
Genetic Variants in the NOD-like Receptor Signaling Pathway Are Associated with HIV-1/AIDS in a Northern Chinese Population

 and
and
Abstract
1. Introduction
2. Results
2.1. Selection of NOD-like Receptor Signaling Pathway
2.2. Selection and Annotation of Functional SNPs
2.3. Sample Characteristics and HWE Test
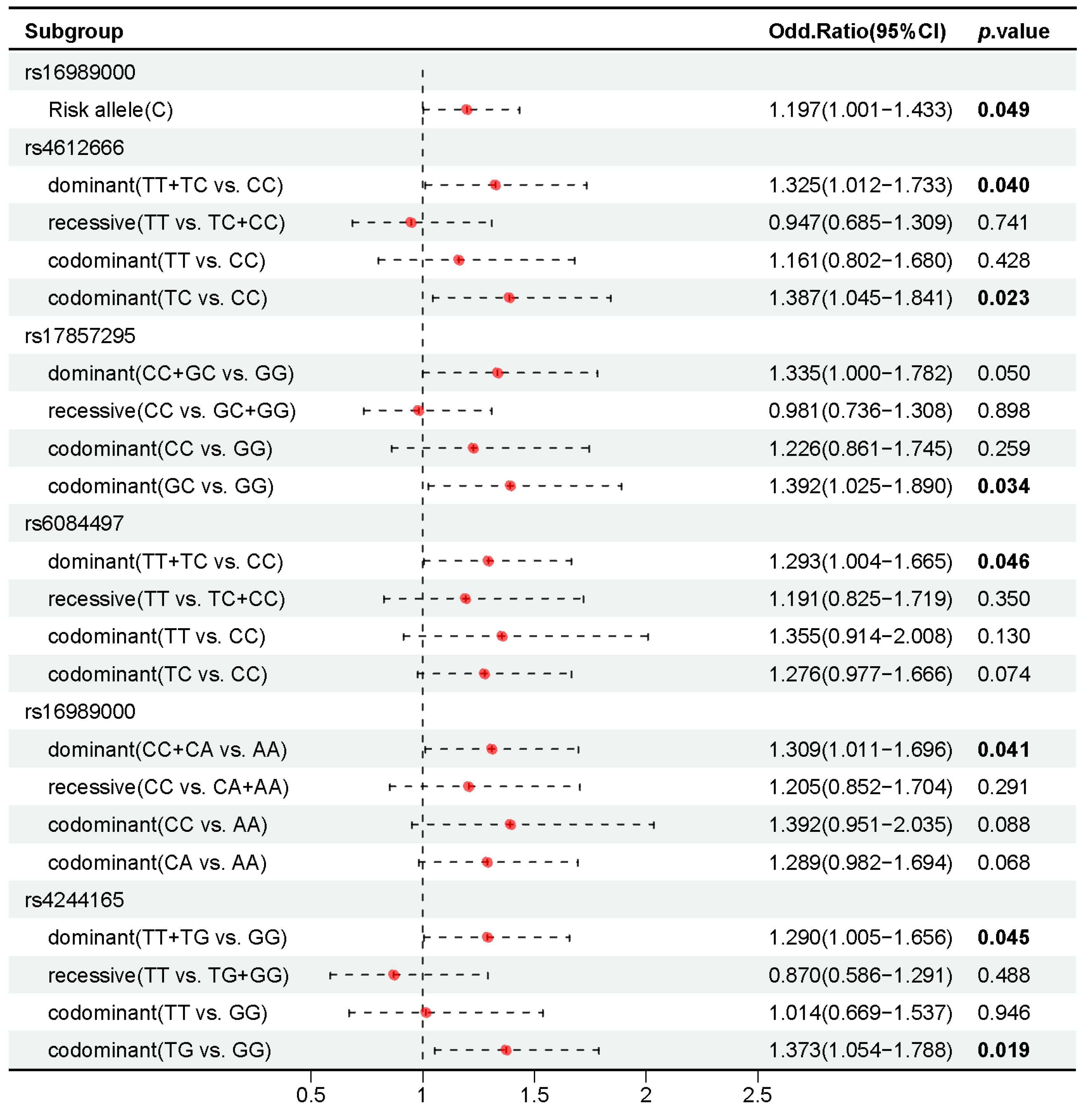
2.4. Association of 37 Candidate SNPs with HIV-1 Infection
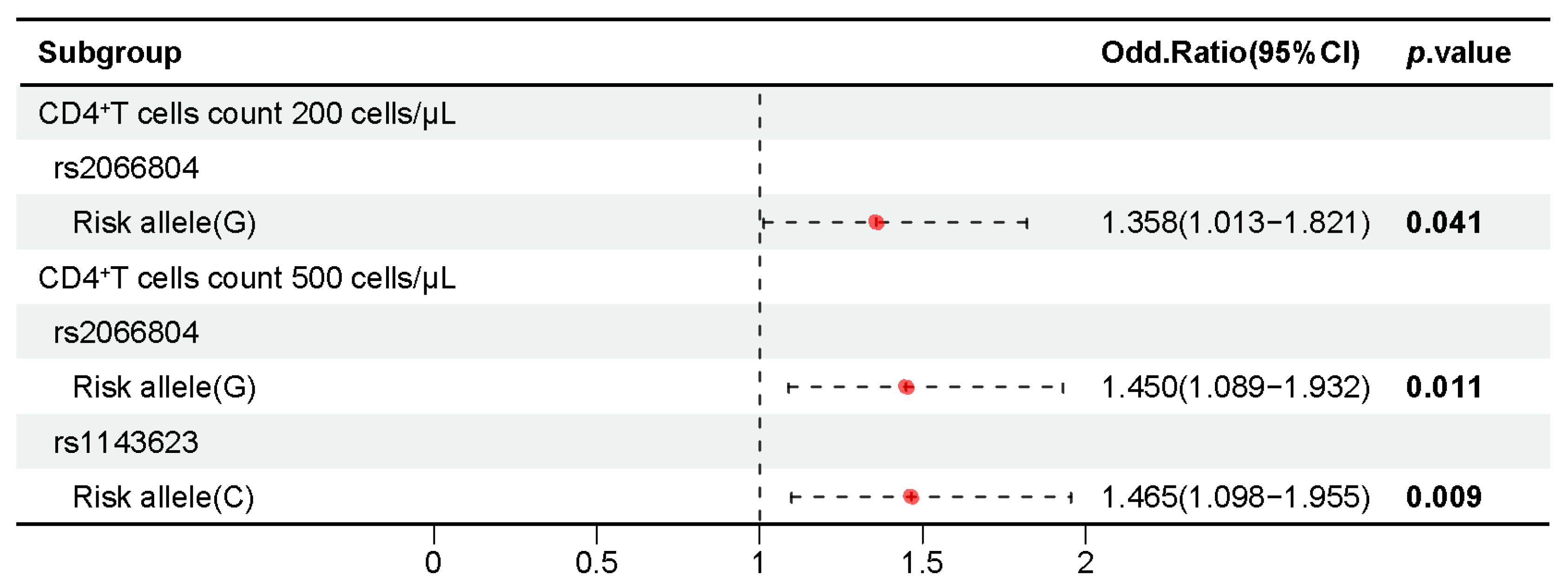
2.5. Association of 37 Candidate SNPs with CD4+ T Cell Counts in People with HIV-1
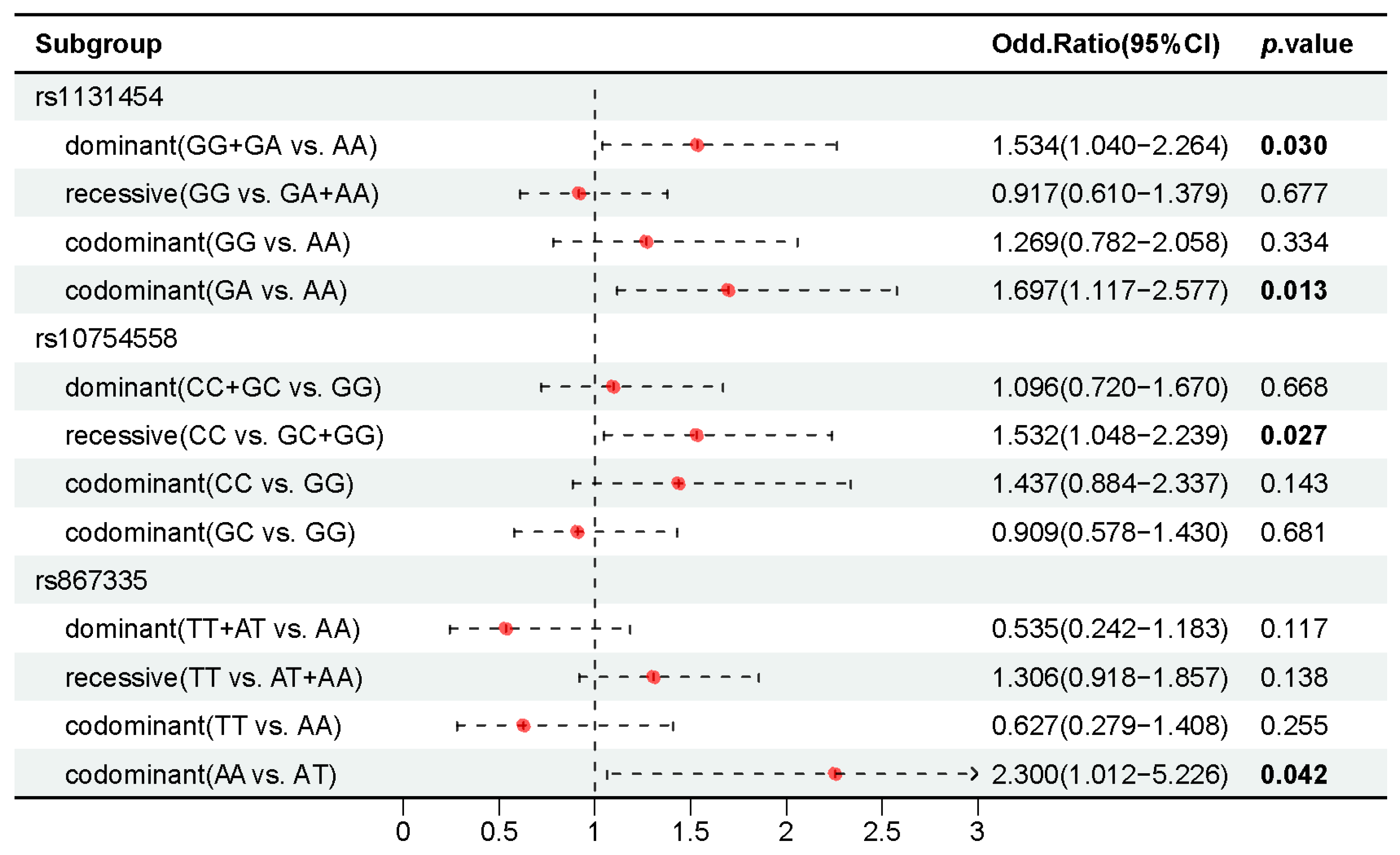
2.6. Association of 37 Candidate SNPs with AIDS Staging in People with HIV-1
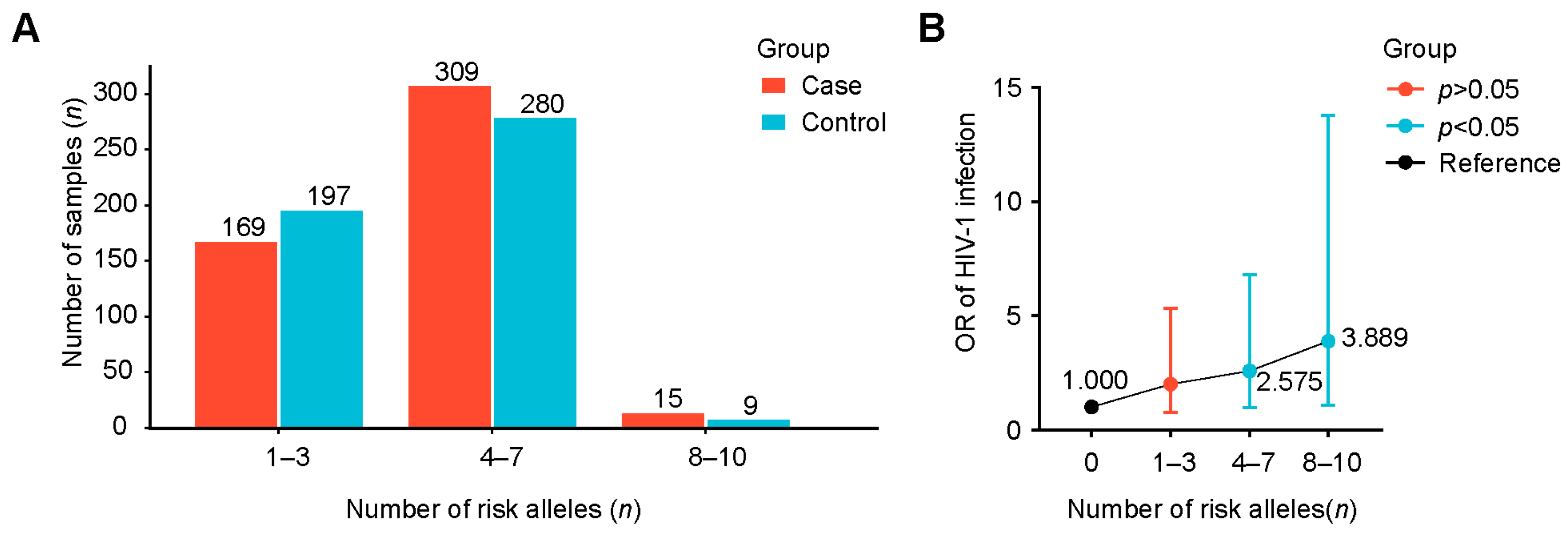
2.7. Interactions of Positively Associated SNPs and Their Combined Effect for AIDS
3. Discussion
4. Materials and Methods
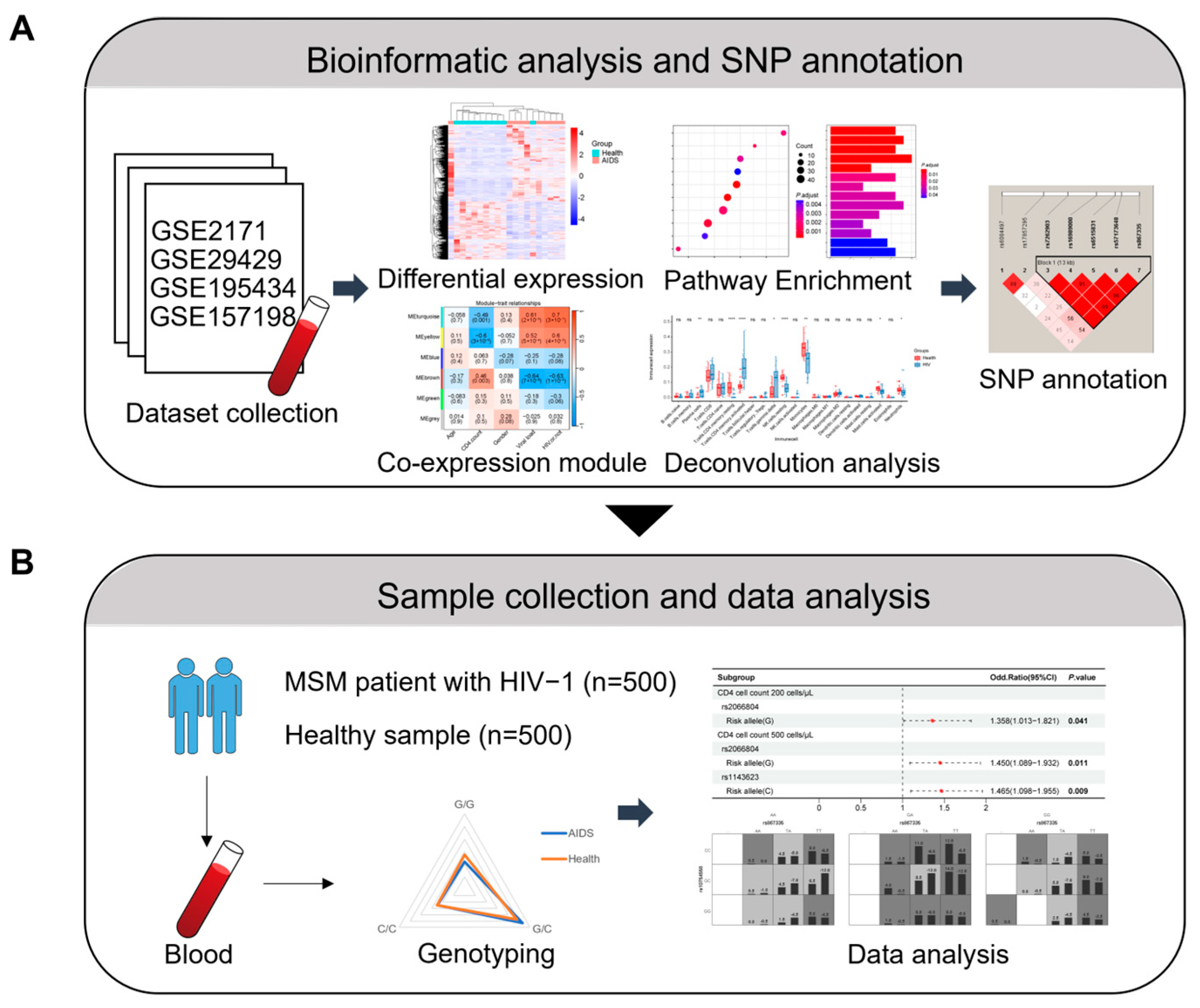
4.1. Dataset Collection and Pre-Processing
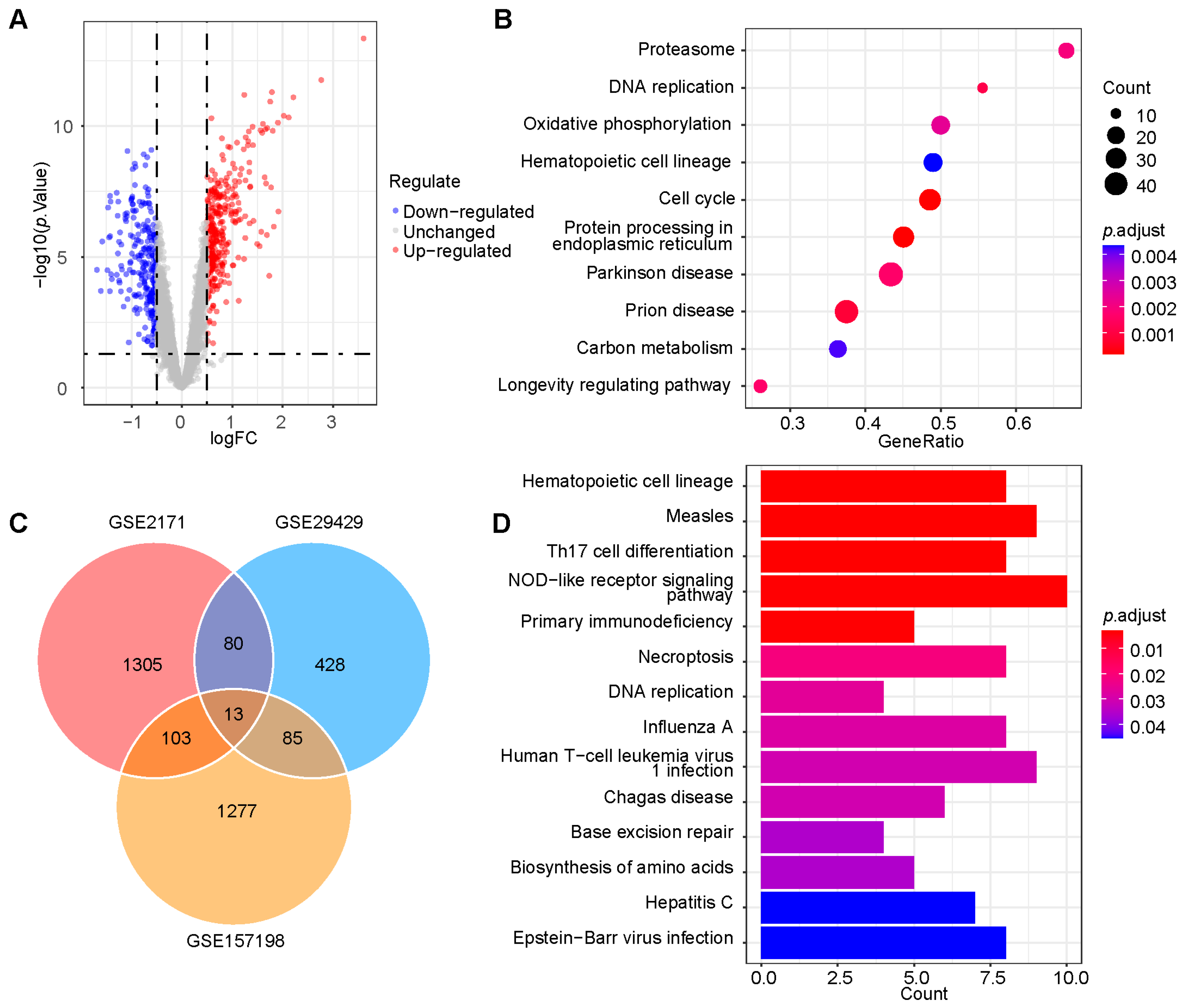
4.2. Differential Expression Analysis and GSEA
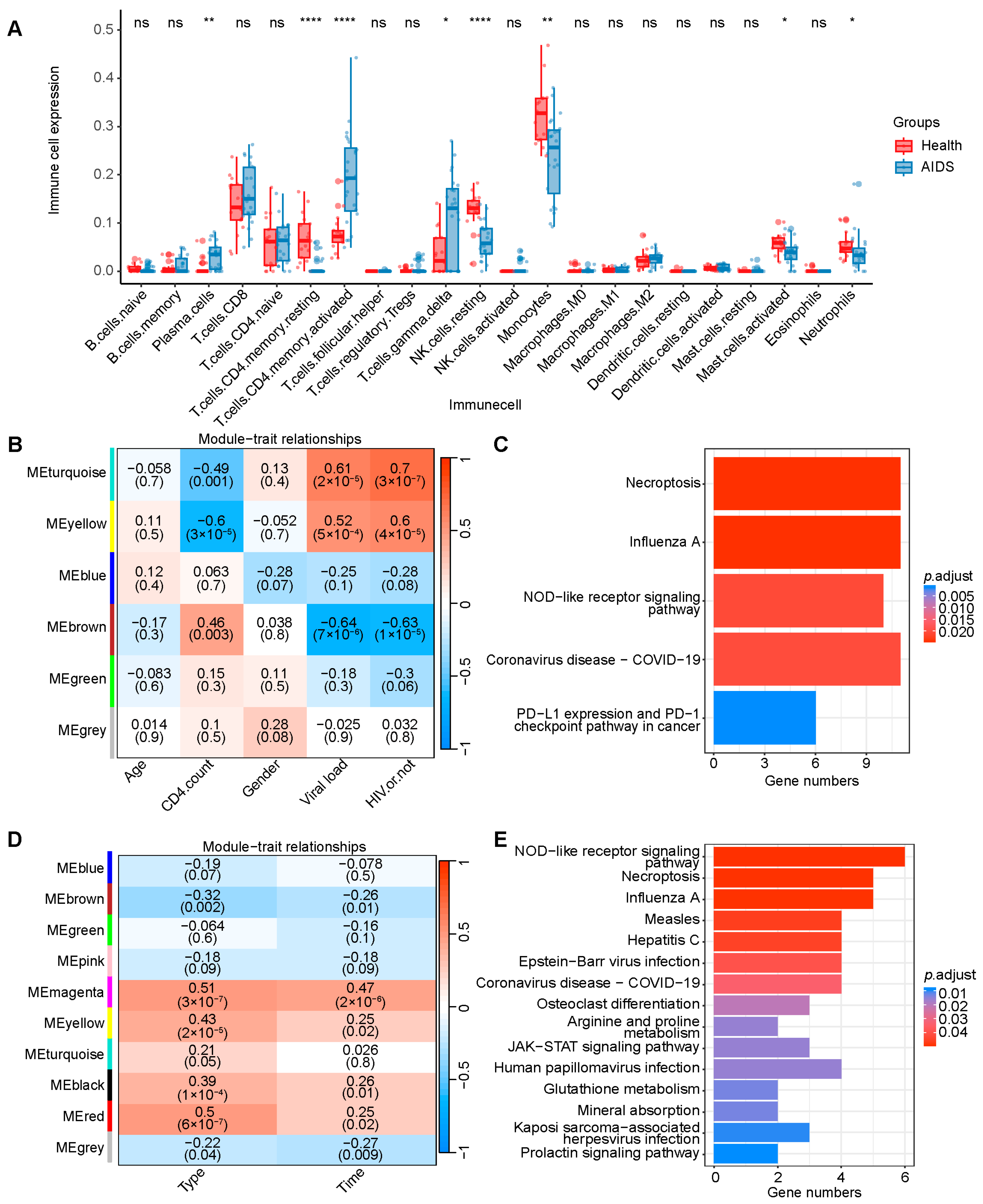
4.3. Cell-Type Deconvolution Analysis
4.4. WGCNA and KEGG Pathway Enrichment
4.5. Selection of SNPs
4.6. Sample Collection and Ethical Statement
4.7. Genotyping
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | Acquired immunodeficiency syndrome |
| ART | Antiretroviral therapy |
| CI | Confidence interval |
| CVC | Cross-validation consistency |
| DEGs | Differentially expressed genes |
| eQTL | Expression quantitative trait loci |
| GSEA | Gene set enrichment analysis |
| HIV | Human immunodeficiency virus |
| HWE | Hardy–Weinberg equilibrium |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LD | Linkage disequilibrium |
| MSM | Men who have sex with men |
| NLR | NOD-like receptor |
| OR | Odds ratio |
| PBMC | Peripheral blood mononuclear cell |
| PCR | Polymerase chain reaction |
| PLWH | People living with HIV |
| PRR | Pattern recognition receptor |
| SNP | Single-nucleotide polymorphism |
| WGCNA | Weighted correlation network analysis |
| WHO | World Health Organization |
References
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [PubMed]
- Mody, A.; Sohn, A.H.; Iwuji, C.; Tan, R.K.J.; Venter, F.; Geng, E.H. HIV epidemiology, prevention, treatment, and implementation strategies for public health. Lancet 2024, 403, 471–492. [Google Scholar] [PubMed]
- Geneva: Joint United Nations Programme on HIV/AIDS. Available online: https://www.unaids.org/en/resources/documents/2024/2024_unaids_data (accessed on 2 December 2024).
- Gandhi, R.T.; Landovitz, R.J.; Sax, P.E.; Smith, D.M.; Springer, S.A.; Günthard, H.F.; Thompson, M.A.; Bedimo, R.J.; Benson, C.A.; Buchbinder, S.P.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV in Adults: 2024 Recommendations of the International Antiviral Society-USA Panel. JAMA 2025, 333, 609–628. [Google Scholar] [PubMed]
- Board, N.L.; Moskovljevic, M.; Wu, F.; Siliciano, R.F.; Siliciano, J.D. Engaging innate immunity in HIV-1 cure strategies. Nat. Rev. Immunol. 2022, 22, 499–512. [Google Scholar]
- Armani-Tourret, M.; Bone, B.; Tan, T.S.; Sun, W.; Bellefroid, M.; Struyve, T.; Louella, M.; Yu, X.G.; Lichterfeld, M. Immune targeting of HIV-1 reservoir cells: A path to elimination strategies and cure. Nat. Rev. Microbiol. 2024, 22, 328–344. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar]
- Sundaram, B.; Tweedell, R.E.; Prasanth Kumar, S.; Kanneganti, T.D. The NLR family of innate immune and cell death sensors. Immunity 2024, 57, 674–699. [Google Scholar]
- Ekabe, C.J.; Clinton, N.A.; Kehbila, J.; Franck, N.C. The Role of Inflammasome Activation in Early HIV Infection. J. Immunol. Res. 2021, 2021, 1487287. [Google Scholar]
- Xia, P.; Xing, X.D.; Yang, C.X.; Liao, X.J.; Liu, F.H.; Huang, H.H.; Zhang, C.; Song, J.W.; Jiao, Y.M.; Shi, M.; et al. Activation-induced pyroptosis contributes to the loss of MAIT cells in chronic HIV-1 infected patients. Mil. Med. Res. 2022, 9, 24. [Google Scholar]
- Jin, X.; Zhou, R.; Huang, Y. Role of inflammasomes in HIV-1 infection and treatment. Trends Mol. Med. 2022, 28, 421–434. [Google Scholar]
- Deng, C.H.; Li, T.Q.; Zhang, W.; Zhao, Q.; Wang, Y. Targeting Inflammasome Activation in Viral Infection: A Therapeutic Solution? Viruses 2023, 15, 1451. [Google Scholar] [CrossRef]
- Min, A.K.; Fortune, T.; Rodriguez, N.; Hedge, E.; Swartz, T.H. Inflammasomes as mediators of inflammation in HIV-1 infection. Transl. Res. 2023, 252, 1–8. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Roberts, R.L.; Van Rij, A.M.; Phillips, L.V.; Young, S.; McCormick, S.P.; Merriman, T.R.; Jones, G.T. Interaction of the inflammasome genes CARD8 and NLRP3 in abdominal aortic aneurysms. Atherosclerosis 2011, 218, 123–126. [Google Scholar] [PubMed]
- Meng, D.M.; Zhou, Y.J.; Wang, L.; Ren, W.; Cui, L.L.; Han, L.; Qu, Z.H.; Li, C.G.; Zhao, J.J. Polymorphisms in the NLRP3 gene and risk of primary gouty arthritis. Mol. Med. Rep. 2013, 7, 1761–1766. [Google Scholar]
- Huffman, J.E.; Butler-Laporte, G.; Khan, A.; Pairo-Castineira, E.; Drivas, T.G.; Peloso, G.M.; Nakanishi, T.; Ganna, A.; Verma, A.; Baillie, J.K.; et al. Multi-ancestry fine mapping implicates OAS1 splicing in risk of severe COVID-19. Nat. Genet. 2022, 54, 125–127. [Google Scholar] [PubMed]
- Bekker, L.G.; Beyrer, C.; Mgodi, N.; Lewin, S.R.; Delany-Moretlwe, S.; Taiwo, B.; Masters, M.C.; Lazarus, J.V. HIV infection. Nat. Rev. Dis. Primers 2023, 9, 42. [Google Scholar]
- Diagnosis of AIDS and HIV Infection. Available online: http://www.nhc.gov.cn/wjw/s9491/201905/6430aa653728439c901a7340796e4723.shtml (accessed on 2 January 2019).
- HIV and AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 20 July 2024).
- Taisheng, L. Chinese Guidelines for Diagnosis and Treatment of Human Immunodeficiency Virus Infection/Acquired Immunodeficiency Syndrome (2024 Edition). Med. J. Peking Union Med. Coll. Hosp. 2024, 137, 2654–2680. [Google Scholar]
- Chakrabarti, A.; Banerjee, S.; Franchi, L.; Loo, Y.M.; Gale, M., Jr.; Núñez, G.; Silverman, R.H. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 2015, 17, 466–477. [Google Scholar] [PubMed]
- Acchioni, C.; Marsili, G.; Perrotti, E.; Remoli, A.L.; Sgarbanti, M.; Battistini, A. Type I IFN—A blunt spear in fighting HIV-1 infection. Cytokine Growth Factor Rev. 2015, 26, 143–158. [Google Scholar]
- Niu, J.; Wu, S.; Chen, M.; Xu, K.; Guo, Q.; Lu, A.; Zhao, L.; Sun, B.; Meng, G. Hyperactivation of the NLRP3 inflammasome protects mice against influenza A virus infection via IL-1β mediated neutrophil recruitment. Cytokine 2019, 120, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.U.; Jeong, Y.J.; Lee, P.; Lee, M.S.; Park, J.H.; Kim, Y.S.; Kim, D.J. Extracellular nucleoprotein exacerbates influenza virus pathogenesis by activating Toll-like receptor 4 and the NLRP3 inflammasome. Cell Mol. Immunol. 2022, 19, 715–725. [Google Scholar] [PubMed]
- de Sá, N.B.R.; de Souza, N.C.S.; Neira-Goulart, M.; Ribeiro-Alves, M.; Da Silva, T.P.; Pilotto, J.H.; Rolla, V.C.; Giacoia-Gripp, C.B.W.; de Oliveira Pinto, L.M.; Scott-Algara, D.; et al. Inflammasome genetic variants are associated with tuberculosis, HIV-1 infection, and TB/HIV-immune reconstitution inflammatory syndrome outcomes. Front. Cell. Infect. Microbiol. 2022, 12, 962059. [Google Scholar]
- de Alencar, J.B.; Zacarias, J.M.V.; Tsuneto, P.Y.; Souza, V.H.; Silva, C.O.E.; Visentainer, J.E.L.; Sell, A.M. Influence of inflammasome NLRP3, and IL1B and IL2 gene polymorphisms in periodontitis susceptibility. PLoS ONE 2020, 15, e0227905. [Google Scholar]
- Bellini, N.; Ye, C.; Ajibola, O.; Murooka, T.T.; Lodge, R.; Cohen, É.A. Downregulation of miRNA-26a by HIV-1 Enhances CD59 Expression and Packaging, Impacting Virus Susceptibility to Antibody-Dependent Complement-Mediated Lysis. Viruses 2024, 16, 1076. [Google Scholar] [CrossRef]
- Pontillo, A.; Brandão, L.A.; Guimarães, R.L.; Segat, L.; Athanasakis, E.; Crovella, S. A 3′ UTR SNP in NLRP3 gene is associated with susceptibility to HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2010, 54, 236–240. [Google Scholar]
- Pontillo, A.; Bricher, P.; Leal, V.N.; Lima, S.; Souza, P.R.; Crovella, S. Role of inflammasome genetics in susceptibility to HPV infection and cervical cancer development. J. Med. Virol. 2016, 88, 1646–1651. [Google Scholar]
- Lu, Q.; Lao, X.; Gan, J.; Du, P.; Zhou, Y.; Nong, W.; Yang, Z. Impact of NLRP3 gene polymorphisms (rs10754558 and rs10733113) on HPV infection and cervical cancer in southern Chinese population. Infect. Agents Cancer 2023, 18, 64. [Google Scholar]
- Liu, K.; Chen, L.; Zhao, G.; Cao, Z.; Li, F.; Lin, L.; Zhu, C.; Xie, Q.; Xu, Y.; Bao, S.; et al. IPS-1 polymorphisms in regulating interferon response in HBV infection. Biosci. Trends 2019, 13, 130–135. [Google Scholar]
- Gringhuis, S.I.; Hertoghs, N.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Sarrami-Forooshani, R.; Sprokholt, J.K.; van Teijlingen, N.H.; Kootstra, N.A.; Booiman, T.; van Dort, K.A.; et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 2017, 18, 225–235. [Google Scholar]
- Andersen, V.; Holst, R.; Kopp, T.I.; Tjønneland, A.; Vogel, U. Interactions between diet, lifestyle and IL10, IL1B, and PTGS2/COX-2 gene polymorphisms in relation to risk of colorectal cancer in a prospective Danish case-cohort study. PLoS ONE 2013, 8, e78366. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Cao, D.; Wang, L. The association between SNPs and hepatitis B virus related acute-on-chronic liver failure. Infect. Genet. Evol. 2020, 86, 104615. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Zhang, D.; Bao, C. Two variants of Interleukin-1B gene are associated with the decreased risk, clinical features, and better overall survival of colorectal cancer: A two-center case-control study. Aging 2018, 10, 4084–4092. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, M.M.; Abuharfeil, N.M.; Darmani, H. The role of IL-1β during human immunodeficiency virus type 1 infection. Rev. Med. Virol. 2023, 33, e2400. [Google Scholar] [CrossRef]
- Fagone, P.; Nunnari, G.; Lazzara, F.; Longo, A.; Cambria, D.; Distefano, G.; Palumbo, M.; Nicoletti, F.; Malaguarnera, L.; Di Rosa, M. Induction of OAS gene family in HIV monocyte infected patients with high and low viral load. Antivir. Res. 2016, 131, 66–73. [Google Scholar] [CrossRef]
- Gauderman, W.J. Sample size requirements for matched case-control studies of gene-environment interaction. Stat. Med. 2002, 21, 35–50. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Case (n = 500) | Control (n = 500) | p Value |
|---|---|---|---|
| Age range (years) | 17–81 | 16–78 | - |
| Mean age ± SD, years | 35.22 ± 11.81 | 36.60 ± 14.32 | 0.649 a |
| Gender n (%) | |||
| Male | 500 (100%) | 500 (100%) | - |
| Female | - | - | - |
| Clinical stages, n (frequency) | |||
| Ⅰ | 125 (25.0%) | - | - |
| Ⅱ | 125 (25.0%) | - | - |
| Ⅲ | 190 (38.0%) | - | - |
| Ⅳ | 60 (12.0%) | - | - |
| CD4+ T cell counts (cells/μL), n (frequency) | |||
| <200 | 124 (24.8%) | - | - |
| 200–349 | 126 (25.2%) | - | - |
| 350–500 | 125 (25.0%) | - | - |
| ≥500 | 125 (25.2%) | - | - |
| Best Combination | Training Accuracy | Testing Accuracy | CV Consistency | Sign Test (p) |
|---|---|---|---|---|
| Infection module | ||||
| rs4244165 | 0.540 | 0.508 | 7/10 | 4(0.828) |
| rs4244165 rs6084497 | 0.555 | 0.477 | 4/10 | 2(0.989) |
| rs4244165 rs6084497 rs4612666 | 0.581 | 0.525 | 5/10 | 6(0.377) |
| rs17857295 rs4244165 rs6084497 rs4612666 | 0.620 | 0.520 | 7/10 | 6(0.377) |
| rs17857295 rs4244165 rs6084497 rs16989000 rs4612666 | 0.670 | 0.567 | 10/10 | 9(0.011) |
| Stage module | ||||
| rs1131454 | 0.556 | 0.494 | 5/10 | 5(0.623) |
| rs867335 rs1131454 | 0.577 | 0.493 | 7/10 | 4(0.828) |
| rs867335 rs10754558 rs1131454 | 0.605 | 0.554 | 10/10 | 9(0.011) |
| Risk Allele (n) | Case with Risk Allele | Case Without Risk Allele | Control with Risk Allele | Control Without Risk Allele | p Value | OR (95%CI) |
|---|---|---|---|---|---|---|
| 1 | 23 | 6 | 35 | 14 | 0.441 | 1.533 (0.515–4.567) |
| 2 | 67 | 6 | 64 | 14 | 0.078 | 2.443 (0.884–6.746) |
| 3 | 79 | 6 | 98 | 14 | 0.210 | 1.881 (0.691–5.119) |
| 4 | 108 | 6 | 111 | 14 | 0.098 | 2.270 (0.842–6.124) |
| 5 | 104 | 6 | 79 | 14 | 0.022 | 3.072 (1.130–8.351) |
| 6 | 55 | 6 | 61 | 14 | 0.148 | 2.104 (0.756–5.854) |
| 7 | 42 | 6 | 29 | 14 | 0.021 | 3.379 (1.163–9.823) |
| 8 | 12 | 6 | 7 | 14 | 0.038 | 4.000 (1.052–15.207) |
| 9 | 3 | 6 | 2 | 14 | 0.312 | 3.500 (0.460–26.616) |
| 10 | 0 | 6 | 0 | 14 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, T.; Yang, Y.; Zhang, X.; You, C.; Wu, J.; Xu, L.; Ji, W.; Jia, X.; Wu, J.; Sun, W.; et al. Genetic Variants in the NOD-like Receptor Signaling Pathway Are Associated with HIV-1/AIDS in a Northern Chinese Population. Int. J. Mol. Sci. 2025, 26, 3484. https://doi.org/10.3390/ijms26083484
Pan T, Yang Y, Zhang X, You C, Wu J, Xu L, Ji W, Jia X, Wu J, Sun W, et al. Genetic Variants in the NOD-like Receptor Signaling Pathway Are Associated with HIV-1/AIDS in a Northern Chinese Population. International Journal of Molecular Sciences. 2025; 26(8):3484. https://doi.org/10.3390/ijms26083484
Chicago/Turabian StylePan, Tingyu, Yi Yang, Xia Zhang, Chenghong You, Jiawei Wu, Lidan Xu, Wei Ji, Xueyuan Jia, Jie Wu, Wenjing Sun, and et al. 2025. "Genetic Variants in the NOD-like Receptor Signaling Pathway Are Associated with HIV-1/AIDS in a Northern Chinese Population" International Journal of Molecular Sciences 26, no. 8: 3484. https://doi.org/10.3390/ijms26083484
APA StylePan, T., Yang, Y., Zhang, X., You, C., Wu, J., Xu, L., Ji, W., Jia, X., Wu, J., Sun, W., Fu, S., Zhang, X., & Qiao, Y. (2025). Genetic Variants in the NOD-like Receptor Signaling Pathway Are Associated with HIV-1/AIDS in a Northern Chinese Population. International Journal of Molecular Sciences, 26(8), 3484. https://doi.org/10.3390/ijms26083484