
Gasdermin-D Genetic Knockout Reduces Inflammasome-Induced Disruption of the Gut-Brain Axis After Traumatic Brain Injury

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
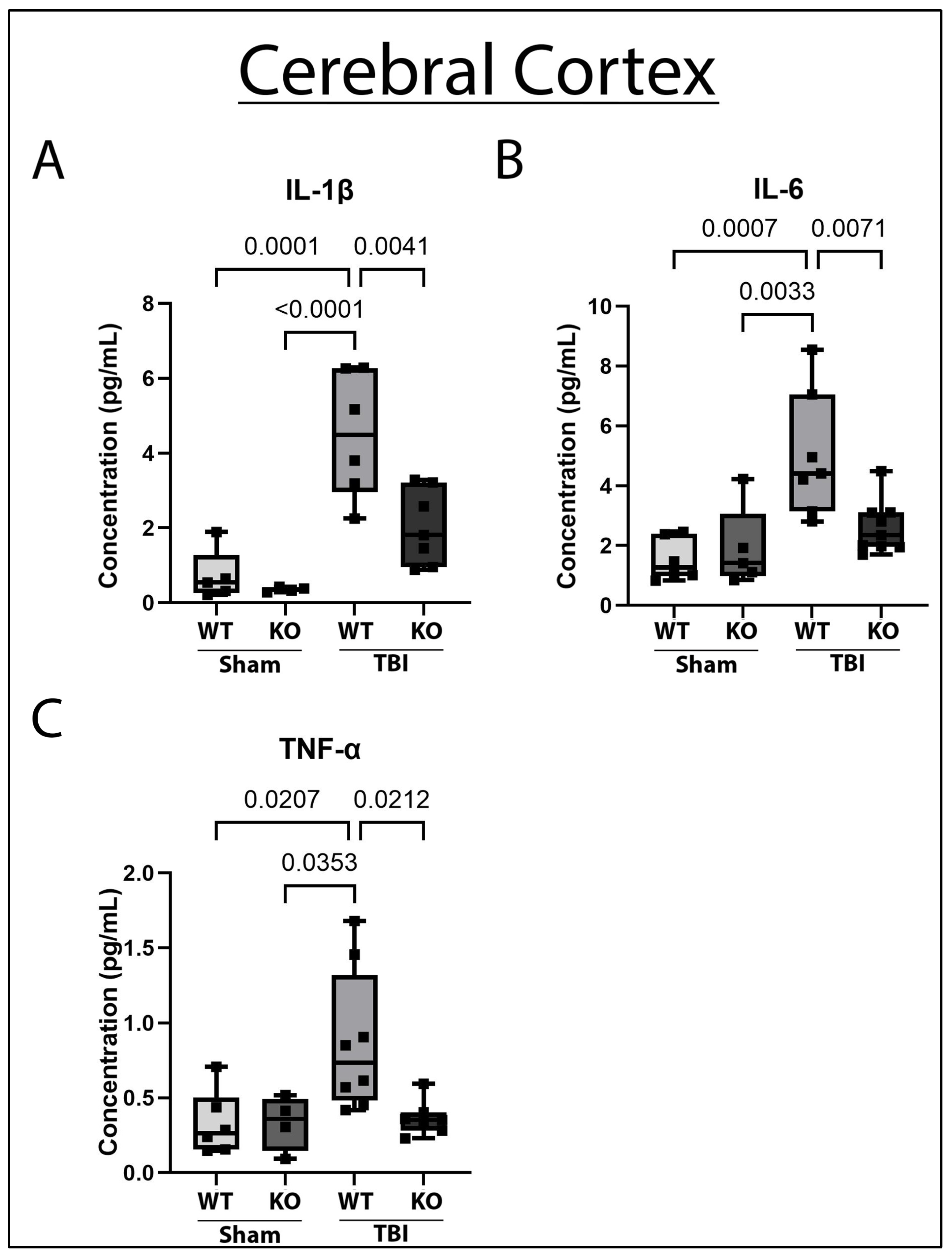
2.1. GSDMD-KO Attenuates Pro-Inflammatory Cytokine Concentration in the Cerebral Cortex 3 Days Post-TBI
2.2. IL-18 and IL-1β Are Decreased in the Gut of GSDMD-KO Mice 3-Days Post-TBI
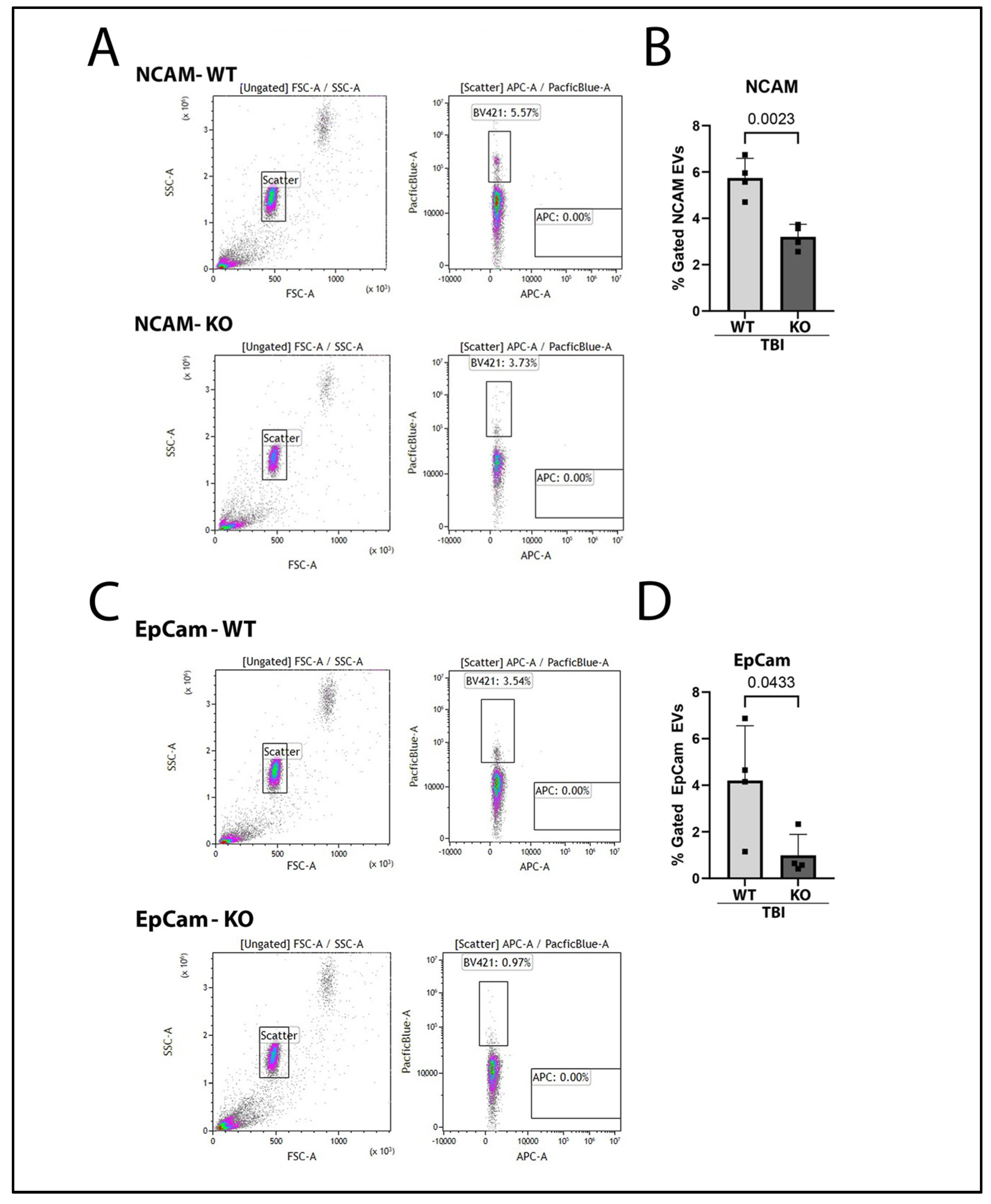
2.3. Brain-Derived and Gut-Derived EVs Are Reduced in the Plasma of GSDMD-KO Mice After TBI
2.4. IL-1β Containing EVs Are Mitigated in GSDMD KO Mice 3 Days After TBI
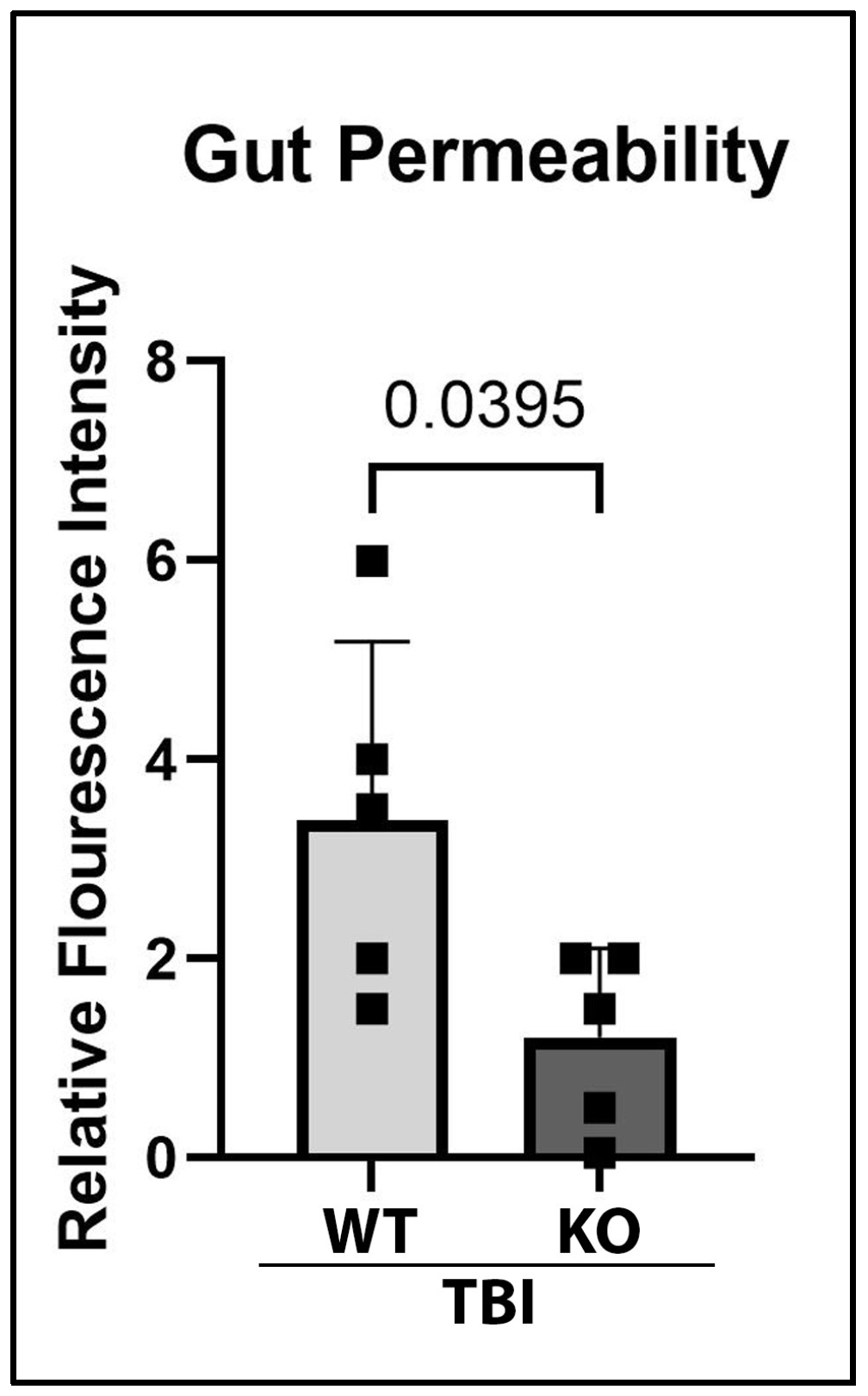
2.5. GSDMD-KO Protects Against Gut Permeability Dysfunction 3 Days After TBI
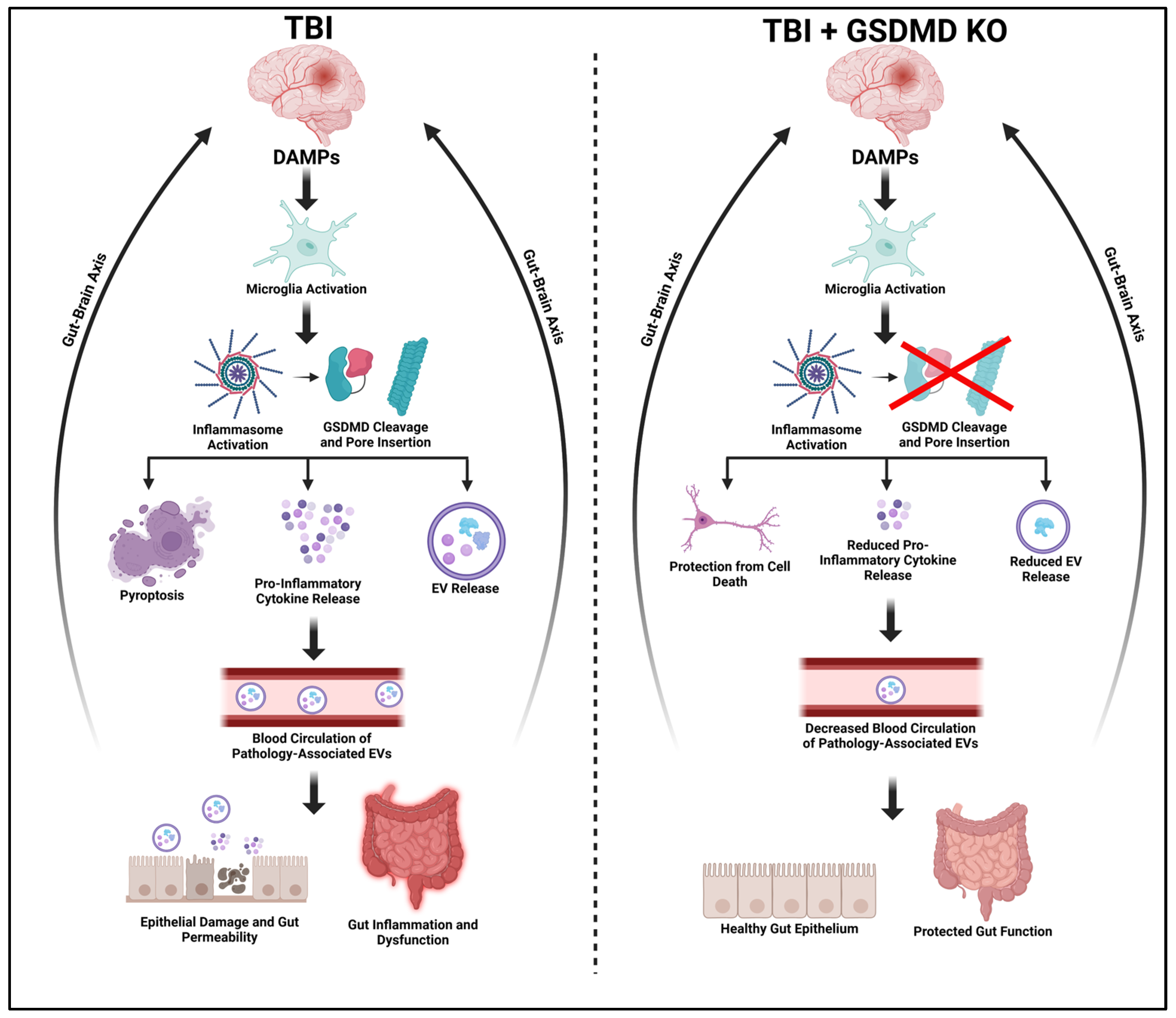
3. Discussion
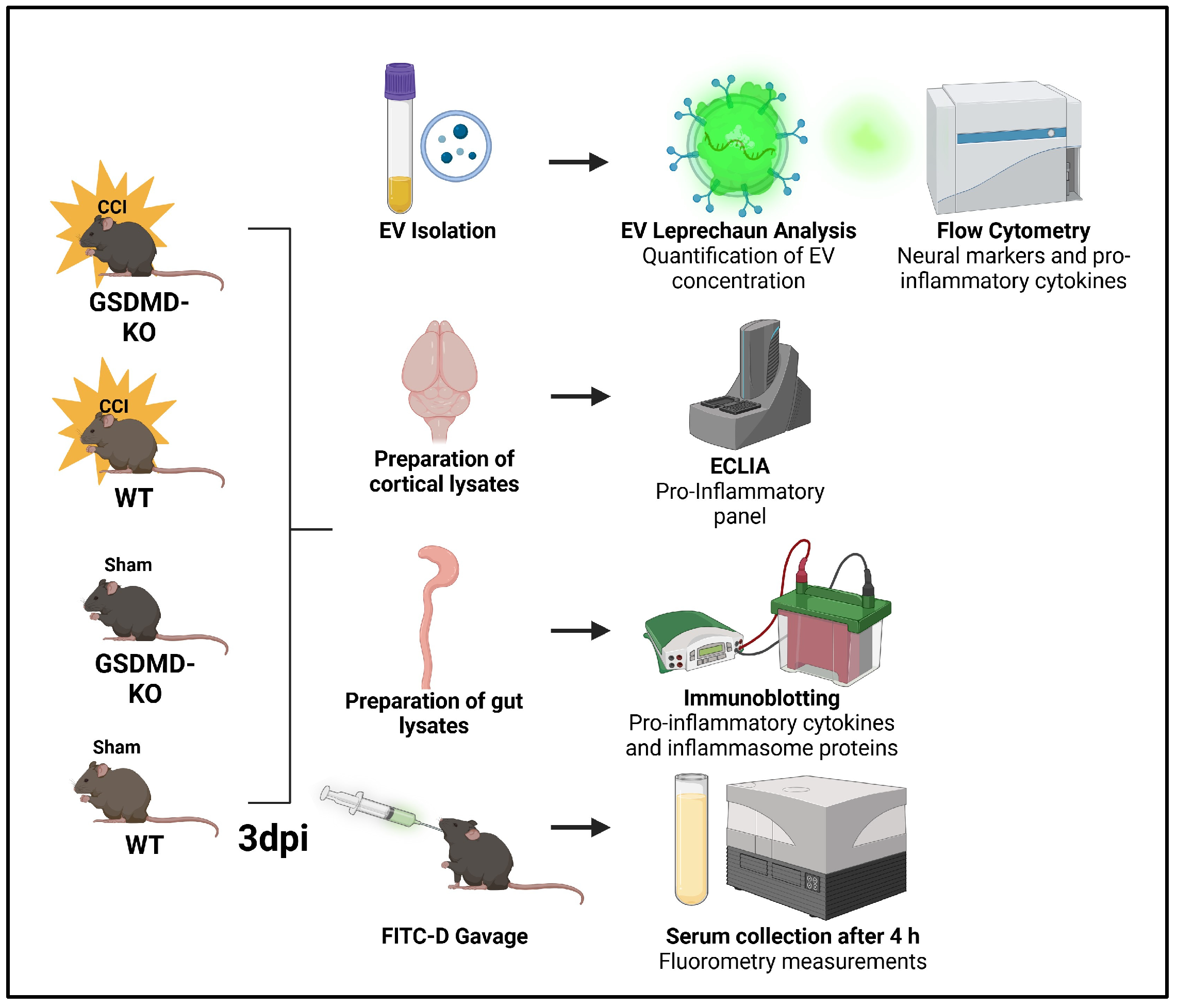
4. Materials and Methods
4.1. Animal Model and Surgical Procedures
4.2. Electrochemiluminescence Immunoassay (ECLIA)
4.3. Immunoblotting
4.4. EV Isolation
4.5. EV Flow Cytometry
4.6. Gut Permeability Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kinoshita, K. Traumatic brain injury: Pathophysiology for neurocritical care. J. Intensive Care 2016, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.D.; Johnson, J.C.; Riordan, W.P.; Cotton, B.A. How we die: The impact of nonneurologic organ dysfunction after severe traumatic brain injury. Am. Surg. 2008, 74, 866–872. [Google Scholar] [CrossRef]
- Harrison-Felix, C.; Whiteneck, G.; Devivo, M.J.; Hammond, F.M.; Jha, A. Causes of death following 1 year postinjury among individuals with traumatic brain injury. J. Head Trauma Rehabil. 2006, 21, 22–33. [Google Scholar] [CrossRef]
- Saxe, J.M.; Ledgerwood, A.M.; Lucas, C.E.; Lucas, W.F. Lower esophageal sphincter dysfunction precludes safe gastric feeding after head injury. J. Trauma Acute Care Surg. 1994, 37, 581–584; discussion 584–586. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Nance, K.; Chen, S. The Gut–Brain Axis. Annu. Rev. Med. 2022, 73, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Hanscom, M.; Loane, D.J.; Shea-Donohue, T. Brain–gut axis dysfunction in the pathogenesis of traumatic brain injury. J. Clin. Investig. 2021, 131, e143777. [Google Scholar] [CrossRef]
- Yuan, C.; He, Y.; Xie, K.; Feng, L.; Gao, S.; Cai, L. Review of microbiota gut–brain axis and innate immunity in inflammatory and infective diseases. Front. Cell. Infect. Microbiol. 2023, 13, 1282431. [Google Scholar] [CrossRef]
- Pellegrini, C.; Antonioli, L.; Calderone, V.; Colucci, R.; Fornai, M.; Blandizzi, C. Microbiota-gut–brain axis in health and disease: Is NLRP3 inflammasome at the crossroads of microbiota-gut–brain communications? Prog. Neurobiol. 2020, 191, 101806. [Google Scholar] [CrossRef]
- Kerr, N.A.; de Rivero Vaccari, J.P.; Abbassi, S.; Kaur, H.; Zambrano, R.; Wu, S.; Dietrich, W.D.; Keane, R.W. Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. J. Neurotrauma 2018, 35, 2067–2076. [Google Scholar] [CrossRef]
- Keane, R.W.; Hadad, R.; Scott, X.O.; Cabrera Ranaldi, E.D.R.; Perez-Barcena, J.; de Rivero Vaccari, J.P. Neural-Cardiac Inflammasome Axis after Traumatic Brain Injury. Pharmaceuticals 2023, 16, 1382. [Google Scholar] [CrossRef]
- Hu, Y.; Jiang, Y.; Li, S.; Ma, X.; Chen, M.; Yang, R.; Wen, S.; Moynagh, P.N.; Wang, B.; Hu, G.; et al. The Gasdermin D N-terminal fragment acts as a negative feedback system to inhibit inflammasome-mediated activation of Caspase-1/11. Proc. Natl. Acad. Sci. USA 2022, 119, e2210809119. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Liu, W.C.; Chen, X.Y.; Wang, X.; Li, J.L.; Zhang, X. Gasdermin D-mediated pyroptosis: Mechanisms, diseases, and inhibitors. Front. Immunol. 2023, 14, 1178662. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.A.; Sanchez, J.; O’Connor, G.; Watson, B.D.; Daunert, S.; Bramlett, H.M.; Dietrich, W.D. Inflammasome-Regulated Pyroptotic Cell Death in Disruption of the Gut–Brain Axis After Stroke. Transl. Stroke Res. 2022, 13, 898–912. [Google Scholar] [CrossRef]
- Sonny, S.; Yuan, H.; Chen, S.; Duncan, M.R.; Chen, P.; Benny, M.; Young, K.; Park, K.K.; Schmidt, A.F.; Wu, S. GSDMD deficiency ameliorates hyperoxia-induced BPD and ROP in neonatal mice. Sci. Rep. 2023, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.P.; de Rivero Vaccari, J.P.; Brand, F., III; Adamczak, S.; Lee, S.W.; Perez-Barcena, J.; Wang, M.Y.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W. Exosome-mediated inflammasome signaling after central nervous system injury. J. Neurochem. 2016, 136 (Suppl. S1), 39–48. [Google Scholar] [CrossRef]
- van der Merwe, Y.; Steketee, M.B. Extracellular Vesicles: Biomarkers, Therapeutics, and Vehicles in the Visual System. Curr. Ophthalmol. Rep. 2017, 5, 276–282. [Google Scholar] [CrossRef]
- Hazelton, I.; Yates, A.; Dale, A.; Roodselaar, J.; Akbar, N.; Ruitenberg, M.J.; Anthony, D.C.; Couch, Y. Exacerbation of Acute Traumatic Brain Injury by Circulating Extracellular Vesicles. J. Neurotrauma 2018, 35, 639–651. [Google Scholar] [CrossRef]
- Kerr, N.A.; de Rivero Vaccari, J.P.; Umland, O.; Bullock, M.R.; Conner, G.E.; Dietrich, W.D.; Keane, R.W. Human Lung Cell Pyroptosis Following Traumatic Brain Injury. Cells 2019, 8, 69. [Google Scholar] [CrossRef]
- Kumar, A.; Stoica, B.A.; Loane, D.J.; Yang, M.; Abulwerdi, G.; Khan, N.; Kumar, A.; Thom, S.R.; Faden, A.I. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflamm. 2017, 14, 47. [Google Scholar] [CrossRef]
- Kim, N.Y.; Lee, H.Y.; Choi, Y.Y.; Mo, S.J.; Jeon, S.; Ha, J.H.; Park, S.D.; Shim, J.J.; Lee, J.; Chung, B.G. Effect of gut microbiota-derived metabolites and extracellular vesicles on neurodegenerative disease in a gut–brain axis chip. Nano Converg. 2024, 11, 7. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic Neutralization of the NLRP1 Inflammasome Reduces the Innate Immune Response and Improves Histopathology after Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2009, 29, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Tehranian, R.; Andell-Jonsson, S.; Beni, S.M.; Yatsiv, I.; Shohami, E.; Bartfai, T.; Lundkvist, J.; Iverfeldt, K. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J. NeuroTrauma 2002, 19, 939–951. [Google Scholar] [CrossRef]
- Clausen, F.; Hånell, A.; Israelsson, C.; Hedin, J.; Ebendal, T.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1β reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2011, 34, 110–123. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, D.; Laskin, D.L.; Jin, Y. Functional Evidence of Pulmonary Extracellular Vesicles in Infectious and Noninfectious Lung Inflammation. J. Immunol. 2018, 201, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Raposo, G.; Candalh, C.; Boussac, M.; Hershberg, R.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 2001, 121, 337–349. [Google Scholar] [CrossRef]
- Ramos-Zaldívar, H.M.; Polakovicova, I.; Salas-Huenuleo, E.; Corvalán, A.H.; Kogan, M.J.; Yefi, C.P.; Andia, M.E. Extracellular vesicles through the blood–brain barrier: A review. Fluids Barriers CNS 2022, 19, 60. [Google Scholar] [CrossRef]
- Rutishauser, U.; Acheson, A.; Hall, A.K.; Mann, D.M.; Sunshine, J. The neural cell adhesion molecule (NCAM) as a regulator of cell–cell interactions. Science 1988, 240, 53–57. [Google Scholar] [CrossRef]
- Nagao, K.; Zhu, J.; Heneghan, M.B.; Hanson, J.C.; Morasso, M.I.; Tessarollo, L.; Mackem, S.; Udey, M.C. Abnormal Placental Development and Early Embryonic Lethality in EpCAM-Null Mice. PLoS ONE 2010, 4, e8543. [Google Scholar] [CrossRef]
- Ma, E.L.; Smith, A.D.; Desai, N.; Cheung, L.; Hanscom, M.; Stoica, B.A.; Loane, D.J.; Shea-Donohue, T.; Faden, A.I. Bidirectional brain–gut interactions and chronic pathological changes after traumatic brain injury in mice. Brain Behav. Immun. 2017, 66, 56–69. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, T.; Fu, J.; Fu, S.; Hu, C.; Sun, B.; Fan, X.; Zhu, J. Lactobacillus acidophilus Exerts Neuroprotective Effects in Mice with Traumatic Brain Injury. J. Nutr. 2019, 149, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Sundman, M.H.; Chen, N.-k.; Subbian, V.; Chou, Y.-h. The bidirectional gut–brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain Behav. Immun. 2017, 66, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.H.; Kerr, N.A.; de Rivero Vaccari, J.P.; Bramlett, H.M.; Keane, R.W.; Dietrich, W.D. Genetic predisposition to Alzheimer’s disease alters inflammasome activity after traumatic brain injury. Transl. Res. 2023, 257, 66–77. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Gasdermin D: The long-awaited executioner of pyroptosis. Cell Res. 2015, 25, 1183–1184. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Nunez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef]
- Kuwar, R.; Rolfe, A.; Di, L.; Xu, H.; He, L.; Jiang, Y.; Zhang, S.; Sun, D. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J. Neuroinflamm. 2019, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Xu, X.; Chen, F.; Kong, X.; Wang, H.; Zhang, J.; Lei, P. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood–brain barrier after traumatic brain injury. Brain Res. 2018, 1697, 10–20. [Google Scholar] [CrossRef]
- Devant, P.; Kagan, J.C. Molecular mechanisms of gasdermin D pore-forming activity. Nat. Immunol. 2023, 24, 1064–1075. [Google Scholar] [CrossRef]
- Hu, R.; Liang, J.; Ding, L.; Zhang, W.; Wang, Y.; Zhang, Y.; Zhang, D.; Pei, L.; Liu, X.; Xia, Z.; et al. Gasdermin D inhibition ameliorates neutrophil mediated brain damage in acute ischemic stroke. Cell Death Discov. 2023, 9, 50. [Google Scholar] [CrossRef]
- Challa, N.V.D.; Chen, S.; Yuan, H.; Duncan, M.R.; Moreno, W.J.; Bramlett, H.; Dietrich, W.D.; Benny, M.; Schmidt, A.F.; Young, K.; et al. GSDMD gene knockout alleviates hyperoxia-induced hippocampal brain injury in neonatal mice. J. Neuroinflamm. 2023, 20, 205. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Liu, W.; Jin, L.; Guo, S.; Fan, H.; Jin, F.; Wei, C.; Fang, D.; Zhang, X.; Su, S.; et al. Dexmedetomidine Inhibits Gasdermin D-Induced Pyroptosis via the PI3K/AKT/GSK3β Pathway to Attenuate Neuroinflammation in Early Brain Injury After Subarachnoid Hemorrhage in Rats. Front. Cell. Neurosci. 2022, 16, 899484. [Google Scholar] [CrossRef]
- Du, H.; Li, C.H.; Gao, R.B.; Cen, X.Q.; Li, P. Ablation of GSDMD Attenuates Neurological Deficits and Neuropathological Alterations After Traumatic Brain Injury. Front. Cell. Neurosci. 2022, 16, 915969. [Google Scholar] [CrossRef] [PubMed]
- Clausen, F.; Marklund, N.; Hillered, L. Acute Inflammatory Biomarker Responses to Diffuse Traumatic Brain Injury in the Rat Monitored by a Novel Microdialysis Technique. J. Neurotrauma 2018, 36, 201–211. [Google Scholar] [CrossRef]
- Woodcock, T.; Morganti-Kossmann, M.C. The Role of Markers of Inflammation in Traumatic Brain Injury. Front. Neurol. 2013, 4, 18. [Google Scholar] [CrossRef]
- Zeiler, F.A.; Thelin, E.P.; Czosnyka, M.; Hutchinson, P.J.; Menon, D.K.; Helmy, A. Cerebrospinal Fluid and Microdialysis Cytokines in Severe Traumatic Brain Injury: A Scoping Systematic Review. Front. Neurol. 2017, 8, 331. [Google Scholar] [CrossRef] [PubMed]
- Hayakata, T.; Shiozaki, T.; Tasaki, O.; Ikegawa, H.; Inoue, Y.; Toshiyuki, F.; Hosotubo, H.; Kieko, F.; Yamashita, T.; Tanaka, H.; et al. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock 2004, 22, 102–107. [Google Scholar] [CrossRef]
- Xu, W.; Yue, S.; Wang, P.; Wen, B.; Zhang, X. Systemic inflammation in traumatic brain injury predicts poor cognitive function. Immun. Inflamm. Dis. 2022, 10, e577. [Google Scholar] [CrossRef]
- Zhou, Y.; Fan, R.; Botchway, B.O.A.; Zhang, Y.; Liu, X. Infliximab Can Improve Traumatic Brain Injury by Suppressing the Tumor Necrosis Factor Alpha Pathway. Mol. Neurobiol. 2021, 58, 2803–2811. [Google Scholar] [CrossRef]
- DeSana, A.J.; Estus, S.; Barrett, T.A.; Saatman, K.E. Acute gastrointestinal permeability after traumatic brain injury in mice precedes a bloom in Akkermansia muciniphila supported by intestinal hypoxia. Sci. Rep. 2024, 14, 2990. [Google Scholar] [CrossRef]
- Bansal, V.; Costantini, T.; Kroll, L.; Peterson, C.; Loomis, W.; Eliceiri, B.; Baird, A.; Wolf, P.; Coimbra, R. Traumatic brain injury and intestinal dysfunction: Uncovering the neuro-enteric axis. J. Neurotrauma 2009, 26, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.W.; Pandya, J.D.; Shear, D.A. Gut Microbiota as a Therapeutic Target to Ameliorate the Biochemical, Neuroanatomical, and Behavioral Effects of Traumatic Brain Injuries. Front. Neurol. 2019, 10, 875. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bao, Z.; Xu, X.; Chao, H.; Lin, C.; Li, Z.; Liu, Y.; Wang, X.; You, Y.; Liu, N.; et al. Extracellular Signal-Regulated Kinase/Nuclear Factor-Erythroid2-like2/Heme Oxygenase-1 Pathway-Mediated Mitophagy Alleviates Traumatic Brain Injury-Induced Intestinal Mucosa Damage and Epithelial Barrier Dysfunction. J. Neurotrauma 2017, 34, 2119–2131. [Google Scholar] [CrossRef]
- Kerr, N.; Lee, S.W.; Perez-Barcena, J.; Crespi, C.; Ibanez, J.; Bullock, M.R.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. Inflammasome proteins as biomarkers of traumatic brain injury. PLoS ONE 2018, 13, e0210128. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Siljander, P.R.M.; Andreu, Z.; Bedina Zavec, A.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Keller, S.; Ridinger, J.; Rupp, A.K.; Janssen, J.W.; Altevogt, P. Body fluid derived exosomes as a novel template for clinical diagnostics. J. Transl. Med. 2011, 9, 86. [Google Scholar] [CrossRef]
- Northrop-Albrecht, E.J.; Taylor, W.R.; Huang, B.Q.; Kisiel, J.B.; Lucien, F. Assessment of extracellular vesicle isolation methods from human stool supernatant. J. Extracell. Vesicles 2022, 11, e12208. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Hilton, H.; Burczynski, M.E. The multifaceted exosome: Biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem. Pharmacol. 2012, 83, 1484–1494. [Google Scholar] [CrossRef]
- Li, L.; Li, F.; Bai, X.; Jia, H.; Wang, C.; Li, P.; Zhang, Q.; Guan, S.; Peng, R.; Zhang, S.; et al. Circulating extracellular vesicles from patients with traumatic brain injury induce cerebrovascular endothelial dysfunction. Pharmacol. Res. 2023, 192, 106791. [Google Scholar] [CrossRef]
- Kerr, N.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Keane, R.W. Neural-respiratory inflammasome axis in traumatic brain injury. Exp. Neurol. 2020, 323, 113080. [Google Scholar] [CrossRef]
- Ali, A.; Zambrano, R.; Duncan, M.R.; Chen, S.; Luo, S.; Yuan, H.; Chen, P.; Benny, M.; Schmidt, A.; Young, K.; et al. Hyperoxia-activated circulating extracellular vesicles induce lung and brain injury in neonatal rats. Sci. Rep. 2021, 11, 8791. [Google Scholar] [CrossRef]
- Cabrera-Pastor, A. Extracellular Vesicles as Mediators of Neuroinflammation in Intercellular and Inter-Organ Crosstalk. Int. J. Mol. Sci. 2024, 25, 7041. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.G.; Anthony, D.C.; Ruitenberg, M.J.; Couch, Y. Systemic Immune Response to Traumatic CNS Injuries—Are Extracellular Vesicles the Missing Link? Front. Immunol. 2019, 10, 2723. [Google Scholar] [CrossRef] [PubMed]
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Hurn, P.D.; Subramanian, S.; Parker, S.M.; Afentoulis, M.E.; Kaler, L.J.; Vandenbark, A.A.; Offner, H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J. Cereb. Blood Flow Metab. 2007, 27, 1798–1805. [Google Scholar] [CrossRef]
- Doyle, K.P.; Quach, L.N.; Solé, M.; Axtell, R.C.; Nguyen, T.V.; Soler-Llavina, G.J.; Jurado, S.; Han, J.; Steinman, L.; Longo, F.M.; et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J. Neurosci. 2015, 35, 2133–2145. [Google Scholar] [CrossRef]
- Chenouard, A.; Chesneau, M.; Braza, F.; Dejoie, T.; Cinotti, R.; Roquilly, A.; Brouard, S.; Asehnoune, K. Phenotype and functions of B cells in patients with acute brain injuries. Mol. Immunol. 2015, 68, 350–356. [Google Scholar] [CrossRef]
- Casili, G.; Impellizzeri, D.; Cordaro, M.; Esposito, E.; Cuzzocrea, S. B-Cell Depletion with CD20 Antibodies as New Approach in the Treatment of Inflammatory and Immunological Events Associated with Spinal Cord Injury. Neurotherapeutics 2016, 13, 880–894. [Google Scholar] [CrossRef]
- Wang, A.A.; Luessi, F.; Neziraj, T.; Pössnecker, E.; Zuo, M.; Engel, S.; Hanuscheck, N.; Florescu, A.; Bugbee, E.; Ma, X.I.; et al. B cell depletion with anti-CD20 promotes neuroprotection in a BAFF-dependent manner in mice and humans. Sci. Transl. Med. 2024, 16, eadi0295. [Google Scholar] [CrossRef]
- Nazmi, A.; Albertsson, A.-M.; Rocha-Ferreira, E.; Zhang, X.; Vontell, R.; Zelco, A.; Rutherford, M.; Zhu, C.; Nilsson, G.; Mallard, C.; et al. Lymphocytes Contribute to the Pathophysiology of Neonatal Brain Injury. Front. Neurol. 2018, 9, 159. [Google Scholar] [CrossRef]
- Xia, J.; Zhang, Y.; Zhao, H.; Wang, J.; Gao, X.; Chen, J.; Fu, B.; Shen, Y.; Miao, F.; Zhang, J.; et al. Non-Invasive Monitoring of CNS MHC-I Molecules in Ischemic Stroke Mice. Theranostics 2017, 7, 2837–2848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Duan, Z.; Cui, Y. CD8+ T cells in brain injury and neurodegeneration. Front. Cell. Neurosci. 2023, 17, 1281763. [Google Scholar] [CrossRef] [PubMed]
- Daglas, M.; Draxler, D.F.; Ho, H.; McCutcheon, F.; Galle, A.; Au, A.E.; Larsson, P.; Gregory, J.; Alderuccio, F.; Sashindranath, M.; et al. Activated CD8 T Cells Cause Long-Term Neurological Impairment after Traumatic Brain Injury in Mice. Cell Rep. 2019, 29, 1178–1191.e1176. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Nguyen, T.; Karnik, S.; Davis, B.C.; Al-Juboori, M.H.; Kacena, M.A.; Obukhov, A.G.; White, F.A. Repeated mild traumatic brain injury in mice elicits long term innate immune cell alterations in blood, spleen, and brain. J. Neuroimmunol. 2023, 380, 578106. [Google Scholar] [CrossRef]
- Miranzadeh Mahabadi, H.; Lin, Y.C.J.; Ogando, N.S.; Moussa, E.W.; Mohammadzadeh, N.; Julien, O.; Alto, N.M.; Noyce, R.S.; Evans, D.H.; Power, C. Monkeypox virus infection of human astrocytes causes gasdermin B cleavage and pyroptosis. Proc. Natl. Acad. Sci. USA 2024, 121, e2315653121. [Google Scholar] [CrossRef]
- Periyasamy, P.; Buch, S. Pore-forming neurons: A new paradigm of pyroptotic cell death in HIV-associated neurocognitive disorder. Brain 2023, 147, 335–336. [Google Scholar] [CrossRef]
- Neel, D.V.; Basu, H.; Gunner, G.; Bergstresser, M.D.; Giadone, R.M.; Chung, H.; Miao, R.; Chou, V.; Brody, E.; Jiang, X.; et al. Gasdermin-E mediates mitochondrial damage in axons and neurodegeneration. Neuron 2023, 111, 1222–1240. [Google Scholar] [CrossRef]
- Chen, S.; Mei, S.; Luo, Y.; Wu, H.; Zhang, J.; Zhu, J. Gasdermin Family: A Promising Therapeutic Target for Stroke. Transl. Stroke Res. 2018, 9, 555–563. [Google Scholar] [CrossRef]
- Cyr, B.; de Rivero Vaccari, J.P. Sex Differences in the Inflammatory Profile in the Brain of Young and Aged Mice. Cells 2023, 12, 1372. [Google Scholar] [CrossRef]
- Cyr, B.; de Rivero Vaccari, J.P. Methods to Study Inflammasome Activation in the Central Nervous System: Immunoblotting and Immunohistochemistry. Methods Mol. Biol. 2023, 2696, 223–238. [Google Scholar] [CrossRef]
- Cyr, B.; Cabrera Ranaldi, E.D.; Hadad, R.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. Extracellular vesicles mediate inflammasome signaling in the brain and heart of Alzheimer’s disease mice. Front. Mol. Neurosci. 2024, 17, 1369781. [Google Scholar] [CrossRef] [PubMed]
- Munley, J.A.; Kirkpatrick, S.L.; Gillies, G.S.; Bible, L.E.; Efron, P.A.; Nagpal, R.; Mohr, A.M. The Intestinal Microbiome after Traumatic Injury. Microorganisms 2023, 11, 1990. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrera Ranaldi, E.d.l.R.M.; Bramlett, H.M.; Umland, O.; Levine, L.I.; Keane, R.W.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Kerr, N.A. Gasdermin-D Genetic Knockout Reduces Inflammasome-Induced Disruption of the Gut-Brain Axis After Traumatic Brain Injury. Int. J. Mol. Sci. 2025, 26, 3512. https://doi.org/10.3390/ijms26083512
Cabrera Ranaldi EdlRM, Bramlett HM, Umland O, Levine LI, Keane RW, de Rivero Vaccari JP, Dietrich WD, Kerr NA. Gasdermin-D Genetic Knockout Reduces Inflammasome-Induced Disruption of the Gut-Brain Axis After Traumatic Brain Injury. International Journal of Molecular Sciences. 2025; 26(8):3512. https://doi.org/10.3390/ijms26083512
Chicago/Turabian StyleCabrera Ranaldi, Erika d. l. R. M., Helen M. Bramlett, Oliver Umland, Leo I. Levine, Robert W. Keane, Juan Pablo de Rivero Vaccari, W. Dalton Dietrich, and Nadine A. Kerr. 2025. "Gasdermin-D Genetic Knockout Reduces Inflammasome-Induced Disruption of the Gut-Brain Axis After Traumatic Brain Injury" International Journal of Molecular Sciences 26, no. 8: 3512. https://doi.org/10.3390/ijms26083512
APA StyleCabrera Ranaldi, E. d. l. R. M., Bramlett, H. M., Umland, O., Levine, L. I., Keane, R. W., de Rivero Vaccari, J. P., Dietrich, W. D., & Kerr, N. A. (2025). Gasdermin-D Genetic Knockout Reduces Inflammasome-Induced Disruption of the Gut-Brain Axis After Traumatic Brain Injury. International Journal of Molecular Sciences, 26(8), 3512. https://doi.org/10.3390/ijms26083512