
Advancing Personalized Medicine in Alzheimer’s Disease: Liquid Biopsy Epigenomics Unveil APOE ε4-Linked Methylation Signatures

 ,
,  , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Results
2.1. Characterization of Subjects and Samples
2.2. cfDNA Concentration and Quality
2.3. Surrogate Variable Analysis
2.4. Differential Methylated Positions
2.5. Correlation with AD Clinical Parameters and Biomarkers
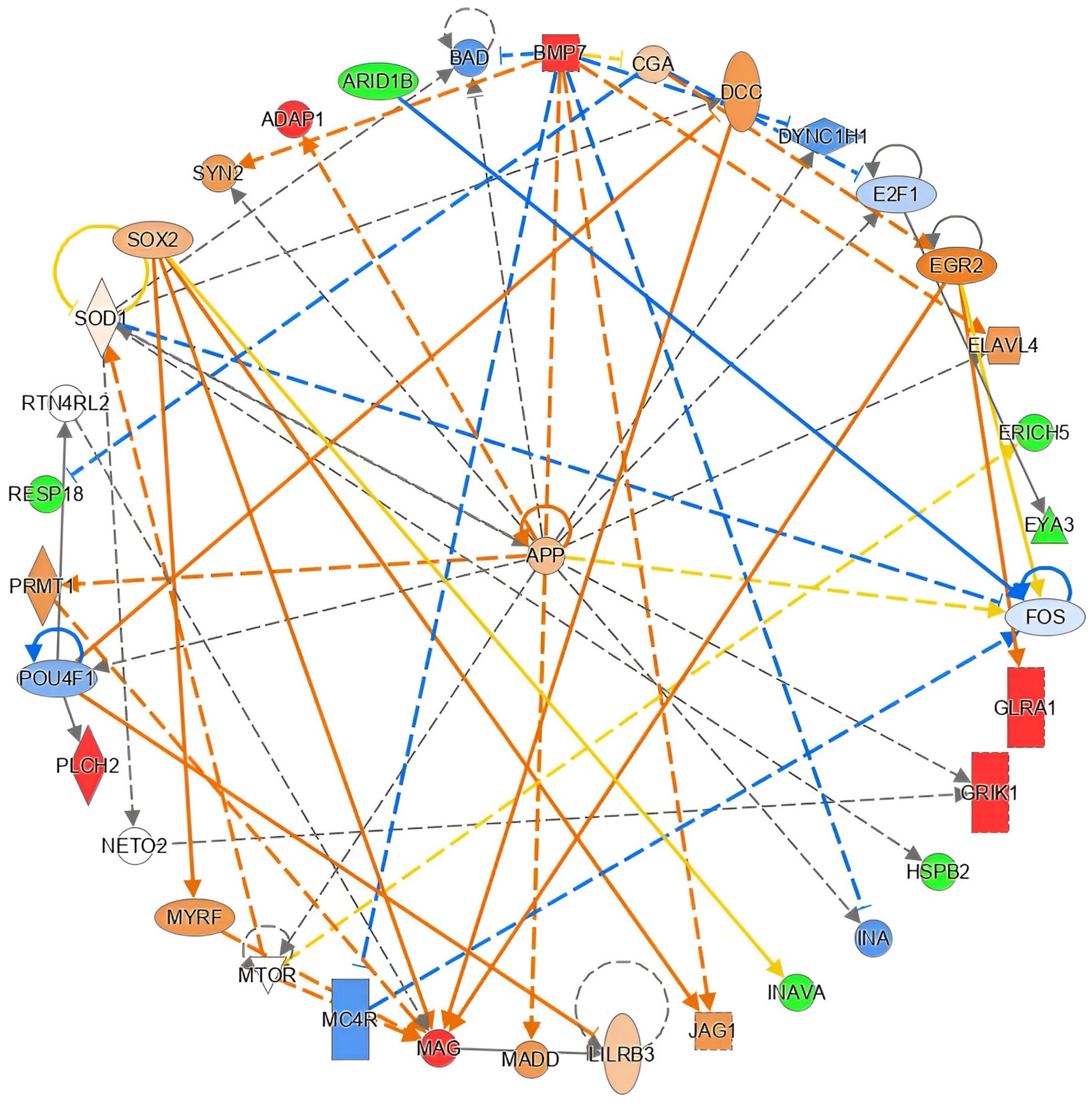
2.6. Functional in Silico Analysis of DMPs
2.7. Differential Methylated Positions According to APOE ε4 Status
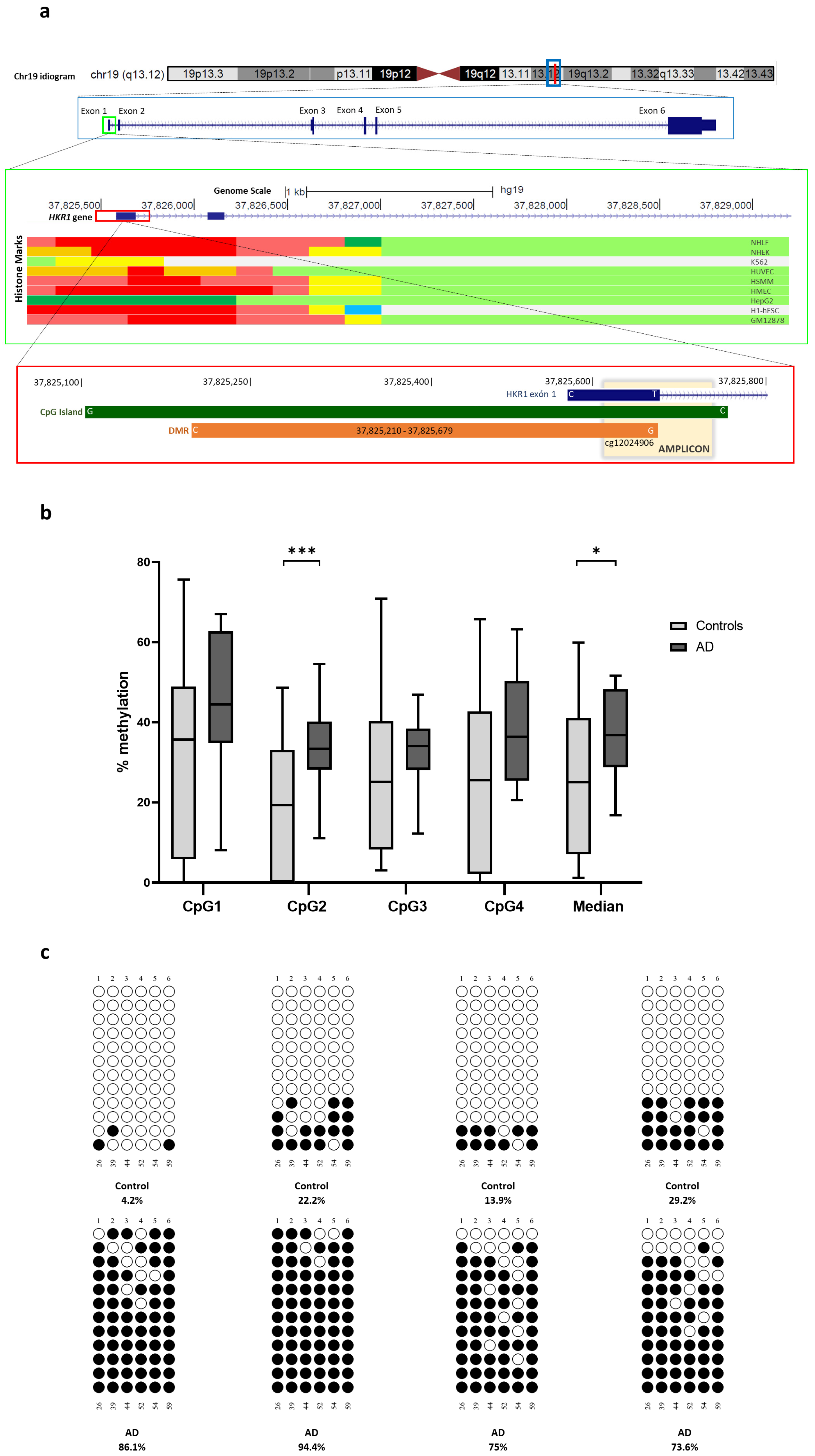
2.8. Differential Methylated Regions
2.9. Orthogonal Validation
3. Discussion
Limitations
4. Materials and Methods
4.1. Study Design
4.2. Subjects’ Characterization
4.3. Blood and Cerebrospinal Fluid (CSF) Samples
4.4. cfDNA Isolation and Quantification
4.5. Characterization of cfDNA: Fragment Size Analysis
4.6. Genome-Wide cfDNA Methylation Analysis
4.7. Array Data Preprocessing
4.8. cfDNA Cell-Type Deconvolution
4.9. Surrogate Variable Analysis
4.10. Probe-Level Differential Methylation Analyses
4.11. Region-Level Differential Methylation Analyses
4.12. Probe Annotation
4.13. Functional In Silico Analysis of DMPs
4.14. Orthogonal Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APOE | Apolipoprotein E |
| cfDNA | Cell-free DNA |
| CpG | Cytosine guanine dinucleotide |
| CSF | Cerebrospinal fluid |
| DMP | Differential methylated position |
| DMR | Differential methylated region |
| GDS | Global Deterioration Scale |
| LOAD | Late-onset Alzheimer’s disease |
| MMSE | Mini-Mental State Examination |
References
- 2024 Alzheimer’s disease facts and figures. Alzheimers Dement. 2024, 20, 3708–3821. [CrossRef] [PubMed]
- Alzheimers Disease International. World Alzheimer Report 2018. 2018. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2018.pdf (accessed on 2 April 2025).
- Alzheimers Disease International. World Alzheimer Report 2021. 2021. Available online: https://www.alzint.org/u/World-Alzheimer-Report-2021.pdf (accessed on 2 April 2025).
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Gräff, J. Epigenetic Alterations in Alzheimer’s Disease. Front. Behav. Neurosci. 2015, 9, 347. [Google Scholar] [CrossRef]
- Zhang, L.; Silva, T.C.; Young, J.I.; Gomez, L.; Schmidt, M.A.; Hamilton-Nelson, K.L.; Kunkle, B.W.; Chen, X.; Martin, E.R.; Wang, L. Epigenome-wide meta-analysis of DNA methylation differences in prefrontal cortex implicates the immune processes in Alzheimer’s disease. Nat. Commun. 2020, 11, 6114. [Google Scholar] [CrossRef]
- Zhang, L.; Young, J.I.; Gomez, L.; Silva, T.C.; Schmidt, M.A.; Cai, J.; Chen, X.; Martin, E.R.; Wang, L. Sex-specific DNA methylation differences in Alzheimer’s disease pathology. Acta Neuropathol. Commun. 2021, 9, 77. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef]
- Gasparoni, G.; Bultmann, S.; Lutsik, P.; Kraus, T.F.J.; Sordon, S.; Vlcek, J.; Dietinger, V.; Steinmaurer, M.; Haider, M.; Mulholland, C.B.; et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 2018, 11, 41. [Google Scholar] [CrossRef]
- Smith, R.G.; Pishva, E.; Shireby, G.; Smith, A.R.; Roubroeks, J.A.Y.; Hannon, E.; Wheildon, G.; Mastroeni, D.; Gasparoni, G.; Riemenschneider, M.; et al. A meta-analysis of epigenome-wide association studies in Alzheimer’s disease highlights novel differentially methylated loci across cortex. Nat. Commun. 2021, 12, 3517. [Google Scholar] [CrossRef]
- Sanchez-Mut, J.V.; Heyn, H.; Vidal, E.; Delgado-Morales, R.; Moran, S.; Sayols, S.; Sandoval, J.; Ferrer, I.; Esteller, M.; Gräff, J. Whole genome grey and white matter DNA methylation profiles in dorsolateral prefrontal cortex. Synapse 2017, 71, e21959. [Google Scholar] [CrossRef]
- Smith, R.G.; Hannon, E.; De Jager, P.L.; Chibnik, L.; Lott, S.J.; Condliffe, D.; Smith, A.R.; Haroutunian, V.; Troakes, C.; Al-Sarraj, S.; et al. Elevated DNA methylation across a 48-kb region spanning the HOXA gene cluster is associated with Alzheimer’s disease neuropathology. Alzheimers Dement. 2018, 14, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Semick, S.A.; Bharadwaj, R.A.; Collado-Torres, L.; Tao, R.; Shin, J.H.; Deep-Soboslay, A.; Weiss, J.R.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E.; et al. Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Keleshian, V.L.; Klein, S.; Rapoport, S.I. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl. Psychiatry 2012, 2, e132. [Google Scholar] [CrossRef] [PubMed]
- Altuna, M.; Urdanoz-Casado, A.; Sanchez-Ruiz de Gordoa, J.; Zelaya, M.V.; Labarga, A.; Lepesant, J.M.J.; Roldan, M.; Blanco-Luquin, I.; Perdones, A.; Larumbe, R.; et al. DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clin. Epigenetics 2019, 11, 91. [Google Scholar] [CrossRef]
- Li, Q.S.; Sun, Y.; Wang, T. Epigenome-wide association study of Alzheimer’s disease replicates 22 differentially methylated positions and 30 differentially methylated regions. Clin. Epigenetics 2020, 12, 149. [Google Scholar] [CrossRef]
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8, 5. [Google Scholar] [CrossRef]
- Perez, R.F.; Alba-Linares, J.J.; Tejedor, J.R.; Fernandez, A.F.; Calero, M.; Roman-Dominguez, A.; Borras, C.; Vina, J.; Avila, J.; Medina, M.; et al. Blood DNA Methylation Patterns in Older Adults With Evolving Dementia. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 1743–1749. [Google Scholar] [CrossRef]
- Lardenoije, R.; Roubroeks, J.A.Y.; Pishva, E.; Leber, M.; Wagner, H.; Iatrou, A.; Smith, A.R.; Smith, R.G.; Eijssen, L.M.T.; Kleineidam, L.; et al. Alzheimer’s disease-associated (hydroxy)methylomic changes in the brain and blood. Clin. Epigenetics 2019, 11, 164. [Google Scholar] [CrossRef]
- Konki, M.; Malonzo, M.; Karlsson, I.K.; Lindgren, N.; Ghimire, B.; Smolander, J.; Scheinin, N.M.; Ollikainen, M.; Laiho, A.; Elo, L.L.; et al. Peripheral blood DNA methylation differences in twin pairs discordant for Alzheimer’s disease. Clin. Epigenetics 2019, 11, 130. [Google Scholar] [CrossRef]
- Chang, L.; Wang, Y.; Ji, H.; Dai, D.; Xu, X.; Jiang, D.; Hong, Q.; Ye, H.; Zhang, X.; Zhou, X.; et al. Elevation of peripheral BDNF promoter methylation links to the risk of Alzheimer’s disease. PLoS ONE 2014, 9, e110773. [Google Scholar] [CrossRef]
- Macías, M.; Alegre, E.; Díaz-Lagares, A.; Patiño, A.; Pérez-Gracia, J.L.; Sanmamed, M.; López-López, R.; Varo, N.; González, A. Liquid Biopsy: From Basic Research to Clinical Practice. Adv. Clin. Chem. 2018, 83, 73–119. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jiang, P.; Chan, K.C.; Wong, J.; Cheng, Y.K.; Liang, R.H.; Chan, W.K.; Ma, E.S.; Chan, S.L.; Cheng, S.H.; et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef]
- Gaitsch, H.; Franklin, R.J.M.; Reich, D.S. Cell-free DNA-based liquid biopsies in neurology. Brain 2022, 146, 1758–1774. [Google Scholar] [CrossRef]
- Southwood, D.; Singh, S.; Chatterton, Z. Brain-derived cell-free DNA. Neural Regen. Res. 2022, 17, 2213–2214. [Google Scholar] [CrossRef]
- Khemka, S.; Sehar, U.; Manna, P.R.; Kshirsagar, S.; Reddy, P.H. Cell-Free DNA As Peripheral Biomarker of Alzheimer’s Disease. Aging Dis. 2024, 16, 787–803. [Google Scholar] [CrossRef]
- Pollard, C.; Aston, K.; Emery, B.R.; Hill, J.; Jenkins, T. Detection of neuron-derived cfDNA in blood plasma: A new diagnostic approach for neurodegenerative conditions. Front. Neurol. 2023, 14, 1272960. [Google Scholar] [CrossRef]
- Noe, C.R.; Noe-Letschnig, M.; Handschuh, P.; Noe, C.A.; Lanzenberger, R. Dysfunction of the Blood-Brain Barrier-A Key Step in Neurodegeneration and Dementia. Front. Aging Neurosci. 2020, 12, 185. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Costa-Pinheiro, P.; Montezuma, D.; Henrique, R.; Jerónimo, C. Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics 2015, 7, 1003–1015. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.M.; Vaher, K.; Bermingham, M.L.; Morris, S.W.; Bretherick, A.D.; Zeng, Y.; Rawlik, K.; Amador, C.; Campbell, A.; Haley, C.S.; et al. Identification of epigenome-wide DNA methylation differences between carriers of APOE epsilon4 and APOE epsilon2 alleles. Genome Med. 2021, 13, 1. [Google Scholar] [CrossRef]
- Darst, R.P.; Pardo, C.E.; Ai, L.; Brown, K.D.; Kladde, M.P. Bisulfite sequencing of DNA. Curr. Protoc. Mol. Biol. 2010, 91, 7.9.1–7.9.17. [Google Scholar] [CrossRef]
- Gallardo-Gomez, M.; Moran, S.; Paez de la Cadena, M.; Martinez-Zorzano, V.S.; Rodriguez-Berrocal, F.J.; Rodriguez-Girondo, M.; Esteller, M.; Cubiella, J.; Bujanda, L.; Castells, A.; et al. A new approach to epigenome-wide discovery of non-invasive methylation biomarkers for colorectal cancer screening in circulating cell-free DNA using pooled samples. Clin. Epigenetics 2018, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Pai, M.C.; Kuo, Y.M.; Wang, I.F.; Chiang, P.M.; Tsai, K.J. The Role of Methylated Circulating Nucleic Acids as a Potential Biomarker in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 2440–2449. [Google Scholar] [CrossRef]
- Mendioroz, M.; Martínez-Merino, L.; Blanco-Luquin, I.; Urdánoz, A.; Roldán, M.; Jericó, I. Liquid biopsy: A new source of candidate biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 763–768. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Aucamp, J.; Pretorius, P.J. Cell-free DNA: Preanalytical variables. Clin. Chim. Acta 2015, 450, 243–253. [Google Scholar] [CrossRef]
- Kresse, S.H.; Brandt-Winge, S.; Pharo, H.; Flatin, B.T.B.; Jeanmougin, M.; Vedeld, H.M.; Lind, G.E. Evaluation of commercial kits for isolation and bisulfite conversion of circulating cell-free tumor DNA from blood. Clin. Epigenetics 2023, 15, 151. [Google Scholar] [CrossRef]
- Alcaide, M.; Cheung, M.; Hillman, J.; Rassekh, S.R.; Deyell, R.J.; Batist, G.; Karsan, A.; Wyatt, A.W.; Johnson, N.; Scott, D.W.; et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci. Rep. 2020, 10, 12564. [Google Scholar] [CrossRef]
- Guemri, J.; Pierre-Jean, M.; Brohard, S.; Oussada, N.; Horgues, C.; Bonnet, E.; Mauger, F.; Deleuze, J.F. Methylated ccfDNA from plasma biomarkers of Alzheimer’s disease using targeted bisulfite sequencing. Epigenomics 2022, 14, 451–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shen, Q.; Xu, S.; Yu, H.; Pei, S.; Zhang, Y.; He, X.; Wang, Q.; Li, D. 5-Hydroxymethylcytosine Signatures in Circulating Cell-Free DNA as Diagnostic Biomarkers for Late-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2022, 85, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Radhakrishna, U.; Gordevicius, J.; Aydas, B.; Yilmaz, A.; Jafar, F.; Imam, K.; Maddens, M.; Challapalli, K.; Metpally, R.P.; et al. Artificial Intelligence and Circulating Cell-Free DNA Methylation Profiling: Mechanism and Detection of Alzheimer’s Disease. Cells 2022, 11, 1744. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Turkoglu, O.; Graham, S.F.; Radhakrishna, U. Alzheimer’s Precision Neurology: Epigenetics of Cytochrome P450 Genes in Circulating Cell-Free DNA for Disease Prediction and Mechanism. Int. J. Mol. Sci. 2023, 24, 2876. [Google Scholar] [CrossRef]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef]
- Konki, M.; Lindgren, N.; Kylaniemi, M.; Venho, R.; Laajala, E.; Ghimire, B.; Lahesmaa, R.; Kaprio, J.; Rinne, J.O.; Lund, R.J. Plasma cell-free DNA methylation marks for episodic memory impairment: A pilot twin study. Sci. Rep. 2020, 10, 14192. [Google Scholar] [CrossRef]
- Huang, C.Y.; Rasband, M.N. Axon initial segments: Structure, function, and disease. Ann. N. Y. Acad. Sci. 2018, 1420, 46–61. [Google Scholar] [CrossRef]
- Grubb, M.S.; Burrone, J. Building and maintaining the axon initial segment. Curr. Opin. Neurobiol. 2010, 20, 481–488. [Google Scholar] [CrossRef]
- Sanchez-Mut, J.V.; Aso, E.; Panayotis, N.; Lott, I.; Dierssen, M.; Rabano, A.; Urdinguio, R.G.; Fernandez, A.F.; Astudillo, A.; Martin-Subero, J.I.; et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 2013, 136, 3018–3027. [Google Scholar] [CrossRef]
- Zhu, D.; Li, C.; Swanson, A.M.; Villalba, R.M.; Guo, J.; Zhang, Z.; Matheny, S.; Murakami, T.; Stephenson, J.R.; Daniel, S.; et al. BAI1 regulates spatial learning and synaptic plasticity in the hippocampus. J. Clin. Investig. 2015, 125, 1497–1508. [Google Scholar] [CrossRef]
- Foraker, J.; Millard, S.P.; Leong, L.; Thomson, Z.; Chen, S.; Keene, C.D.; Bekris, L.M.; Yu, C.E. The APOE Gene is Differentially Methylated in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Panitch, R.; Sahelijo, N.; Hu, J.; Nho, K.; Bennett, D.A.; Lunetta, K.L.; Au, R.; Stein, T.D.; Farrer, L.A.; Jun, G.R. APOE genotype-specific methylation patterns are linked to Alzheimer disease pathology and estrogen response. Transl. Psychiatry 2024, 14, 129. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Zhuang, H.; Crain, B.; Doré, S. Expression and localization of prostaglandin transporter in Alzheimer disease brains and age-matched controls. J. Neuroimmunol. 2008, 195, 81–87. [Google Scholar] [CrossRef]
- Gagliardi, M.; Strazzullo, M.; Matarazzo, M.R. DNMT3B Functions: Novel Insights From Human Disease. Front. Cell Dev. Biol. 2018, 6, 140. [Google Scholar] [CrossRef]
- Rajendran, G.; Shanmuganandam, K.; Bendre, A.; Muzumdar, D.; Goel, A.; Shiras, A. Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J. Neurooncol. 2011, 104, 483–494. [Google Scholar] [CrossRef]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G.H. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef]
- de Bem, C.M.; Pezzi, J.C.; Borba, E.M.; Chaves, M.L.; de Andrade, F.M.; Fiegenbaum, M.; Camozzato, A. The synergistic risk effect of apolipoprotein epsilon4 and DNA (cytosine-5-)-methyltransferase 3 beta (DNMT3B) haplotype for Alzheimer’s disease. Mol. Biol. Rep. 2016, 43, 653–658. [Google Scholar] [CrossRef]
- Tannorella, P.; Stoccoro, A.; Tognoni, G.; Petrozzi, L.; Salluzzo, M.G.; Ragalmuto, A.; Siciliano, G.; Haslberger, A.; Bosco, P.; Bonuccelli, U.; et al. Methylation analysis of multiple genes in blood DNA of Alzheimer’s disease and healthy individuals. Neurosci. Lett. 2015, 600, 143–147. [Google Scholar] [CrossRef]
- Zeng, Q.; Chen, X.; Ning, C.; Zhu, Q.; Yao, Y.; Zhao, Y.; Luan, F. Methylation of the genes ROD1, NLRC5, and HKR1 is associated with aging in Hainan centenarians. BMC Med. Genom. 2018, 11, 7. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef] [PubMed]
- Olmedillas-López, S.; Olivera-Salazar, R.; García-Arranz, M.; García-Olmo, D. Current and Emerging Applications of Droplet Digital PCR in Oncology: An Updated Review. Mol. Diagn. Ther. 2022, 26, 61–87. [Google Scholar] [CrossRef] [PubMed]
- Nidadavolu, L.S.; Feger, D.; Wu, Y.; Grodstein, F.; Gross, A.L.; Bennett, D.A.; Walston, J.D.; Oh, E.S.; Abadir, P.M. Circulating Cell-Free Genomic DNA Is Associated with an Increased Risk of Dementia and with Change in Cognitive and Physical Function. J. Alzheimers Dis. 2022, 89, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Wenz, H.M.; Dailey, D.; Johnson, M.D. Development of a high-throughput capillary electrophoresis protocol for DNA fragment analysis. Methods Mol. Biol. 2001, 163, 3–17. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Reisberg, B.; Ferris, S.H.; de Leon, M.J.; Crook, T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am. J. Psychiatry 1982, 139, 1136–1139. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Stevenson, M.; Nunes, T.; Sanchez, J.; Thornton, R.; Reiczigel, J.; Robison-Cox, J.; Sebastiani, P. EpiR: An R Package for the Analysis of Epidemiological Data. 2013. Available online: https://www.researchgate.net/publication/303185003_EpiR_An_R_package_for_the_analysis_of_epidemiological_data (accessed on 2 April 2025).
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017, 45, e22. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Triche, T.J., Jr.; Laird, P.W.; Shen, H. SeSAMe: Reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018, 46, e123. [Google Scholar] [CrossRef] [PubMed]
- Triche, T.J., Jr.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef]
- Andrews, S.V.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Fallin, M.D. “Gap hunting” to characterize clustered probe signals in Illumina methylation array data. Epigenetics Chromatin 2016, 9, 56. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Xu, Z.; Niu, L.; Li, L.; Taylor, J.A. ENmix: A novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016, 44, e20. [Google Scholar] [CrossRef]
- Salas, L.A.; Koestler, D.C.; Butler, R.A.; Hansen, H.M.; Wiencke, J.K.; Kelsey, K.T.; Christensen, B.C. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol. 2018, 19, 64. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; Schwartz, D.A.; Yang, I.V.; Kechris, K.J. Comb-p: Software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 2012, 28, 2986–2988. [Google Scholar] [CrossRef]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef]
- Kumaki, Y.; Oda, M.; Okano, M. QUMA: Quantification tool for methylation analysis. Nucleic Acids Res. 2008, 36, W170–W175. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Phenotypical Features | Controls (n = 35) | Patients with AD (n = 35) | p-Value |
|---|---|---|---|
| Median (IQR) | |||
| Age (years) | 77 (72–80) | 79 (76–83) | 0.213 |
| MMSE | 30 (29–30) | 22 (19–26) | 0.000 |
| GDS | 1 (1–1) | 4 (4–4) | 0.000 |
| cfDNA amount (ng) | 96 (47–212) | 81 (34–241) | 0.445 |
| N (%) | |||
| Gender | 0.811 | ||
| Female | 17 (49) | 18 (51) | |
| Male | 18 (51) | 17 (49) | |
| APOE genotype | 0.001 | ||
| ε4 non-carriers | 31 (89) | 15 (43) | |
| ε4 carriers | 3 (9) | 20 (57) | |
| pTau181 (pg/mL) | 1.5 (1.2–1.8) | 3.0 (2.1–3.9) | 0.000 |
| DMP | Genomic Coordinates | Gene ID | Relation to CpG Context | Relation to Gene Structure | p-Value | β-Difference | |
|---|---|---|---|---|---|---|---|
| cg26023019 | chr21 | 31311859 | GRIK1 | Island | 1stExon | 0.013 | 0.155 |
| cg19665696 | chr7 | 949154 | ADAP1 | Island | Body | 0.021 | 0.151 |
| cg25069157 | chr6 | 44102572 | TMEM63B | OpenSea | Body | 0.040 | 0.148 |
| cg13578160 | chr7 | 72813978 | OpenSea | 0.007 | 0.139 | ||
| cg11955641 | chr5 | 151304999 | GLRA1 | S_Shore | TSS1500 | 0.001 | 0.139 |
| cg09465533 | chr3 | 32327675 | CMTM8 | OpenSea | Body | 0.001 | 0.137 |
| cg20601028 | chr20 | 22738632 | OpenSea | 0.006 | 0.133 | ||
| cg24245216 | chr19 | 7004657 | OpenSea | 0.044 | 0.130 | ||
| cg23506049 | chr12 | 103228185 | OpenSea | 0.039 | 0.129 | ||
| cg21550804 | chr8 | 74282865 | OpenSea | 0.035 | 0.125 | ||
| cg22597210 | chr19 | 10172841 | C3P1 | Island | Body | 0.008 | 0.125 |
| cg17857094 | chr6 | 30907280 | DPCR1 | OpenSea | TSS1500 | 0.029 | 0.119 |
| cg00055434 | chr1 | 2415344 | PLCH2 | Island | Body | 0.006 | 0.117 |
| cg27416647 | chr15 | 96630572 | OpenSea | 0.027 | 0.117 | ||
| cg11646124 | chr1 | 182140416 | OpenSea | 0.008 | 0.113 | ||
| cg22238209 | chr19 | 35800743 | MAG | Island | Body | 0.017 | 0.113 |
| cg07983614 | chr16 | 84587903 | OpenSea | 0.024 | 0.111 | ||
| cg21764456 | chr16 | 10777077 | TEKT5 | OpenSea | Body | 0.037 | 0.110 |
| cg07812827 | chr8 | 74282708 | OpenSea | 0.010 | 0.110 | ||
| cg06572225 | chr11 | 7748353 | OpenSea | 0.021 | 0.110 | ||
| cg03463818 | chr8 | 94766468 | TMEM67 | N_Shore | TSS1500 | 0.034 | 0.108 |
| cg26802564 | chr1 | 30446406 | OpenSea | 0.006 | 0.107 | ||
| cg18056749 | chr20 | 55836268 | BMP7 | N_Shelf | Body | 0.043 | 0.106 |
| cg27454064 | chr12 | 64215611 | Island | 0.017 | 0.105 | ||
| cg24699005 | chr19 | 1192342 | N_Shelf | 0.022 | 0.104 | ||
| cg26861034 | chr22 | 26908874 | TFIP11 | S_Shore | TSS1500 | 0.002 | 0.104 |
| cg10411590 | chr13 | 21900810 | S_Shore | 0.032 | 0.102 | ||
| cg00796424 | chr12 | 54365966 | HOXC11 | N_Shore | TSS1500 | 0.004 | 0.101 |
| cg12906062 | chr13 | 105462162 | OpenSea | 0.042 | −0.100 | ||
| cg00242341 | chr11 | 72447419 | ARAP1 | OpenSea | 5′UTR | 0.039 | −0.100 |
| cg18955367 | chr19 | 49002338 | LMTK3 | Island | Body | 0.030 | −0.100 |
| cg26003334 | chr7 | 100661866 | LOC102724094 | OpenSea | TSS1500 | 0.004 | −0.101 |
| cg16419584 | chr10 | 129947858 | Island | 0.027 | −0.101 | ||
| cg13063165 | chr15 | 79093076 | ADAMTS7 | S_Shore | Body | 0.017 | −0.101 |
| cg01495416 | chr8 | 59085270 | OpenSea | 0.027 | −0.101 | ||
| cg02258724 | chr19 | 53832577 | Island | 0.020 | −0.101 | ||
| cg04772328 | chr2 | 170549930 | C2orf77 | N_Shore | Body | 0.002 | −0.101 |
| cg16045681 | chr1 | 31575570 | OpenSea | 0.002 | −0.102 | ||
| cg21210642 | chr9 | 100881995 | TRIM14 | S_Shore | TSS1500 | 0.008 | −0.102 |
| cg24463437 | chr1 | 28396758 | EYA3 | OpenSea | Body | 0.001 | −0.102 |
| cg20212912 | chr4 | 147557774 | N_Shore | 0.007 | −0.102 | ||
| cg06311780 | chr18 | 6633544 | OpenSea | 0.012 | −0.103 | ||
| cg21010821 | chr11 | 111782679 | HSPB2 | OpenSea | TSS1500 | 0.004 | −0.103 |
| cg14891200 | chr2 | 220197664 | RESP18 | S_Shore | Body | 0.001 | −0.103 |
| cg14520947 | chr1 | 225942842 | OpenSea | 0.003 | −0.103 | ||
| cg15086439 | chr1 | 236563070 | EDARADD | S_Shelf | Body | 0.040 | −0.103 |
| cg04855678 | chr3 | 195946921 | OSTalpha | OpenSea | Body | 0.010 | −0.104 |
| cg19146301 | chr1 | 235100790 | LOC101927851 | OpenSea | TSS1500 | 0.004 | −0.104 |
| cg13443570 | chr8 | 99098126 | ERICH5 | OpenSea | Body | 0.042 | −0.105 |
| cg02784823 | chr19 | 49000897 | LMTK3 | Island | Body | 0.041 | −0.105 |
| cg06398054 | chr1 | 53092881 | OpenSea | 0.019 | −0.106 | ||
| cg20062681 | chr11 | 94988642 | OpenSea | 0.001 | −0.106 | ||
| cg09484559 | chr17 | 12692246 | RICH2 | N_Shore | TSS1500 | 0.018 | −0.106 |
| cg08986575 | chr5 | 173235445 | OpenSea | 0.049 | −0.106 | ||
| cg27159720 | chr9 | 7971612 | OpenSea | 0.002 | −0.106 | ||
| cg07870920 | chr4 | 121569769 | OpenSea | 0.049 | −0.106 | ||
| cg02938172 | chr17 | 185152 | RPH3AL | Island | 5′UTR | 0.021 | −0.106 |
| cg27087112 | chr2 | 114737475 | LOC100499194 | Island | Body | 0.000 | −0.107 |
| cg03174228 | chr9 | 124658583 | TTLL11 | N_Shore | Body | 0.010 | −0.107 |
| cg24984452 | chr1 | 95261186 | LINC01057 | OpenSea | Body | 0.035 | −0.107 |
| cg17566325 | chr12 | 133022423 | N_Shore | 0.006 | −0.107 | ||
| cg03465894 | chr11 | 106342311 | OpenSea | 0.022 | −0.108 | ||
| cg15410835 | chr8 | 143125637 | OpenSea | 0.001 | −0.108 | ||
| cg24760557 | chr10 | 31986724 | OpenSea | 0.016 | −0.108 | ||
| cg18625538 | chr6 | 87832609 | Island | 0.045 | −0.109 | ||
| cg21933626 | chr3 | 123026636 | ADCY5 | OpenSea | Body | 0.004 | −0.109 |
| cg05800368 | chr9 | 124658957 | TTLL11 | Island | Body | 0.036 | −0.109 |
| cg02774630 | chr2 | 154727554 | GALNT13 | N_Shore | TSS1500 | 0.002 | −0.109 |
| cg06878111 | chr10 | 9999498 | OpenSea | 0.003 | −0.110 | ||
| cg23213894 | chr11 | 7691961 | CYB5R2 | N_Shelf | Body | 0.010 | −0.111 |
| cg06957053 | chr7 | 137533035 | DGKI | S_Shore | TSS1500 | 0.024 | −0.111 |
| cg10531073 | chr22 | 38485757 | BAIAP2L2 | S_Shore | Body | 0.005 | −0.112 |
| cg24104237 | chr3 | 45649408 | LIMD1 | OpenSea | Body | 0.010 | −0.112 |
| cg10289324 | chr18 | 60710970 | OpenSea | 0.007 | −0.113 | ||
| cg15243027 | chr2 | 32784469 | BIRC6-AS2 | OpenSea | Body | 0.028 | −0.113 |
| cg10092377 | chr1 | 200880981 | C1orf106 | Island | Body | 0.001 | −0.117 |
| cg01583753 | chr2 | 39470725 | N_Shore | 0.018 | −0.117 | ||
| cg16127514 | chr10 | 29273678 | OpenSea | 0.042 | −0.117 | ||
| cg26496930 | chr14 | 70186565 | OpenSea | 0.041 | −0.117 | ||
| cg10305928 | chr10 | 62426219 | ANK3 | OpenSea | Body | 0.043 | −0.117 |
| cg14310109 | chr6 | 157297510 | ARID1B | OpenSea | Body | 0.005 | −0.119 |
| cg16520701 | chr8 | 34606956 | OpenSea | 0.010 | −0.120 | ||
| cg16210088 | chr22 | 31318349 | C22orf27 | Island | Body | 0.009 | −0.122 |
| cg24448113 | chr5 | 140475611 | PCDHB2 | Island | 1stExon | 0.013 | −0.124 |
| cg06862049 | chr19 | 49001890 | LMTK3 | Island | Body | 0.023 | −0.125 |
| cg12172631 | chr19 | 54584915 | TARM1 | OpenSea | TSS1500 | 0.021 | −0.126 |
| cg20548231 | chr22 | 31318373 | C22orf27 | Island | Body | 0.001 | −0.127 |
| cg09544050 | chr8 | 143580965 | BAI1 | Island | Body | 0.003 | −0.128 |
| cg18815398 | chr20 | 61506981 | N_Shore | 0.000 | −0.128 | ||
| cg26651782 | chr19 | 51505779 | KLK9 | N_Shore | 3′UTR | 0.005 | −0.128 |
| cg24061197 | chr2 | 220108496 | GLB1L | S_Shore | 5′UTR | 0.043 | −0.128 |
| cg06260707 | chr1 | 42945689 | OpenSea | 0.022 | −0.128 | ||
| cg15059639 | chr2 | 171220061 | MYO3B | OpenSea | Body | 0.022 | −0.128 |
| cg24658778 | chr6 | 152897280 | SYNE1 | OpenSea | Body | 0.013 | −0.130 |
| cg20920357 | chr4 | 116877727 | OpenSea | 0.002 | −0.133 | ||
| cg02256650 | chr22 | 31317287 | MORC2-AS1 | N_Shore | TSS1500 | 0.013 | −0.133 |
| cg26140120 | chr8 | 124219575 | FAM83A | Island | Body | 0.014 | −0.135 |
| cg06452258 | chr2 | 60597809 | OpenSea | 0.049 | −0.136 | ||
| cg08431893 | chr21 | 44864600 | Island | 0.004 | −0.139 | ||
| cg16467921 | chr8 | 128801108 | OpenSea | 0.004 | −0.139 | ||
| cg04248279 | chr17 | 184833 | RPH3AL | N_Shore | 5′UTR | 0.007 | −0.142 |
| cg24135491 | chr17 | 4487099 | SMTNL2 | N_Shore | TSS200 | 0.015 | −0.147 |
| No | DMP | β-Difference | Gene ID | MMSE | GDS | Aβ42 | Aβ40 | Ratio Aβ42/Aβ40 | pTau | t-Tau | Plasma pTau181 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | cg26023019 | 0.155 | GRIK1 | −0.155 | 0.224 | −0.044 | −0.032 | −0.102 | 0.290 | 0.096 | 0.148 |
| 2 | cg19665696 | 0.151 | ADAP1 | −0.204 | 0.328 ** | −0.180 | 0.093 | −0.549 * | 0.075 | −0.162 | 0.329 * |
| 3 | cg25069157 | 0.148 | TMEM63B | −0.242 | −0.204 | 0.156 | 0.071 | 0.039 | −0.164 | 0.048 | 0.378 ** |
| 4 | cg13578160 | 0.139 | −0.336 * | 0.338 ** | −0.050 | −0.003 | 0.086 | −0.050 | 0.033 | 0.117 | |
| 5 | cg11955641 | 0.139 | GLRA1 | −0.278 * | 0.452 ** | 0.275 | 0.444 + | −0.081 | 0.403 + | 0.268 | 0.327 * |
| 6 | cg09465533 | 0.137 | CMTM8 | −0.304 * | 0.413 ** | −0.331 | −0.233 | −0.149 | 0.107 | −0.117 | 0.141 |
| 7 | cg20601028 | 0.133 | −0.317 * | 0.352 ** | −0.036 | −0.024 | −0.116 | 0.221 | −0.053 | 0.330 * | |
| 8 | cg24245216 | 0.130 | −0.349 ** | 0.273 * | −0.353 | −0.659 ** | 0.332 | −0.427 + | −0.513 * | 0.229 + | |
| 9 | cg23506049 | 0.129 | −0.502 ** | 0.373 ** | 0.177 | −0.029 | 0.159 | −0.117 | −0.313 | 0.199 | |
| 10 | cg21550804 | 0.125 | −0.167 | 0.137 | 0.174 | 0.093 | 0.302 | 0.302 | 0.347 | 0.283 * |
| No | DMP | β-Difference | Gene ID | MMSE | GDS | Aβ42 | Aβ40 | Ratio Aβ42/Aβ40 | pTau | t-Tau | Plasma pTau181 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | cg24135491 | −0.147 | SMTNL2 | 0.349 ** | −0.463 ** | −0.302 | −0.205 | −0.092 | −0.155 | −0.308 | −0.357 ** |
| 2 | cg04248279 | −0.142 | RPH3AL | 0.202 | −0.233 | 0.188 | −0.008 | 0.176 | −0.326 | −0.317 | −0.361 ** |
| 3 | cg16467921 | −0.139 | 0.258 | −0.346 ** | −0.048 | 0.092 | −0.448 * | 0.301 | 0.134 | −0.227 + | |
| 4 | cg08431893 | −0.139 | 0.278 * | −0.338 ** | −0.170 | −0.198 | 0.041 | −0.119 | −0.122 | −0.265 + | |
| 5 | cg06452258 | −0.136 | 0.341 * | 0.430 ** | −0.084 | 0.039 | −0.101 | 0.039 | 0.122 | −0.231 + | |
| 6 | cg26140120 | −0.135 | FAM83A | 0.517 ** | −0.373 ** | 0.135 | 0.092 | 0.156 | 0.032 | 0.233 | −0.217 |
| 7 | cg02256650 | −0.133 | MORC2-AS1 | 0.299 * | −0.409 ** | −0.092 | −0.026 | 0.080 | 0.036 | 0.189 | −0.253 + |
| 8 | cg20920357 | −0.133 | 0.040 | −0.312 * | 0.397 + | 0.365 | 0.165 | 0.071 | 0.095 | −0.194 | |
| 9 | cg24658778 | −0.130 | SYNE1 | 0.311 * | −0.296 * | −0.003 | 0.185 | −0.096 | 0.253 | 0.098 | −0.159 |
| 10 | cg15059639 | −0.128 | MYO3B | 0.203 | −0.252 * | 0.092 | 0.173 | 0.003 | 0.105 | 0.021 | −0.223 + |
| Genomic Location | FDR | Sidak | CpGs in DMR | Gene ID | Relation to CpG Context | Relation to Gene Structure | β-Difference | ||
|---|---|---|---|---|---|---|---|---|---|
| chr8 | 69243284 | 69243293 | 2.41099 × 10−6 | 0.021 | cg06125462; cg03357798; cg19469068 | C8orf34; C8orf34-AS1 | Island | 1stExon; Intergenic | 0.038 |
| chr11 | 89867808 | 89868104 | 6.55924 × 10−6 | 0.004 | cg14304817; cg09584827; cg12403137; cg05236757; cg05500015; cg21500966 | NAALAD2 | OpenSea | 5′UTR; TSS200; Body; 1stExon | 0.013 |
| chr19 | 55972854 | 55973234 | 2.06981× 10−5 | 0.026 | cg12195369; cg02394686; cg23051792; cg18297529; cg19384825;cg13475732 | ISOC2 | Island | 5′UTR; TSS200 | −0.004 |
| chr2 | 168366268 | 168366363 | 7.3012 × 10−6 | 0.021 | cg19288676; cg18683711 | OpenSea | Intergenic | −0.016 | |
| chr19 | 1854548 | 1854819 | 6.55924 × 10−6 | 0.005 | cg08287334; cg22672431; cg08525314; cg01373896 | KLF16 | Island | Body | −0.026 |
| chr6 | 116381903 | 116382179 | 3.57858 × 10−6 | 0.002 | cg15226275; cg05304507; cg26893134; cg26557270; cg18764771 | FRK | OpenSea | TSS200; 1stExon; TSS1500 | −0.027 |
| chr5 | 1316037 | 1316264 | 8.74148 × 10−6 | 0.017 | cg11624060; cg26209169 | OpenSea | Intergenic | −0.033 | |
| chr1 | 155007165 | 155007254 | 8.18998 × 10−6 | 0.038 | cg08472142; cg21596858 | DCST1;DCST2 | OpenSea | Body | −0.038 |
| chr5 | 178692690 | 178692806 | 8.18998 × 10−6 | 0.028 | cg06495631; cg01231141; cg10213542 | ADAMTS2 | OpenSea | Body | −0.040 |
| chr6 | 30103458 | 30103699 | 7.53684 × 10−6 | 0.010 | cg12758147; cg12612406; cg13044052 | TRIM40 | OpenSea | TSS1500 | −0.044 |
| chr20 | 31366408 | 31366486 | 8.18998 × 10−6 | 0.040 | cg00300969;cg09135144; cg24403338; cg17475857 | DNMT3B | OpenSea | 5′UTR; TSS1500 | −0.046 |
| chr11 | 72533201 | 72533487 | 8.18998 × 10−6 | 0.011 | cg13771313; cg24878173; cg04006327 | ATG16L2 | Island | Body | −0.057 |
| chr19 | 7983876 | 7984171 | 1.60512 × 10−7 | 0.000 | cg26284544; cg10073052; cg00654322; cg03685315; cg03840143 | SNAPC2 | Island; S_Shore | TSS1500 | −0.058 |
| chr10 | 123070326 | 123070392 | 7.53684 × 10−6 | 0.036 | cg16273546;cg21380925 | OpenSea | Intergenic | −0.064 | |
| chr6 | 30419490 | 30419576 | 1.45527 × 10−15 | 0.000 | cg26570901; cg26715559; cg12078775; cg27572120;cg11491998 | Island | Intergenic | −0.074 | |
| chr5 | 1867977 | 1868261 | 8.18998 × 10−6 | 0.012 | cg15595755; cg04156016; cg14773178 | OpenSea | Intergenic | −0.084 | |
| chr2 | 220108093 | 220108496 | 2.16849 × 10−6 | 0.000 | cg20314884; cg04945312; cg10602248; cg02258512; cg24061197; cg09715285 | GLB1L | Island; S_Shore | Body; 5′UTR; 1stExon | −0.095 |
| Genomic Location | FDR | Sidak | CpGs in DMR | Gene ID | Relation to CpG Context | Relation to Gene Structure | β-Difference | ||
|---|---|---|---|---|---|---|---|---|---|
| chr13 | 21900391 | 21900591 | 2.318 × 10−6 | 4.081 × 10−3 | cg13903179; cg19500098; cg00035636; cg04632378 | Island | Intergenic | 0.100 | |
| chr17 | 45949676 | 45949878 | 2.318 × 10−6 | 4.887 × 10−3 | cg01135546; cg09065876; cg16913064; cg23008871; cg05123976 | Island | Intergenic | 0.037 | |
| chr19 | 37825210 | 37825679 | 1.743 × 10−8 | 7.935 × 10−6 | cg26734888; cg23756236; cg12948621; cg12024906; cg08565796; cg24834889; cg14166009; cg10237978; cg05280698; cg13687570 | HKR1 | Island | TSS1500; TSS200; 1stExon | 0.021 |
| chr3 | 133748504 | 133748812 | 4.661 × 10−10 | 1.615 × 10−7 | cg19591206; cg12118082; cg26556923; cg02496728 | SLCO2A1 | Island | 1stExon; Body | 0.010 |
| Gene ID | Sample Source | Technique | AD Methylome Study |
|---|---|---|---|
| HKR1 | Hippocampus | Infinium HumanMethylation450 BeadChip | PMID: 31217032 |
| Prefrontal cortex | Infinium MethylationEPIC BeadChip | PMID: 33257653 | |
| Blood | Infinium HumanMethylation450 BeadChip | PMID: 29394898 | |
| ZNF154 | Blood | Infinium HumanMethylation450 BeadChip | PMID: 31775875 |
| HOXA5 | Superior temporal gyrus and inferior frontal gyrus | Infinium MethylationEPIC BeadChip | PMID: 33069246 |
| Prefrontal cortex | Infinium HumanMethylation450 BeadChip | PMID: 33902726 | |
| Superior temporal gyrus | Infinium HumanMethylation450 BeadChip | PMID: 29550519 | |
| TRIM40 | Superior temporal gyrus | Infinium HumanMethylation450 BeadChip | PMID: 26803900 |
| Prefrontal cortex | Infinium MethylationEPIC BeadChip | PMID: 33257653 | |
| ATG16L2 | Hippocampus | Infinium HumanMethylation450 BeadChip | PMID: 31217032 |
| Dorsolateral prefrontal cortex | Infinium HumanMethylation450 BeadChip | PMID: 25129075 | |
| Prefrontal cortex | Infinium HumanMethylation450 BeadChip | PMID: 33902726 | |
| Prefrontal cortex | Infinium MethylationEPIC BeadChip | PMID: 33257653 | |
| ADAMST2 | Prefrontal cortex | Infinium HumanMethylation450 BeadChip | PMID: 35982059 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macías, M.; Alba-Linares, J.J.; Acha, B.; Blanco-Luquin, I.; Fernández, A.F.; Álvarez-Jiménez, J.; Urdánoz-Casado, A.; Roldan, M.; Robles, M.; Cabezon-Arteta, E.; et al. Advancing Personalized Medicine in Alzheimer’s Disease: Liquid Biopsy Epigenomics Unveil APOE ε4-Linked Methylation Signatures. Int. J. Mol. Sci. 2025, 26, 3419. https://doi.org/10.3390/ijms26073419
Macías M, Alba-Linares JJ, Acha B, Blanco-Luquin I, Fernández AF, Álvarez-Jiménez J, Urdánoz-Casado A, Roldan M, Robles M, Cabezon-Arteta E, et al. Advancing Personalized Medicine in Alzheimer’s Disease: Liquid Biopsy Epigenomics Unveil APOE ε4-Linked Methylation Signatures. International Journal of Molecular Sciences. 2025; 26(7):3419. https://doi.org/10.3390/ijms26073419
Chicago/Turabian StyleMacías, Mónica, Juan José Alba-Linares, Blanca Acha, Idoia Blanco-Luquin, Agustín F. Fernández, Johana Álvarez-Jiménez, Amaya Urdánoz-Casado, Miren Roldan, Maitane Robles, Eneko Cabezon-Arteta, and et al. 2025. "Advancing Personalized Medicine in Alzheimer’s Disease: Liquid Biopsy Epigenomics Unveil APOE ε4-Linked Methylation Signatures" International Journal of Molecular Sciences 26, no. 7: 3419. https://doi.org/10.3390/ijms26073419
APA StyleMacías, M., Alba-Linares, J. J., Acha, B., Blanco-Luquin, I., Fernández, A. F., Álvarez-Jiménez, J., Urdánoz-Casado, A., Roldan, M., Robles, M., Cabezon-Arteta, E., Alcolea, D., Gordoa, J. S. R. d., Corroza, J., Cabello, C., Erro, M. E., Jericó, I., Fraga, M. F., & Mendioroz, M. (2025). Advancing Personalized Medicine in Alzheimer’s Disease: Liquid Biopsy Epigenomics Unveil APOE ε4-Linked Methylation Signatures. International Journal of Molecular Sciences, 26(7), 3419. https://doi.org/10.3390/ijms26073419