

Harnessing Nanopore Sequencing to Investigate the Epigenomic Landscape in Molar Incisor Hypomineralization—A Pilot Study

 , , , , , , , and
, , , , , , , and 
Abstract
1. Introduction
2. Results
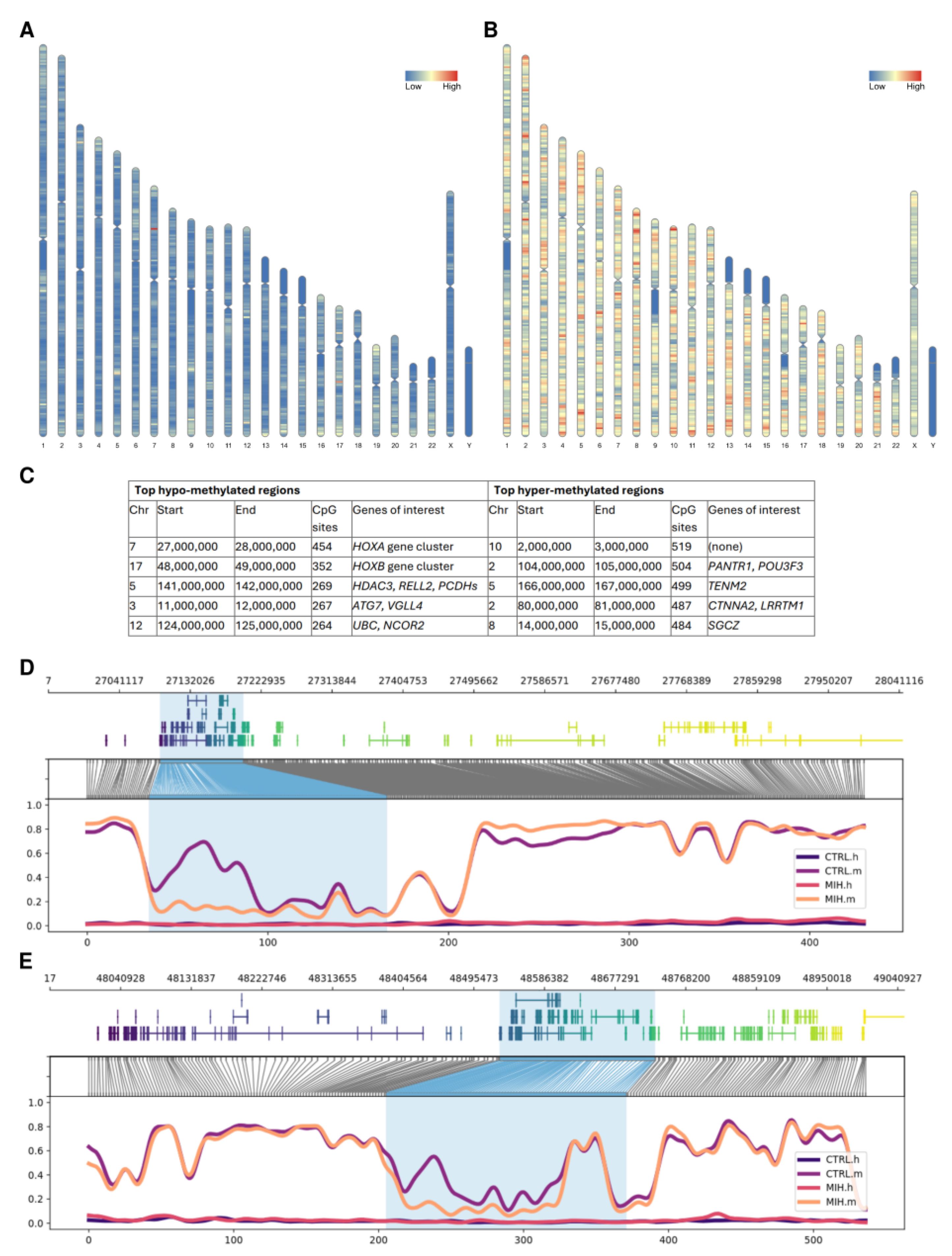
2.1. Genome-Wide Distribution of Methylation Events
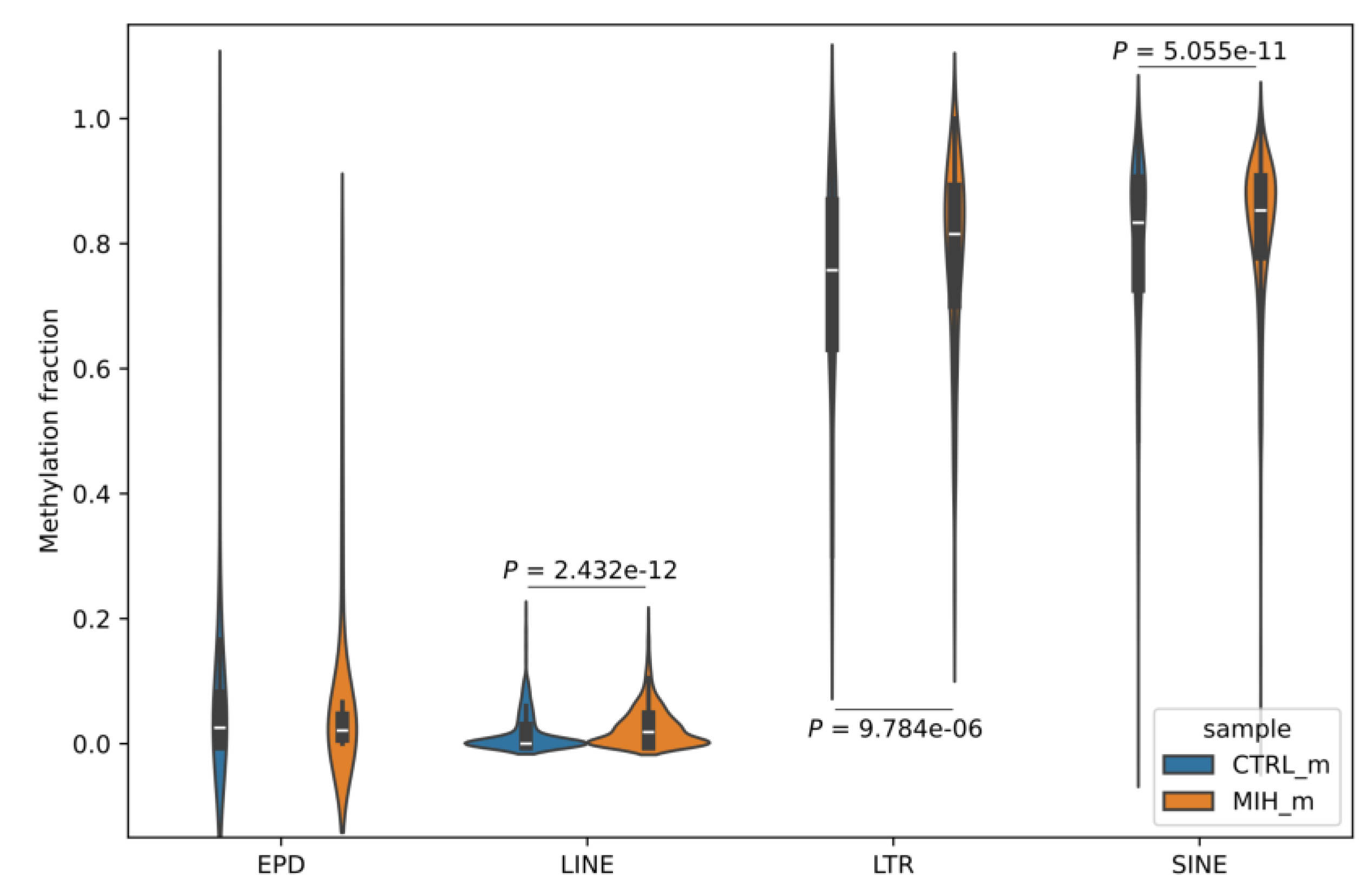
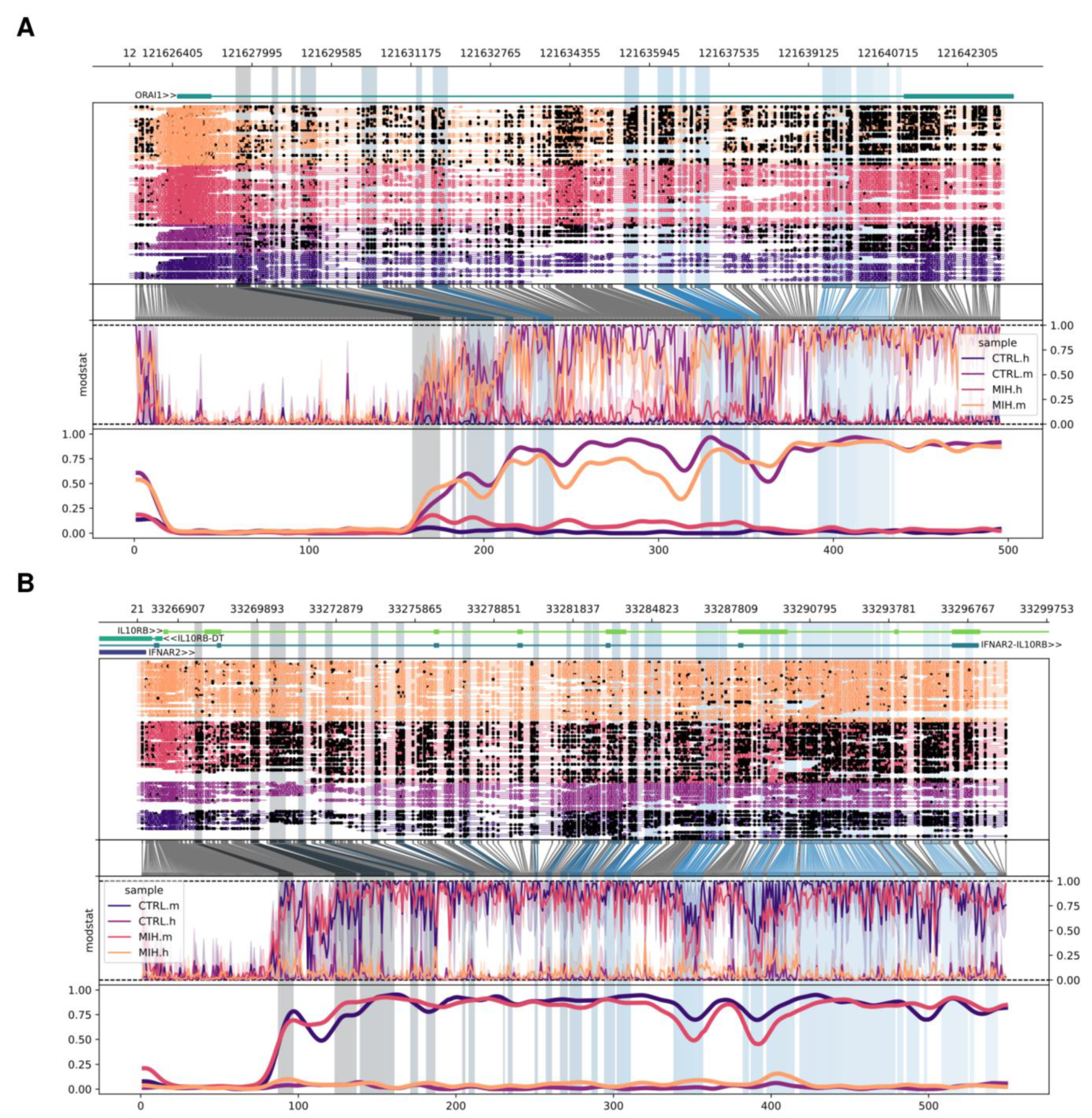
2.2. Differences in Methylation Signal of Promoters and Transposable Elements Within MIH-Associated Genes
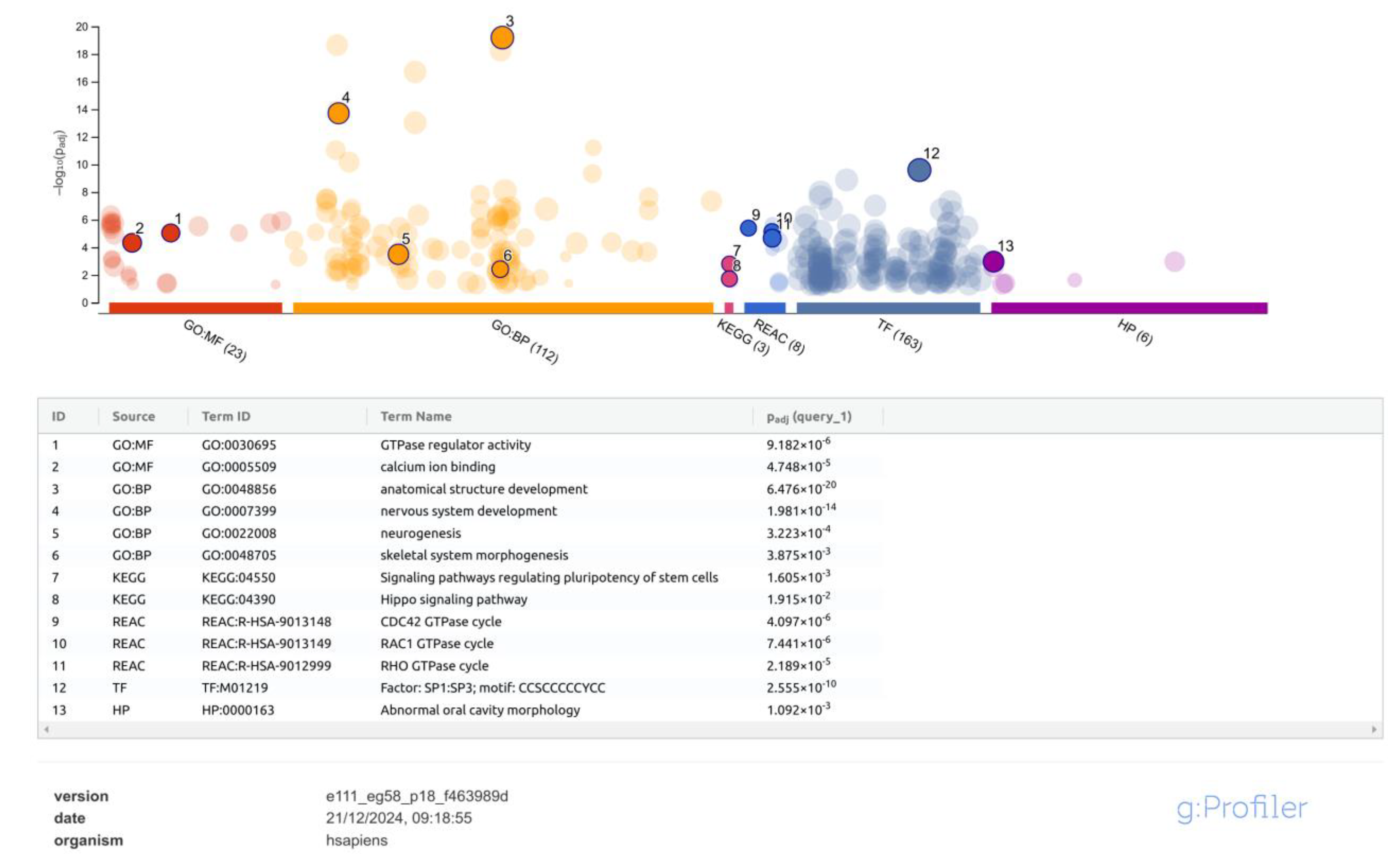
2.3. Identification of Differentially Methylated Regions and Functional Profiling
3. Discussion
4. Materials and Methods
4.1. Ethical Considerations
4.2. Clinical Examination
4.3. Sample Collection
4.4. DNA Isolation from Teeth
4.5. ONT Sequencing
4.6. Bioinformatic Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopes, L.B.; Machado, V.; Mascarenhas, P.; Mendes, J.J.; Botelho, J. The Prevalence of Molar-Incisor Hypomineralization: A Systematic Review and Meta-Analysis. Sci. Rep. 2021, 11, 22405. [Google Scholar] [CrossRef] [PubMed]
- Pentapati, K.C.; Yeturu, S.K.; Siddiq, H. Systematic Review and Meta-analysis of the Prevalence of Molar-incisor Hypomineralization. J. Int. Oral Health 2017, 9, 243–250. [Google Scholar] [CrossRef]
- Weerheijm, K.L.; Duggal, M.; Mejàre, I.; Papagiannoulis, L.; Koch, G.; Martens, L.C.; Hallonsten, A.L. Judgement Criteria for Molar Incisor Hypomineralisation (MIH) in Epidemiologic Studies: A Summary of the European Meeting on MIH Held in Athens, 2003. Eur. J. Paediatr. Dent. 2023, 4, 110–113. [Google Scholar]
- Lygidakis, N.A.; Garot, E.; Somani, C.; Taylor, G.D.; Rouas, P.; Wong, F.S.L. Best Clinical Practice Guidance for Clinicians Dealing with Children Presenting with Molar-Incisor-Hypomineralisation (MIH): An Updated European Academy of Paediatric Dentistry Policy Document. Eur. Arch. Paediatr. Dent. 2022, 23, 3–21. [Google Scholar] [CrossRef]
- Garot, E.; Rouas, P.; Somani, C.; Taylor, G.D.; Wong, F.; Lygidakis, N.A. An Update of the Aetiological Factors Involved in Molar Incisor Hypomineralisation (MIH): A Systematic Review and Meta-Analysis. Eur. Arch. Paediatr. Dent. 2022, 23, 23–38. [Google Scholar] [CrossRef]
- Bekes, K. Molar Incisor Hypomineralization, 1st ed.; Quintessence Publishing: Berlin, Germany, 2022; ISBN 978-1-78698-124-0. [Google Scholar]
- Bezamat, M.; Souza, J.F.; Silva, F.M.F.; Corrêa, E.G.; Fatturi, A.L.; Brancher, J.A.; Carvalho, F.M.; Cavallari, T.; Bertolazo, L.; Machado-Souza, C.; et al. Gene-Environment Interaction in Molar-Incisor Hypomineralization. PLoS ONE 2021, 16, e0241898. [Google Scholar] [CrossRef]
- Silva, M.J.; Scurrah, K.J.; Craig, J.M.; Manton, D.J.; Kilpatrick, N. Etiology of Molar Incisor Hypomineralization—A Systematic Review. Community Dent. Oral Epidemiol. 2016, 44, 342–353. [Google Scholar] [CrossRef]
- da Silva Figueira, R.; Muniz, F.W.M.G.; Costa, L.C.; de Moura, M.S.; de Deys Moura, L.D.F.A.; de Oliveira, B.M.; Lima, C.C.B.; Rösing, C.K.; de Lima, M.D.D.M. Association between Genetic Factors and Molar-Incisor Hypomineralisation or Hypomineralised Second. Primary Molar: A Systematic Review. Arch. Oral Biol. 2023, 152, 105716. [Google Scholar] [CrossRef]
- Tynior, W.; Ilczuk-Rypuła, D.; Hudy, D.; Strzelczyk, J.K. Is Aberrant DNA Methylation a Key Factor in Molar Incisor Hypomineralization? Curr. Issues Mol. Biol. 2022, 44, 2868–2878. [Google Scholar] [CrossRef]
- Bird, A.; Taggart, M.; Frommer, M.; Miller, O.J.; Macleod, D. A Fraction of the Mouse Genome That Is Derived from Islands of Nonmethylated, CpG-Rich DNA. Cell 1985, 40, 91–99. [Google Scholar] [CrossRef]
- Horst, O.V.; Horst, J.A.; Samudrala, R.; Dale, B.A. Caries Induced Cytokine Network in the Odontoblast Layer of Human Teeth. BMC Immunol. 2011, 12, 9. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, Y. DNA Methylation in Human Diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Suryadeva, S.; Begum, M. Role of Homeobox Genes in Tooth Morphogenesis: A Review. J. Clin. Diagn. Res. 2015, 9, ZE09–ZE12. [Google Scholar] [CrossRef] [PubMed]
- Lagosz, K.B.; Bysiek, A.; Macina, J.M.; Bereta, G.P.; Kantorowicz, M.; Lipska, W.; Sochalska, M.; Gawron, K.; Kaczmarzyk, T.; Chomyszyn-Gajewska, M.; et al. HDAC3 Regulates Gingival Fibroblast Inflammatory Responses in Periodontitis. J. Dent. Res. 2020, 99, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulos, G.; Smith, C.E.L.; Brookes, S.J.; El-Asrag, M.E.; Brown, C.J.; Patel, A.; Murillo, G.; O’Connell, M.J.; Inglehearn, C.F.; Mighell, A.J. New Missense Variants in RELT Causing Hypomineralised Amelogenesis Imperfecta. Clin. Genet. 2020, 97, 688–695. [Google Scholar] [CrossRef]
- Heymann, R.; Kallenbach, S.; Alonso, S.; Carroll, P.; Mitsiadis, T.A. Dynamic Expression Patterns of the New Protocadherin Families CNRs and Pcdh-γ during Mouse Odontogenesis: Comparison with Reelin Expression. Mech. Dev. 2001, 106, 181–184. [Google Scholar] [CrossRef]
- El Hajj, N.; Dittrich, M.; Haaf, T. Epigenetic Dysregulation of Protocadherins in Human Disease. Semin. Cell Dev. Biol. 2017, 69, 172–182. [Google Scholar] [CrossRef]
- Sukseree, S.; Schwarze, U.Y.; Gruber, R.; Gruber, F.; Quiles Del Rey, M.; Mancias, J.D.; Bartlett, J.D.; Tschachler, E.; Eckhart, L. ATG7 Is Essential for Secretion of Iron from Ameloblasts and Normal Growth of Murine Incisors during Aging. Autophagy 2020, 16, 1851–1857. [Google Scholar] [CrossRef]
- Silva, M.; Mohandas, N.; Craig, J.; Manton, D.; Saffery, R.; Southey, M.; Burgner, D.; Lucas, J.; Kilpatrick, N.; Hopper, J.; et al. DNA Methylation in Childhood Dental Caries and Hypomineralization. J. Dent. 2022, 117, 103913. [Google Scholar] [CrossRef]
- Donnelly, C.R.; Shah, A.A.; Suh, E.B.; Pierchala, B.A. Ret Signaling Is Required for Tooth Pulp Innervation during Organogenesis. J. Dent. Res. 2019, 98, 705–712. [Google Scholar] [CrossRef]
- Paluvai, H.; Shanmukha, K.D.; Tyedmers, J.; Backs, J. Insights into the Function of HDAC3 and NCoR1/NCoR2 Co-Repressor Complex in Metabolic Diseases. Front. Mol. Biosci. 2023, 10, 1190094. [Google Scholar] [CrossRef]
- Hu, H.; Duan, Y.; Wang, K.; Fu, H.; Liao, Y.; Wang, T.; Zhang, Z.; Kang, F.; Zhang, B.; Zhang, H.; et al. Dental Niche Cells Directly Contribute to Tooth Reconstitution and Morphogenesis. Cell Rep. 2022, 41, 111737. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Li, X.; McEvilly, R.J.; Rosenfeld, M.G.; Lufkin, T.; Rubenstein, J.L.R. Dlx Genes Pattern Mammalian Jaw Primordium by Regulating Both Lower Jaw-Specific and Upper Jaw-Specific Genetic Programs. Development 2008, 135, 2905–2916. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.W.; Shin, M.S.; Ahn, Y.B.; Lee, H.J.; Kim, H.D. Genomewide Association Study on Chronic Periodontitis in Korean Population: Results from the Yangpyeong Health Cohort. J. Clin. Periodontol. 2015, 42, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Marconcini, S.; Denaro, M.; Cosola, S.; Gabriele, M.; Toti, P.; Mijiritsky, E.; Proietti, A.; Basolo, F.; Giammarinaro, E.; Covani, U. Myofibroblast Gene Expression Profile after Tooth Extraction in the Rabbit. Materials 2019, 12, 3697. [Google Scholar] [CrossRef]
- Miyata, K.; Chiba, Y.; Marchelina, T.; Inada, S.; Oka, S.; Saito, K.; Yamada, A.; Fukumoto, S. Single-Cell RNA-Sequence of Dental Epithelium Reveals Responsible Genes of Dental Anomalies in Human. Pediatr. Dent. J. 2023, 33, 102–115. [Google Scholar] [CrossRef]
- Laajala, A.; Pesonen, P.; Alaraudanjoki, V.; Anttonen, V.; Laitala, M.L. Genome-Wide Association Study Identifies Novel Caries-Associated Loci Showing Sex-Specificity-A Study on the Northern Finland Birth Cohort 1966. Eur. J. Oral Sci. 2023, 131, e12953. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, J.; Kim, S.O.; Song, J.S.; Lee, J.H.; Lee, S.I.; Jung, H.S.; Choi, B.J. Comparative Gene-Expression Analysis of the Dental Follicle and Periodontal Ligament in Humans. PLoS ONE 2013, 8, e84201. [Google Scholar] [CrossRef]
- Luo, Y.; Lu, X.; Xie, H. Dynamic Alu Methylation during Normal Development, Aging, and Tumorigenesis. Biomed. Res. Int. 2014, 2014, 784706. [Google Scholar] [CrossRef]
- Gebrie, A. Transposable Elements as Essential Elements in the Control of Gene Expression. Mob. DNA 2023, 14, 9. [Google Scholar] [CrossRef]
- Biz, M.T.; Marques, M.R.; Crema, V.O.; Moriscot, A.S.; Dos Santos, M.F. GTPases RhoA and Rac1 Are Important for Amelogenin and DSPP Expression during Differentiation of Ameloblasts and Odontoblasts. Cell Tissue Res. 2010, 340, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Yu, H.; Zhang, X.; Wang, B.; Yuan, Y.; Zhang, Q.; Ye, R.; Gong, P.; Wu, Y. The Versatile Hippo Pathway in Oral-Maxillofacial Development and Bone Remodeling. Dev. Biol. 2018, 440, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Ni, D. The Hippo Pathway in Oral Diseases and Treatments: A Review. Medicine 2024, 103, e40553. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Yu, S.; Chen, S.; Liu, H.; Chen, Z. SP1 Regulates KLF4 via SP1 Binding Motif Governed by DNA Methylation during Odontoblastic Differentiation of Human Dental Pulp Cells. J. Cell Biochem. 2019, 120, 14688–14699. [Google Scholar] [CrossRef]
- Zhao, M.; Gupta, V.; Raj, L.; Roussel, M.; Bei, M. A Network of Transcription Factors Operates during Early Tooth Morphogenesis. Mol. Cell Biol. 2013, 33, 3099–3112. [Google Scholar] [CrossRef]
- De Coster, W.; Rademakers, R. NanoPack2: Population-Scale Evaluation of Long-Read Sequencing Data. Bioinformatics 2023, 39, btad311. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Hao, Z.; Lv, D.; Ge, Y.; Shi, J.; Weijers, D.; Yu, G.; Chen, J. RIdeogram: Drawing SVG Graphics to Visualize and Map Genome-Wide Data on the Idiograms. PeerJ Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef]
- Cheetham, S.W.; Kindlova, M.; Ewing, A.D. Methylartist: Tools for Visualizing Modified Bases from Nanopore Sequence Data. Bioinformatics 2022, 38, 3109–3112. [Google Scholar] [CrossRef]
- Jühling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S. Metilene: Fast and Sensitive Calling of Differentially Methylated Regions from Bisulfite Sequencing Data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. G:Profiler-Interoperable Web Service for Functional Enrichment Analysis and Gene Identifier Mapping (2023 Update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| Age: 6–16 | Birth defects |
| The condition of the first permanent molar did not allow for conservative and endodontic treatment | Genetic disorders |
| Parent or legal guardian agreement | Cranio-mandibular disorders and developmental defects of tooth (apart from MIH) |
| MIH | Control | ||
|---|---|---|---|
| Gender | female | female | |
| Age | 9 | 15 | |
| MIH TNI Index | 4c | 0 | |
| EAPD Criteria | Demarcated opacities | Present | No |
| Post-eruptive enamel breakdown | Present | No | |
| Atypical restorations | Yes | No | |
| Extractions of molars due to MIH | No | No | |
| Failure of eruption of molar or an incisor | No | No | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salatino, S.; Cuber, P.; Tynior, W.; Gustave, C.; Hudy, D.; Chan, Y.-T.; Raczkowska-Siostrzonek, A.; Misra, R.; Aleksandrowicz, D.; Nałęcz, D.; et al. Harnessing Nanopore Sequencing to Investigate the Epigenomic Landscape in Molar Incisor Hypomineralization—A Pilot Study. Int. J. Mol. Sci. 2025, 26, 3401. https://doi.org/10.3390/ijms26073401
Salatino S, Cuber P, Tynior W, Gustave C, Hudy D, Chan Y-T, Raczkowska-Siostrzonek A, Misra R, Aleksandrowicz D, Nałęcz D, et al. Harnessing Nanopore Sequencing to Investigate the Epigenomic Landscape in Molar Incisor Hypomineralization—A Pilot Study. International Journal of Molecular Sciences. 2025; 26(7):3401. https://doi.org/10.3390/ijms26073401
Chicago/Turabian StyleSalatino, Silvia, Piotr Cuber, Wojciech Tynior, Carla Gustave, Dorota Hudy, Yuen-Ting Chan, Agnieszka Raczkowska-Siostrzonek, Raju Misra, Dagmara Aleksandrowicz, Dariusz Nałęcz, and et al. 2025. "Harnessing Nanopore Sequencing to Investigate the Epigenomic Landscape in Molar Incisor Hypomineralization—A Pilot Study" International Journal of Molecular Sciences 26, no. 7: 3401. https://doi.org/10.3390/ijms26073401
APA StyleSalatino, S., Cuber, P., Tynior, W., Gustave, C., Hudy, D., Chan, Y.-T., Raczkowska-Siostrzonek, A., Misra, R., Aleksandrowicz, D., Nałęcz, D., & Strzelczyk, J. K. (2025). Harnessing Nanopore Sequencing to Investigate the Epigenomic Landscape in Molar Incisor Hypomineralization—A Pilot Study. International Journal of Molecular Sciences, 26(7), 3401. https://doi.org/10.3390/ijms26073401