
Microglia/Macrophages in Autoimmune Demyelinating Encephalomyelitis (Multiple Sclerosis/Neuromyelitis Optica)
Abstract
1. Introduction
2. Origins and Classification of CNS Microglia and Peripheral Macrophages
2.1. Nomenclature of Microglia and Macrophages
2.2. Fate Mapping and Origins
2.3. Surface Markers for Microglia and Macrophages
2.4. Classification via Single-Cell Analysis
3. Mechanisms of Activation and Phagocytosis Regulation in Microglia and Macrophages
3.1. Activation Pathways and Molecular Signals
3.2. Transcriptional Regulation of Functional States
3.3. Regulation of Phagocytosis
3.3.1. Phagocytosis-Promoting Molecules
3.3.2. Phagocytosis-Inhibiting Molecules
3.4. Integrated Roles in Disease and Repair
4. Interactions Between Microglia/Macrophages and Neural Cells
4.1. Interactions with Neurons
4.2. Interactions with Astrocytes
4.3. Interactions with Oligodendrocytes
4.4. Modulatory Effects on Neural Cell Functions
4.5. Microglia and Connexins (Cx)
5. Multiple Sclerosis: Disease Mechanisms and Microglia/Macrophage Involvement
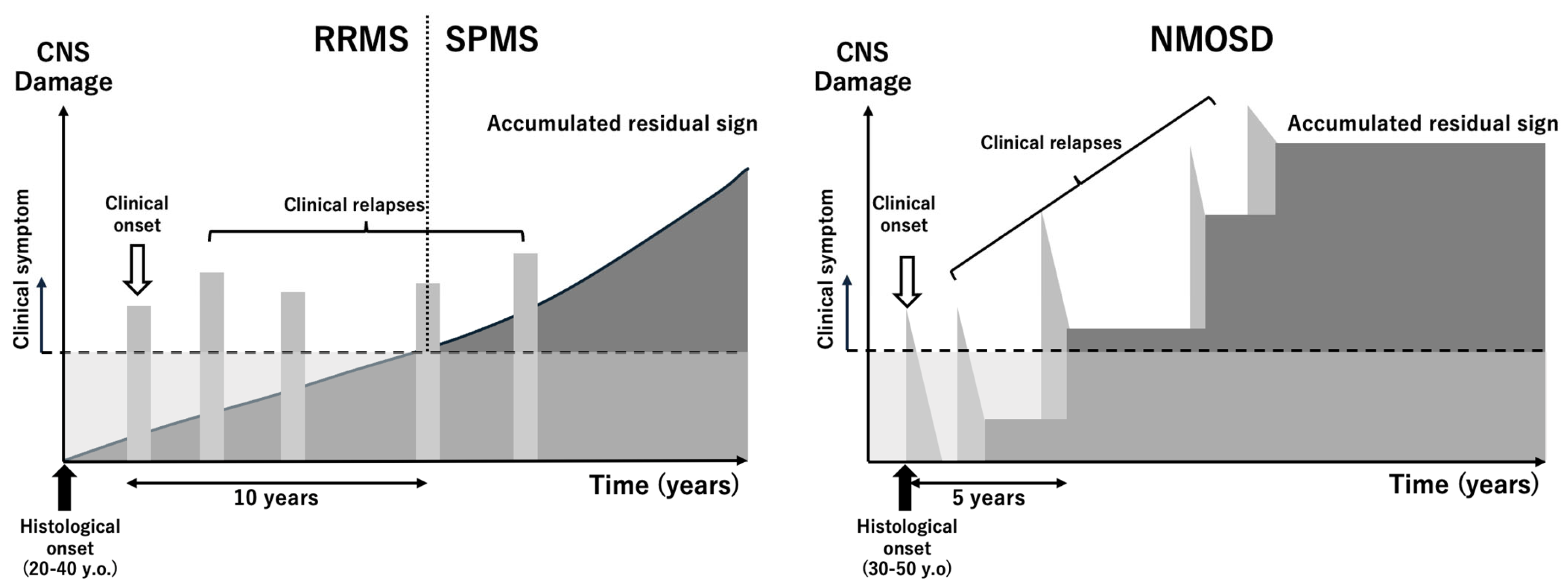
5.1. Disease Overview
5.2. Animal Models of MS
5.2.1. Experimental Autoimmune Encephalomyelitis (EAE)
5.2.2. Toxin-Induced Demyelination Models
5.2.3. Viral Models
5.2.4. Transgenic Models
5.2.5. Strengths and Limitations
5.3. Roles of Microglia and Macrophages in MS Pathogenesis
5.3.1. Contribution to Inflammation and Demyelination
5.3.2. Chronic Lesion Dynamics
5.3.3. Neurodegenerative Processes
5.3.4. Role in Repair and Remyelination
5.3.5. Emerging Therapeutic Implications
5.4. Mechanisms of Action of MS Therapies Through Microglia/Macrophage Modulation
5.4.1. Anti-Inflammatory Therapies
5.4.2. Remyelination-Promoting Strategies
5.4.3. Neuroprotection and Microglial Modulation
5.4.4. Emerging Therapies
5.4.5. Limitations and Future Directions
6. Neuromyelitis Optica: Disease Mechanisms and Microglia/Macrophage Involvement
6.1. Disease Overview
6.2. Animal Models of NMOSD
6.2.1. Passive Transfer Models
6.2.2. Active Immunization Models
6.2.3. Limitations of Current Models
6.2.4. Advances in Model Development
6.3. Roles of Microglia and Macrophages in NMOSD Pathogenesis
6.3.1. Microglial Activation in NMOSD
6.3.2. Macrophage Recruitment and Activation
6.3.3. Complement-Mediated Effects
6.4. Therapeutic Agents for NMOSD and Their Effects on Microglia and Macrophages
6.4.1. Complement Inhibition
6.4.2. IL-6 Receptor Blockade
6.4.3. B-Cell Depletion Therapies
6.4.4. Intravenous (IV)Ig Therapy
6.4.5. Steroid Pulse Therapy
6.4.6. Plasma Exchange
6.4.7. Emerging Therapeutic Targets
6.4.8. Mechanistic Insights
7. Similarities and Differences in Microglial and Macrophage Actions in MS and NMOSD
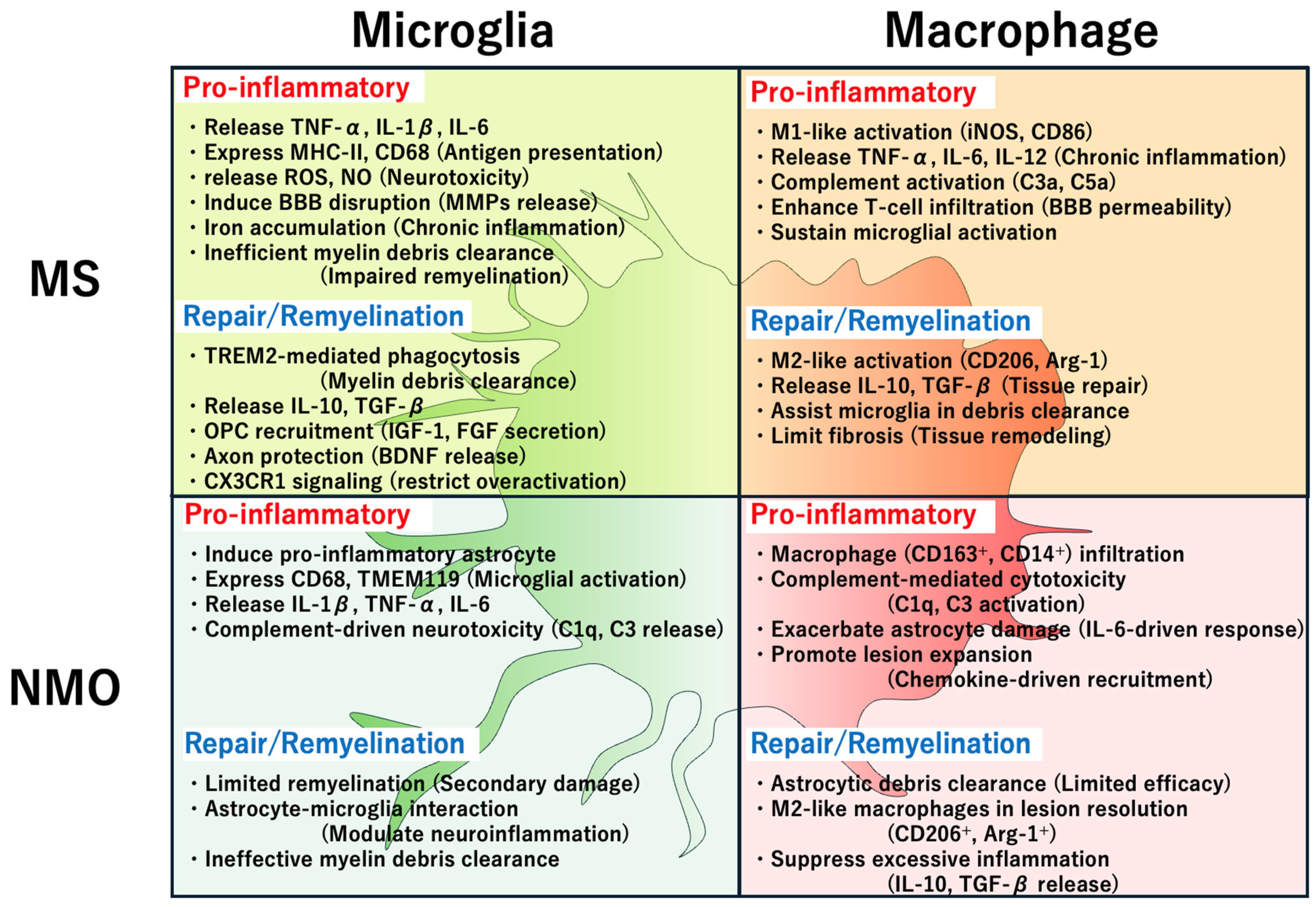
7.1. Similarities
7.1.1. Pro-Inflammatory Activation
7.1.2. BBB Disruption
7.1.3. Phagocytosis and Debris Clearance
7.1.4. Complement Activation
7.1.5. Dual Role in Pathogenesis and Repair
7.2. Differences
7.2.1. Primary Targets of Damage
7.2.2. Role of Complement
7.2.3. Inflammatory Profile
7.2.4. Recruitment of Peripheral Macrophages
7.2.5. Disease-Specific Phenotypes
7.2.6. Therapeutic Implications
7.3. Clinical Implications
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Tremblay, M.E.; Lecours, C.; Samson, L.; Sanchez-Zafra, V.; Sierra, A. From the Cajal alumni Achucarro and Rio-Hortega to the rediscovery of never-resting microglia. Front. Neuroanat. 2015, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, R. Elucidation of the role of microglia-macrophage-based neuroinflammation in neurological diseases. Neurol. Clin. Neurosci. 2024, 12, 329–339. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Masuda, T.; Amann, L.; Prinz, M. Novel insights into the origin and development of CNS macrophage subsets. Clin. Transl. Med. 2022, 12, e1096. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.E17. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef]
- Ydens, E.; Amann, L.; Asselbergh, B.; Scott, C.L.; Martens, L.; Sichien, D.; Mossad, O.; Blank, T.; De Prijck, S.; Low, D.; et al. Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat. Neurosci. 2020, 23, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Amit, I.; Schwartz, M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 2018, 21, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef]
- Hickman, S.E.; El Khoury, J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 495–498. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Crocker, P.R.; McMillan, S.J.; Richards, H.E. CD33-related siglecs as potential modulators of inflammatory responses. Ann. N. Y. Acad. Sci. 2012, 1253, 102–111. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Yamasaki, R.; Kira, J.I. Multiple Sclerosis. Adv. Exp. Med. Biol. 2019, 1190, 217–247. [Google Scholar]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Rajendran, R.; Bottiger, G.; Stadelmann, C.; Karnati, S.; Berghoff, M. FGF/FGFR Pathways in Multiple Sclerosis and in Its Disease Models. Cells 2021, 10, 884. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, R. Connexins in health and disease. Clin. Exp. Neuroimmunol. 2018, 9, 30–36. [Google Scholar] [CrossRef]
- Yamasaki, R. Connexins Control Glial Inflammation in Various Neurological Diseases. Int. J. Mol. Sci. 2023, 24, 16879. [Google Scholar] [CrossRef]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef]
- Zirngibl, M.; Assinck, P.; Sizov, A.; Caprariello, A.V.; Plemel, J.R. Oligodendrocyte death and myelin loss in the cuprizone model: An updated overview of the intrinsic and extrinsic causes of cuprizone demyelination. Mol. Neurodegener. 2022, 17, 34. [Google Scholar] [CrossRef]
- Yamazaki, R.; Ohno, N. The Mouse Model of Internal Capsule Demyelination: A Novel Tool for Investigating Motor Functional Changes Caused by Demyelination and for Evaluating Drugs That Promote Remyelination. Acta Histochem. Cytochemica. 2024, 57, 1–5. [Google Scholar] [CrossRef]
- Oleszak, E.L.; Chang, J.R.; Friedman, H.; Katsetos, C.D.; Platsoucas, C.D. Theiler’s virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 2004, 17, 174–207. [Google Scholar] [CrossRef]
- Mendel, I.; Natarajan, K.; Ben-Nun, A.; Shevach, E.M. A novel protective model against experimental allergic encephalomyelitis in mice expressing a transgenic TCR-specific for myelin oligodendrocyte glycoprotein. J. Neuroimmunol. 2004, 149, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Siriratnam, P.; Huda, S.; Butzkueven, H.; van der Walt, A.; Jokubaitis, V.; Monif, M. A comprehensive review of the advances in neuromyelitis optica spectrum disorder. Autoimmun. Rev. 2023, 22, 103465. [Google Scholar] [CrossRef]
- Duan, T.; Verkman, A.S. Experimental animal models of aquaporin-4-IgG-seropositive neuromyelitis optica spectrum disorders: Progress and shortcomings. Brain Pathol. 2020, 30, 13–25. [Google Scholar] [CrossRef]
- Serizawa, K.; Miyake, S.; Katsura, Y.; Yorozu, K.; Kurasawa, M.; Tomizawa-Shinohara, H.; Yasuno, H.; Matsumoto, Y. Intradermal AQP4 peptide immunization induces clinical features of neuromyelitis optica spectrum disorder in mice. J. Neuroimmunol. 2023, 380, 578109. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yamasaki, R.; Yamaguchi, H.; Nagata, S.; Une, H.; Cui, Y.; Masaki, K.; Nakamuta, Y.; Iinuma, K.; Watanabe, M.; et al. Oligodendroglial connexin 47 regulates neuroinflammation upon autoimmune demyelination in a novel mouse model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 2160–2169. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077. [Google Scholar] [CrossRef]
- Prineas, J.W.; Parratt, J.D.E. Multiple Sclerosis: Microglia, Monocytes, and Macrophage-Mediated Demyelination. J. Neuropathol. Exp. Neurol. 2021, 80, 975–996. [Google Scholar] [CrossRef]
- Kamma, E.; Lasisi, W.; Libner, C.; Ng, H.S.; Plemel, J.R. Central nervous system macrophages in progressive multiple sclerosis: Relationship to neurodegeneration and therapeutics. J. Neuroinflamm. 2022, 19, 45. [Google Scholar] [CrossRef]
- Yong, V.W. Microglia in multiple sclerosis: Protectors turn destroyers. Neuron 2022, 110, 3534–3548. [Google Scholar] [CrossRef]
- Ivan, D.C.; Berve, K.C.; Walthert, S.; Monaco, G.; Borst, K.; Bouillet, E.; Ferreira, F.; Lee, H.; Steudler, J.; Buch, T.; et al. Insulin-like growth factor-1 receptor controls the function of CNS-resident macrophages and their contribution to neuroinflammation. Acta Neuropathol. Commun. 2023, 11, 35. [Google Scholar] [CrossRef]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.C.; Thompson, K.K.; West, B.L.; Tsirka, S.E. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp. Neurol. 2018, 307, 24–36. [Google Scholar] [CrossRef]
- Kramer, J.; Bar-Or, A.; Turner, T.J.; Wiendl, H. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat. Rev. Neurol. 2023, 19, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Siponimod: A Review in Secondary Progressive Multiple Sclerosis. CNS Drugs 2020, 34, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- de Seze, J.; Maillart, E.; Gueguen, A.; Laplaud, D.A.; Michel, L.; Thouvenot, E.; Zephir, H.; Zimmer, L.; Biotti, D.; Liblau, R. Anti-CD20 therapies in multiple sclerosis: From pathology to the clinic. Front. Immunol. 2023, 14, 1004795. [Google Scholar] [CrossRef]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Espiritu, A.I.; Remalante-Rayco, P.P.M. High-dose biotin for multiple sclerosis: A systematic review and meta-analyses of randomized controlled trials. Mult. Scler. Relat. Disord. 2021, 55, 103159. [Google Scholar] [CrossRef]
- Vermersch, P.; Brieva-Ruiz, L.; Fox, R.J.; Paul, F.; Ramio-Torrenta, L.; Schwab, M.; Moussy, A.; Mansfield, C.; Hermine, O.; Maciejowski, M.; et al. Efficacy and Safety of Masitinib in Progressive Forms of Multiple Sclerosis: A Randomized, Phase 3, Clinical Trial. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1148. [Google Scholar] [CrossRef]
- Giovannoni, G. Targeting Epstein-Barr virus in multiple sclerosis: When and how? Curr. Opin. Neurol. 2024, 37, 228–236. [Google Scholar] [CrossRef]
- Ladakis, D.C.; Harrison, K.L.; Smith, M.D.; Solem, K.; Gadani, S.; Jank, L.; Hwang, S.; Farhadi, F.; Dewey, B.E.; Fitzgerald, K.C.; et al. Bile acid metabolites predict multiple sclerosis progression and supplementation is safe in progressive disease. Med 2024, 6, 100522. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Lucchinetti, C.F. Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2022, 387, 631–639. [Google Scholar] [CrossRef]
- Zhang, H.; Verkman, A.S. Longitudinally extensive NMO spinal cord pathology produced by passive transfer of NMO-IgG in mice lacking complement inhibitor CD59. J. Autoimmun. 2014, 53, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, S.; Masaki, K.; Suzuki, S.O.; Yamasaki, R.; Watanabe, M.; Koyama, S.; Isobe, N.; Matsushita, T.; Takahashi, K.; Tabira, T.; et al. Distinct microglial and macrophage distribution patterns in the concentric and lamellar lesions in Balo’s disease and neuromyelitis optica spectrum disorders. Brain Pathol. 2020, 30, 1144–1157. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Wang, H.; Chen, J. Microglial/macrophage activation in the cerebrospinal fluid of neuromyelitis optica spectrum disorders. Brain Behav. 2022, 12, e2798. [Google Scholar] [CrossRef]
- Stathopoulos, P.; Dalakas, M.C. The role of complement and complement therapeutics in neuromyelitis optica spectrum disorders. Expert. Rev. Clin. Immunol. 2022, 18, 933–945. [Google Scholar] [CrossRef]
- Shi, M.; Chu, F.; Jin, T.; Zhu, J. Progress in treatment of neuromyelitis optica spectrum disorders (NMOSD): Novel insights into therapeutic possibilities in NMOSD. CNS Neurosci. Ther. 2022, 28, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Makkawi, S.; Salamatullah, H.K.; Alkhiri, A.; Faidah, D.E.; Afif, L.M.; Bukhari, J.I.; Abulaban, A.; Al Malik, Y.; Levy, M. Role of C5 inhibitors in neuromyelitis optica spectrum disorders with seropositive anti-aquaporin-4 antibody: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2024, 85, 105524. [Google Scholar] [CrossRef]
- Noll, G.; de Lima, M.M.; Mantovani, G.P.; Pineda, F.G.; Silva, Y.P.; Marcarini, P.G.; Reis, L.; Konzen, V.R.; Finkelsztejn, A. Interleukin-6 inhibitors for neuromyelitis optica spectrum disorder (NMOSD): A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2024, 92, 106156. [Google Scholar] [CrossRef]
- Bennett, J.L.; Aktas, O.; Rees, W.A.; Smith, M.A.; Gunsior, M.; Yan, L.; She, D.; Cimbora, D.; Pittock, S.J.; Weinshenker, B.G.; et al. Association between B-cell depletion and attack risk in neuromyelitis optica spectrum disorder: An exploratory analysis from N-MOmentum, a double-blind, randomised, placebo-controlled, multicentre phase 2/3 trial. EBioMedicine 2022, 86, 104321. [Google Scholar] [CrossRef]
- Magraner, M.J.; Coret, F.; Casanova, B. The effect of intravenous immunoglobulin on neuromyelitis optica. Neurologia 2013, 28, 65–72. [Google Scholar] [CrossRef]
- Wang, S.; Xue, M.; Wang, J.; Wu, R.; Shao, Y.; Luo, K.; Liu, J.; Zhu, M. Effects of intravenous pulse methylprednisolone in neuromyelitis optica during the acute phase. Ann. Clin. Transl. Neurol. 2024, 11, 2731–2744. [Google Scholar] [CrossRef]
- Huang, X.; Wu, J.; Xiao, Y.; Zhang, Y. Timing of plasma exchange for neuromyelitis optica spectrum disorders: A meta-analysis. Mult. Scler. Relat. Disord. 2021, 48, 102709. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Bosco, D.B.; Ying, Y.; Tian, D.S.; Wu, L.J. The Emerging Role of Microglia in Neuromyelitis Optica. Front. Immunol. 2021, 12, 616301. [Google Scholar] [CrossRef] [PubMed]
- Owens, T.; Benmamar-Badel, A.; Wlodarczyk, A.; Marczynska, J.; Morch, M.T.; Dubik, M.; Arengoth, D.S.; Asgari, N.; Webster, G.; Khorooshi, R. Protective roles for myeloid cells in neuroinflammation. Scand. J. Immunol. 2020, 92, e12963. [Google Scholar] [CrossRef]
- Lopes Pinheiro, M.A.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta 2016, 1862, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. Chemokines and chemokine receptors: Standing at the crossroads of immunobiology and neurobiology. Immunity 2009, 31, 711–721. [Google Scholar] [CrossRef]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef]
- Scheu, S.; Ali, S.; Ruland, C.; Arolt, V.; Alferink, J. The C-C Chemokines CCL17 and CCL22 and Their Receptor CCR4 in CNS Autoimmunity. Int. J. Mol. Sci. 2017, 18, 2306. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cell Type | State | Surface Markers | Features/Functions |
|---|---|---|---|
| Microglia | Steady state | TMEM119, P2RY12, CX3CR1 | Surveillance of the CNS, neuroprotection, and synaptic remodeling |
| Activated state | MHC class II, CD68, CD86 | Antigen presentation, secretion of pro-inflammatory cytokines, neurotoxicity | |
| Macrophages | Steady state | CD45 (high expression), CD68, CD206 | Antigen presentation, immune surveillance, and tissue repair |
| Activated state | M1-like: CD86, iNOS | Pro-inflammatory, pathogen clearance, tissue destruction | |
| M2-like: CD206, Arginase-1 | Anti-inflammatory, tissue repair, resolution of inflammation |
| Disease | Model Name | Key Features | References |
|---|---|---|---|
| MS | EAE (Experimental Autoimmune Encephalomyelitis) | T cell-mediated inflammation and demyelination; most widely used MS model | [27] |
| Cuprizone Model | Oligodendrocyte toxicity-induced demyelination; models remyelination | [28] | |
| Lysolecithin Model | Localized demyelination via lipid disruption; used for focal lesion studies | [29] | |
| TMEV (Theiler’s Murine Encephalomyelitis Virus) Model | Virus-induced chronic demyelination; models progressive MS | [30] | |
| Transgenic MOG-TCR Model | Spontaneous MS-like autoimmunity; models T cell responses | [31] | |
| NMO | Passive Transfer Model | AQP4-IgG transfer induces astrocyte injury; requires BBB disruption | [32] |
| Active Immunization Model | AQP4 peptide immunization mimics chronic inflammation | [33] | |
| Intrathecal AQP4-IgG Model | Direct administration of AQP4-IgG and complement; astrocytopathy model | [34] | |
| CD59-Knockout Model | Complement regulatory deficiency increases AQP4-IgG pathology | [34] |
| Disease | Drug | Mechanism of Action |
|---|---|---|
| MS | Ocrelizumab | Anti-CD20 monoclonal antibody; B-cell depletion |
| Natalizumab | Anti-α4 integrin monoclonal antibody; inhibits immune cell migration | |
| Fingolimod | S1P receptor modulator; prevents lymphocyte egress | |
| Siponimod | Selective S1P1/S1P5 modulator; reduces neuroinflammation | |
| Tolebrutinib | BTK inhibitor; suppresses microglial activation | |
| Clemastine fumarate | Promotes OPC differentiation and remyelination | |
| Masitinib | Tyrosine kinase inhibitor; reduces microglial activation | |
| NMO | Eculizumab | C5 complement inhibitor; prevents complement-mediated astrocyte injury |
| Satralizumab | IL-6 receptor antagonist; reduces pro-inflammatory signaling | |
| Inebilizumab | Anti-CD19 monoclonal antibody; depletes B cells | |
| Rituximab | Anti-CD20 monoclonal antibody; depletes B cells | |
| IVIg | Modulates Fc receptor activation; reduces inflammation | |
| PLEX | Removes pathogenic autoantibodies and immune complexes | |
| CSF1R inhibitors | CSF1R blockade; reduces microglial/macrophage activation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamasaki, R. Microglia/Macrophages in Autoimmune Demyelinating Encephalomyelitis (Multiple Sclerosis/Neuromyelitis Optica). Int. J. Mol. Sci. 2025, 26, 3585. https://doi.org/10.3390/ijms26083585
Yamasaki R. Microglia/Macrophages in Autoimmune Demyelinating Encephalomyelitis (Multiple Sclerosis/Neuromyelitis Optica). International Journal of Molecular Sciences. 2025; 26(8):3585. https://doi.org/10.3390/ijms26083585
Chicago/Turabian StyleYamasaki, Ryo. 2025. "Microglia/Macrophages in Autoimmune Demyelinating Encephalomyelitis (Multiple Sclerosis/Neuromyelitis Optica)" International Journal of Molecular Sciences 26, no. 8: 3585. https://doi.org/10.3390/ijms26083585
APA StyleYamasaki, R. (2025). Microglia/Macrophages in Autoimmune Demyelinating Encephalomyelitis (Multiple Sclerosis/Neuromyelitis Optica). International Journal of Molecular Sciences, 26(8), 3585. https://doi.org/10.3390/ijms26083585