vitro inhibition Mechanism of action | [87] |
| L-Leucyl-AMS | GrsA SrfA-C | N.A. | B. brevis B. subtilis | In vitro inhibition Mechanism of action | [87] |
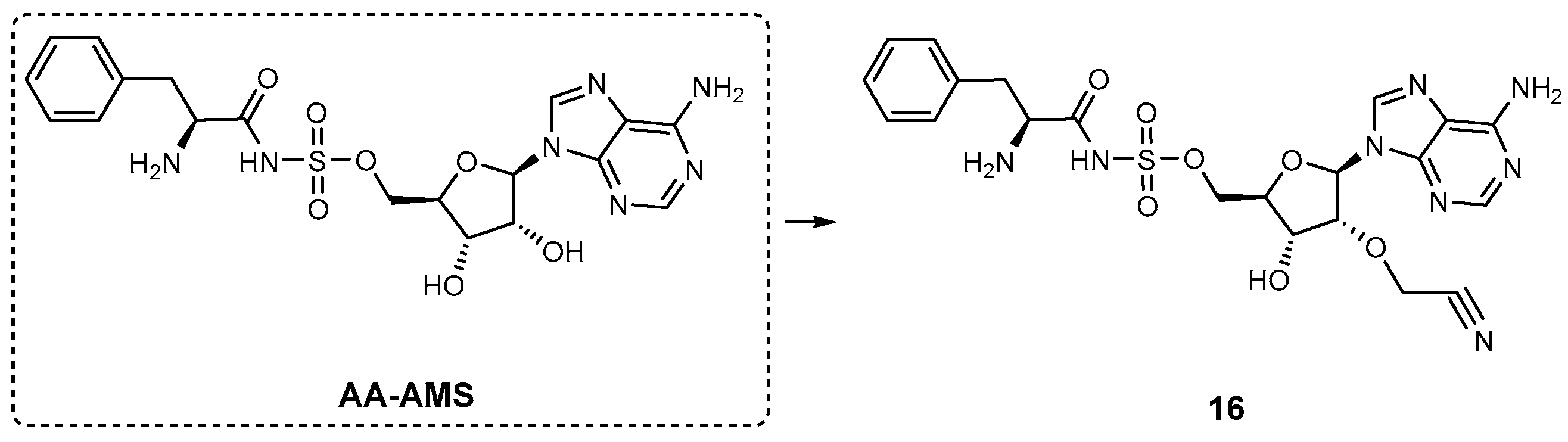
| AA-AMS | GrsA | N.A. | A. miglanus | In vitro inhibition and PK studies | [88] |
| 16 | GrsA | N.A. | A. miglanus | In vitro inhibition | [88] |
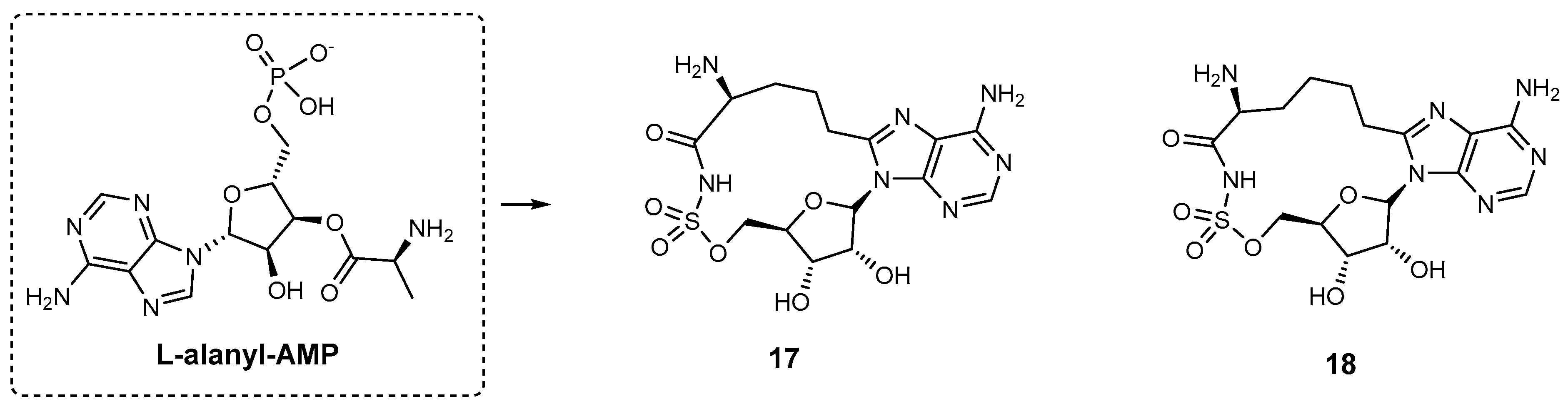
| 17 | HMWP2 | N.A. | Y. pestis | In vitro inhibition | [91] |
| 18 | HMWP2 | N.A. | Y. pestis | In vitro inhibition | [91] |
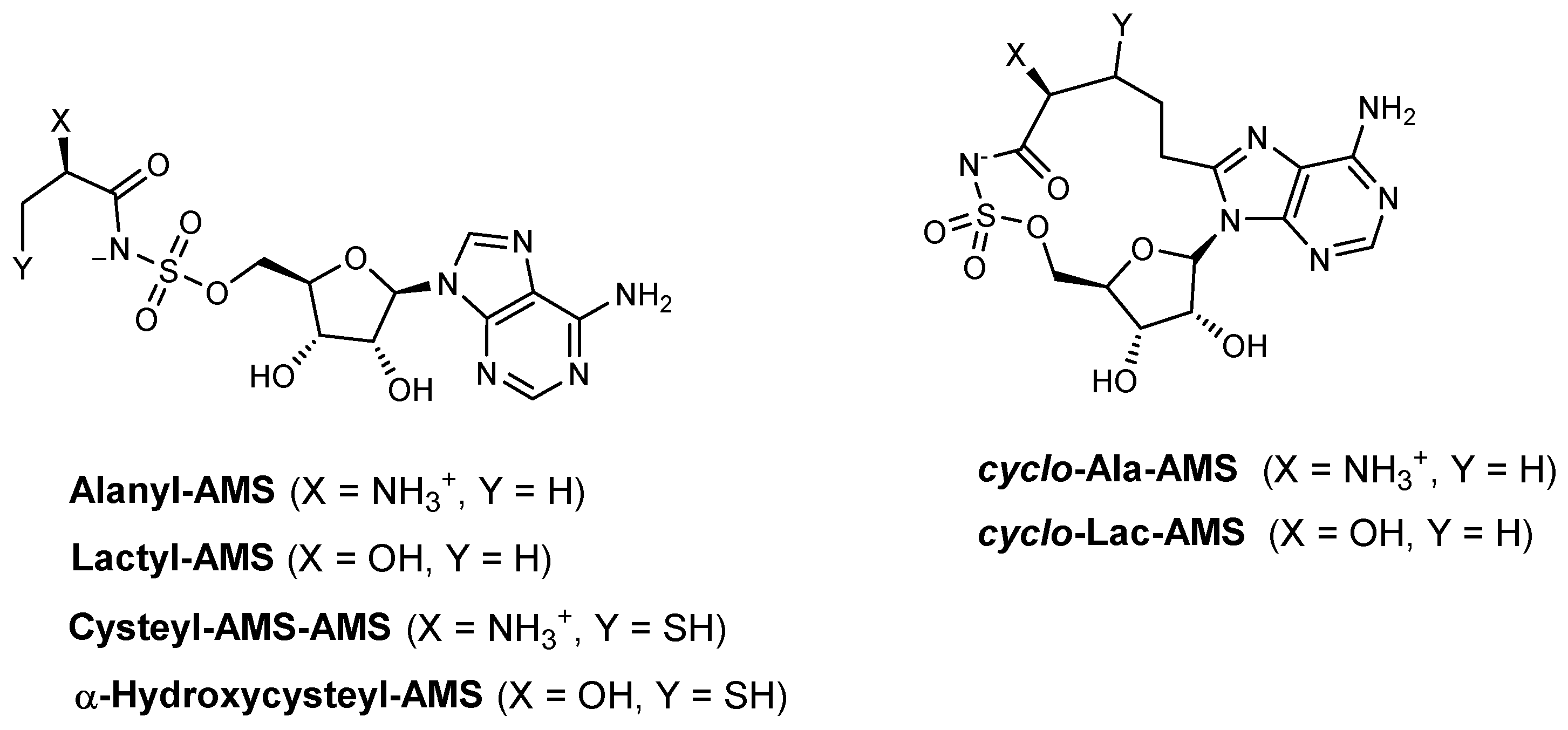
| Alanyl-AMS | HMWP2 (ATP–32PPi isotope exchange assay) | N.A. | Y. pestis | In vitro inhibition Mechanism of action | [91] |
| HMWP2 (MesG assay) | 1.15 | Y. pestis | In vitro inhibition Mechanism of action | [90] | |
| IVT (ATP–32PPi isotope exchange assay) | 0.16 | Y. pestis | In vitro inhibition Mechanism of action | [91] | |
| IVT (MesG assay) | 0.118 | Y. pestis | In vitro inhibition Mechanism of action | [90] | |
| Lactyl-AMS | HMWP2 (MesG assay) IVT (MesG assay) | >1000 166.4 | Y. pestis | In vitro inhibition Mechanism of action | [90] |
| Cysteyl-AMS | HMWP2 (MesG assay) IVT (MesG assay) | 0.04 3.27 | Y. pestis | In vitro inhibition Mechanism of action | [90] |
| α-Hydroxycysteyl-AMS | HMWP2 (MesG assay) IVT (MesG assay) | 16.53 221.5 | Y. pestis | In vitro inhibition Mechanism of action | [90] |
| Cyclo-alanylAMS | HMWP2 (ATP–32PPi isotope exchange assay) | N.A. | Y. pestis | In vitro inhibition Mechanism of action | [91] |
| HMWP2 (MesG assay) | 0.34 | Y. pestis | In vitro inhibition Mechanism of action | [90] | |
| IVT (ATP–32PPi isotope exchange assay) | >250 | Y. pestis | In vitro inhibition Mechanism of action | [91] | |
| IVT (MesG assay) | >250 | Y. pestis | In vitro inhibition Mechanism of action | [90] | |
| Cyclo-lactyl-AMS | HMWP2 (MesG assay) IVT (MesG assay) | >1000 >250 | Y. pestis | In vitro inhibition Mechanism of action | [90] |
| 19 | Mab-SaS | 5.3 | M. abscessus | In vitro inhibition Cytotoxicity studies | [29] |
| 20 | Mab-SaS | 2.6 | M. abscessus | In vitro inhibition Cytotoxicity studies | [94] |
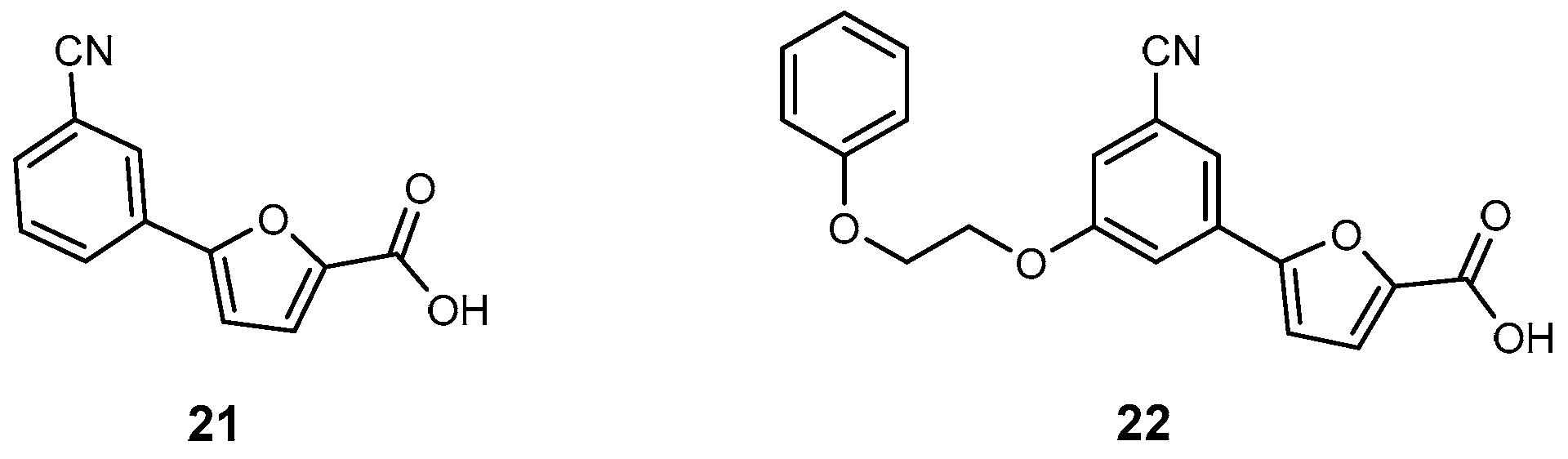
| 21 | MbtI | 6.3 | M. tuberculosis | In vitro inhibition | [95] |
| 22 | MbtI | 12 | M. tuberculosis | In vitro inhibition | [96] |
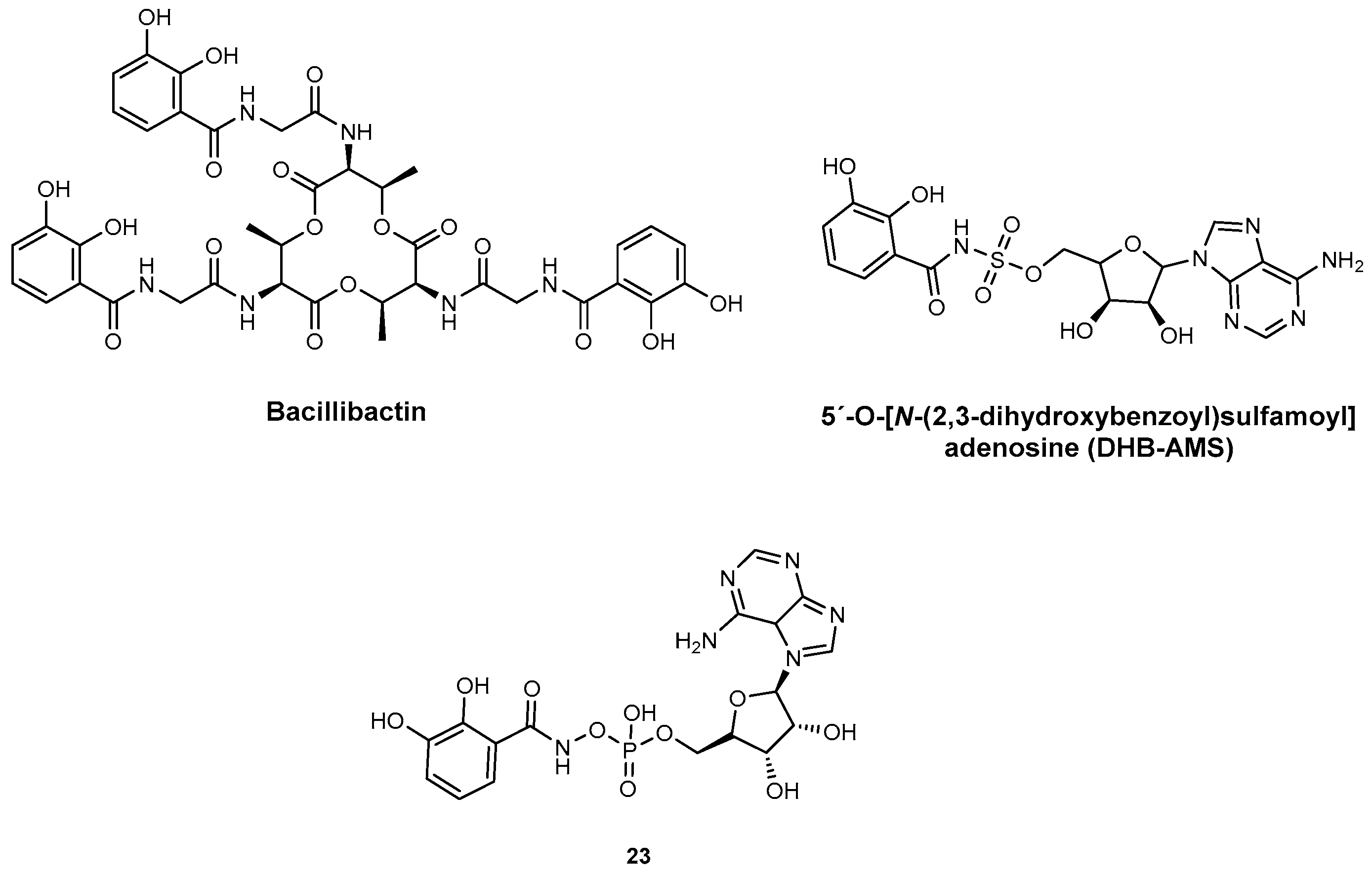
| DHB-AMS | DhbE YbtE | N.A. | B. subtilis Y. pestis | In vitro inhibition and PK studies | [82] |
| 23 | DhbE YbtE | N.A. | B. subtilis Y. pestis | In vitro inhibition | [98] |
| Inhibitors targeting PKS biosynthesis | |||||
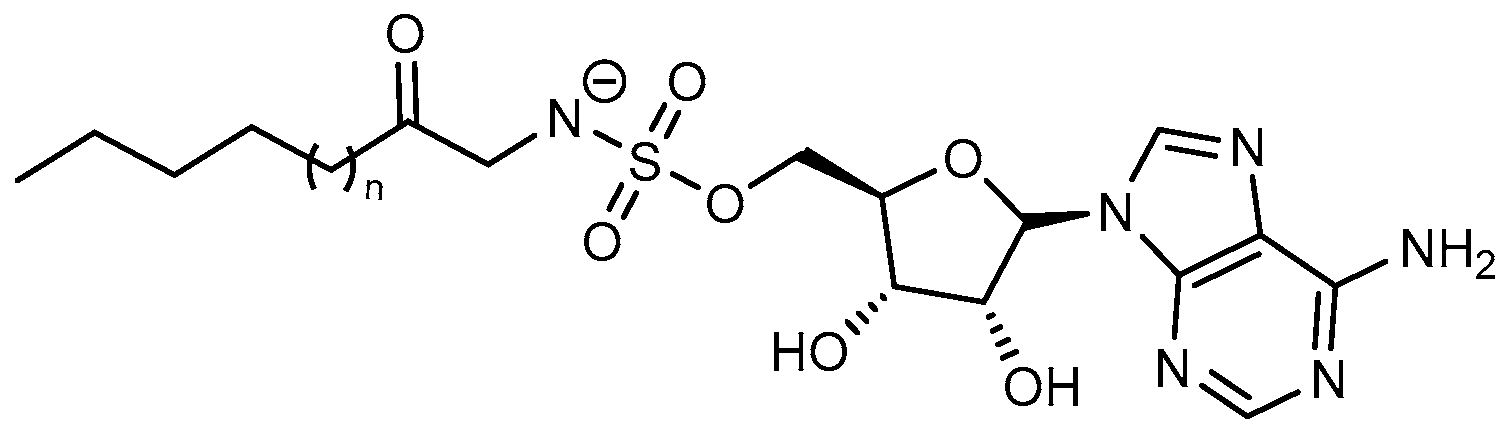
| Hexanoyl-AMS | FAAL28 FACL19 | N.A. | M. tuberculosis | In vitro inhibition Mechanism of action | [105] |
| Lauryl-AMS | FAAL28 FACL19 | M. tuberculosis | In vitro inhibition Mechanism of action | [105] | |
| Arachidyl-AMS | FAAL28 FACL19 | M. tuberculosis | In vitro inhibition Mechanism of action | [105] | |
| Inhibitors targeting NIS biosynthesis | |||||
| Baulamycin A | SbnE AsbA AsbB | 4.8 μM 180 μM >1000 | S. aureus B. anthracis B. anthracis | In vitro inhibition Mechanism of action | [107] |
| Baulamycin B | SbnE AsbA AsbB | 19 μM 200 μM >1000 μM | S. aureus B. anthracis B. anthracis | In vitro inhibition Mechanism of action | [107] |
| Inhibition of siderophore biosynthesis in pathogenic fungi | |||||
| Celastrol | SidA | 11 μM | A. fumigatus | In vitro inhibition Mechanism of action | [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, B.M.; Pinto, E.; Sousa, E.; Resende, D.I.S.P. Targeting Siderophore Biosynthesis to Thwart Microbial Growth. Int. J. Mol. Sci. 2025, 26, 3611. https://doi.org/10.3390/ijms26083611
Rocha BM, Pinto E, Sousa E, Resende DISP. Targeting Siderophore Biosynthesis to Thwart Microbial Growth. International Journal of Molecular Sciences. 2025; 26(8):3611. https://doi.org/10.3390/ijms26083611
Chicago/Turabian StyleRocha, Beatriz M., Eugénia Pinto, Emília Sousa, and Diana I. S. P. Resende. 2025. "Targeting Siderophore Biosynthesis to Thwart Microbial Growth" International Journal of Molecular Sciences 26, no. 8: 3611. https://doi.org/10.3390/ijms26083611
APA StyleRocha, B. M., Pinto, E., Sousa, E., & Resende, D. I. S. P. (2025). Targeting Siderophore Biosynthesis to Thwart Microbial Growth. International Journal of Molecular Sciences, 26(8), 3611. https://doi.org/10.3390/ijms26083611