
The Clinical Outcomes Among Patients Under 60 Years Old with Lynch Syndrome: Variations Based on Different Mutation Patterns

 ,
,  ,
,
Abstract
1. Introduction
2. Results
3. Discussion
3.1. Summary of Evidence
3.2. Limitations
4. Materials and Methods
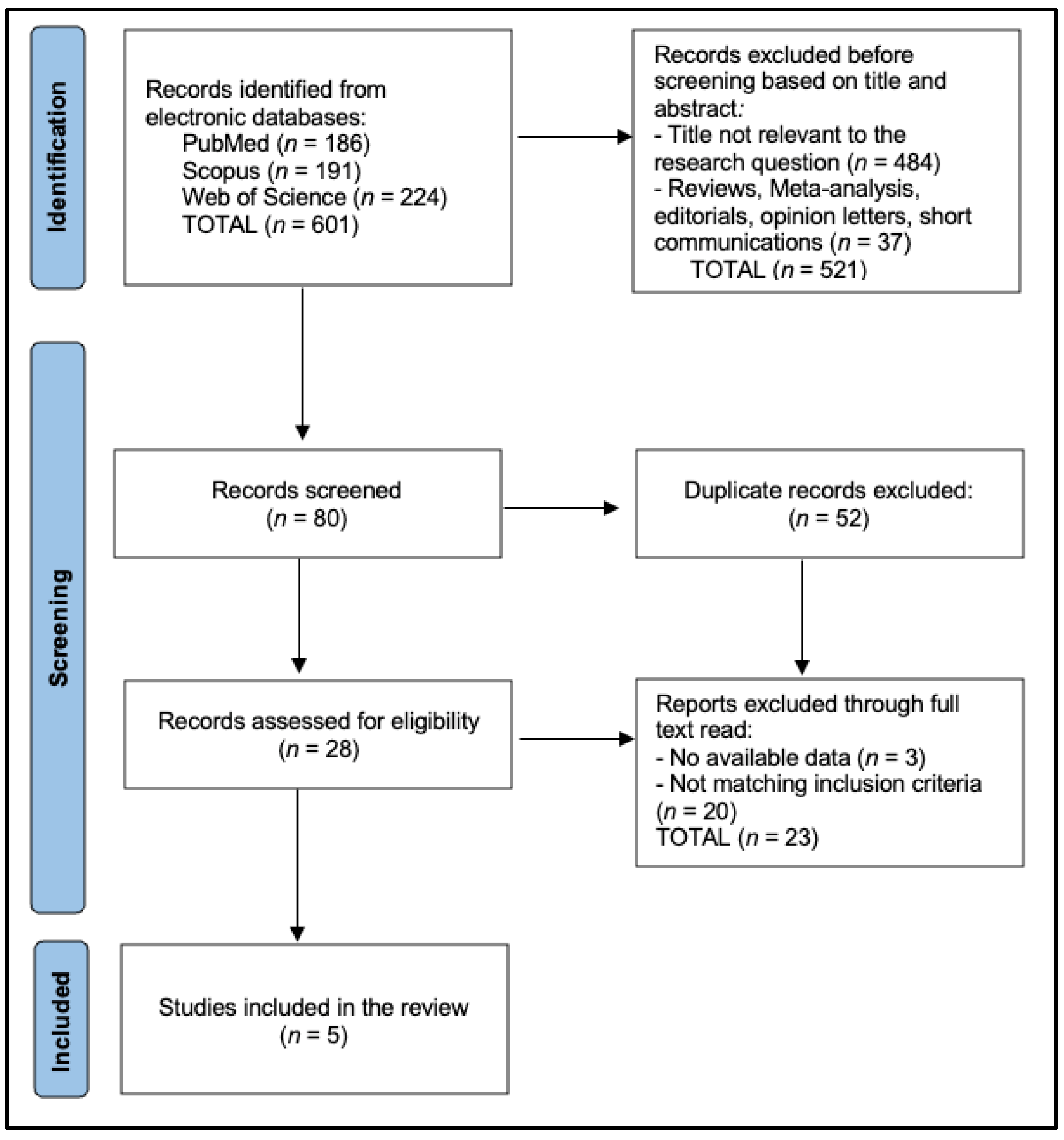
4.1. Search Strategy and Eligibility Criteria
4.2. Data Extraction and Quality Assessment
4.3. Data Synthesis and Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [PubMed]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [PubMed]
- Stoffel, E.M.; Mangu, P.B.; Gruber, S.B.; Hamilton, S.R.; Kalady, M.F.; Lau, M.W.; Lu, K.H.; Roach, N.; Limburg, P.J. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the Familial Risk–Colorectal Cancer: European Society for Medical Oncology Clinical Practice Guidelines. J. Clin. Oncol. 2015, 33, 209–217. [Google Scholar]
- Vasen, H.F.A.; Mecklin, J.P.; Meera Khan, P.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum 1991, 34, 424–425. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Clendenning, M.; Sotamaa, K.; Prior, T.; Westman, J.A.; et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J. Clin. Oncol. 2008, 26, 5783–5788. [Google Scholar] [CrossRef]
- Kastrinos, F.; Steyerberg, E.W.; Mercado, R.; Balmaña, J.; Holter, S.; Gallinger, S.; Haile, R.; Bapat, B.; LeMarchand, L.; Casey, G.; et al. The PREMM1,2,6 model predicts risk of MLH1, MSH2, and MSH6 gene mutations in Lynch syndrome. J. Clin. Oncol. 2009, 27, 2724–2729. [Google Scholar]
- Heald, B.; Plesec, T.; Liu, X.; Pai, R.; Patil, D.; Moline, J.; Sharp, R.R.; Burke, C.A.; Kalady, M.F.; Church, J.; et al. Implementation of universal screening for Lynch syndrome: Recommendations based on a single-center experience. J. Clin. Oncol. 2013, 31, 1336–1340. [Google Scholar]
- Moreira, L.; Balaguer, F.; Lindor, N.; de la Chapelle, A.; Hampel, H.; Engel, C.; Hopper, J.; Le Marchand, L.; Haile, R.; Gallinger, S.; et al. Identification of Lynch syndrome among patients with colorectal cancer. J. Clin. Oncol. 2012, 30, 1724–1730. [Google Scholar]
- Koopman, M.; Kortman, G.A.; Mekenkamp, L.; Ligtenberg, M.J.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.; Van Krieken, J.H. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [PubMed]
- Hampel, H.; Pearlman, R.; Beightol, M.; Zhao, W.; Jones, D.; Frankel, W.L.; Goodfellow, P.J.; Yilmaz, A.; Miller, K.; Bacher, J.; et al. Assessment of tumor sequencing as a replacement for Lynch syndrome screening and current molecular tests for hereditary colorectal cancer. JAMA Oncol. 2018, 4, 806–813. [Google Scholar]
- Gupta, S.; Provenzale, D.; Llor, X.; Halverson, A.L.; Grady, W.; Chung, D.C.; Haraldsdottir, S.; Markowitz, A.J.; Slavin, T.P., Jr.; Hampel, H.; et al. NCCN Guidelines Insights: Genetic/familial high-risk assessment: Colorectal, version 3.2017. J. Natl. Compr. Canc. Netw. 2017, 15, 1465–1475. [Google Scholar]
- Ladabaum, U.; Wang, G.; Terdiman, J.; Blanco, A.; Kuppermann, M.; Boland, C.R.; Ford, J.; Elkin, E.; Phillips, K.A. Strategies to identify the Lynch syndrome among patients with colorectal cancer: A cost-effectiveness analysis. Ann. Intern. Med. 2011, 155, 69–79. [Google Scholar] [PubMed]
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 404–412. [Google Scholar]
- Cohen, R.; Hain, E.; Buhard, O.; Guilloux, A.; Bardier, A.; Kaci, R.; Schischmanoff, O.; Adler, L.; Blanchet, B.; Tougeron, D.; et al. Association of primary resistance to immune checkpoint inhibitors in metastatic colorectal cancer with misdiagnosis of mismatch repair status. JAMA Oncol. 2018, 4, 979–983. [Google Scholar]
- de Leon, M.P.; Di Gregorio, C.; Silvestri, F.; Fornasarig, M.; Marra, G.; Bertario, L.; Ponz de Leon, M.; Gafà, R.; Cecconi, M.; Ponz, D.; et al. Histopathological aspects of MSI-H colorectal carcinomas in patients at high risk for Lynch syndrome lacking germline mutations in mismatch repair genes. J. Pathol. 2010, 221, 153–159. [Google Scholar]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar]
- Vonica, R.C.; Butuca, A.; Vonica-Tincu, A.L.; Morgovan, C.; Pumnea, M.; Cipaian, R.C.; Curca, R.O.; Batar, F.; Vornicu, V.; Solomon, A.; et al. The Descriptive and Disproportionality Assessment of EudraVigilance Database Reports on Capecitabine Induced Cardiotoxicity. Cancers 2024, 16, 3847. [Google Scholar] [CrossRef]
- Schofield, L.; Watson, N.; Grieu, F.; Li, W.Q.; Zeps, N.; Harvey, J.; Stewart, C.; Abdo, M.; Goldblatt, J.; Iacopetta, B. Population-based detection of Lynch syndrome in young colorectal cancer patients using microsatellite instability as the initial test. Int. J. Cancer 2009, 124, 1097–1102. [Google Scholar] [PubMed]
- Buchanan, D.D.; Clendenning, M.; Rosty, C.; Eriksen, S.V.; Walsh, M.D.; Walters, R.J.; Thibodeau, S.N.; Stewart, J.; Preston, S.; Win, A.K.; et al. Tumour testing to identify Lynch syndrome in two Australian colorectal cancer cohorts. J. Gastroenterol. Hepatol. 2017, 32, 427–438. [Google Scholar]
- Schofield, L.; Grieu, F.; Goldblatt, J.; Amanuel, B.; Iacopetta, B. A state-wide population-based program for detection of Lynch syndrome based upon immunohistochemical and molecular testing of colorectal tumours. Fam. Cancer 2012, 11, 1–6. [Google Scholar] [PubMed]
- Ward, R.L.; Turner, J.; Williams, R.; Pekarsky, B.; Packham, D.; Velickovic, M.; Meagher, A.; O’Connor, T.; Hawkins, N.J. Routine testing for mismatch repair deficiency in sporadic colorectal cancer is justified. J. Pathol. 2005, 207, 377–384. [Google Scholar] [CrossRef]
- Watson, N.; Grieu, F.; Morris, M.; Harvey, J.; Stewart, C.; Schofield, L.; Goldblatt, J.; Iacopetta, B. Heterogeneous staining for mismatch repair proteins during population-based prescreening for hereditary nonpolyposis colorectal cancer. Am. J. Pathol. 2007, 171, 383–392. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals with Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zumstein, V.; Vinzens, F.; Zettl, A.; Heinimann, K.; Koeberle, D.; von Flüe, M.; Bolli, M. Systematic immunohistochemical screening for Lynch syndrome in colorectal cancer: A single centre experience of 486 patients. Swiss Med. Wkly. 2016, 146, w14315. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hoodfar, E.; Jiang, S.F.; Udaltsova, N.; Pham, N.P.; Jodesty, Y.; Armstrong, M.A.; Hung, Y.Y.; Baker, R.J.; Postlethwaite, D.; et al. Comparison of Universal Versus Age-Restricted Screening of Colorectal Tumors for Lynch Syndrome Using Mismatch Repair Immunohistochemistry: A Cohort Study. Ann. Intern Med. 2019, 171, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Porkka, N.; Lahtinen, L.; Ahtiainen, M.; Böhm, J.P.; Kuopio, T.; Eldfors, S.; Mecklin, J.P.; Seppälä, T.T.; Peltomäki, P. Epidemiological, clinical and molecular characterization of Lynch-like syndrome: A population-based study. Int. J. Cancer 2019, 145, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Sarode, V.R.; Robinson, L. Screening for Lynch Syndrome by Immunohistochemistry of Mismatch Repair Proteins: Significance of Indeterminate Result and Correlation with Mutational Studies. Arch. Pathol. Lab. Med. 2019, 143, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Therkildsen, C.; Jensen, L.H.; Rasmussen, M.; Bernstein, I. An Update on Immune Checkpoint Therapy for the Treatment of Lynch Syndrome. Clin. Exp. Gastroenterol. 2021, 14, 181–197. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bellone, S.; Roque, D.M.; Siegel, E.R.; Buza, N.; Hui, P.; Bonazzoli, E.; Guglielmi, A.; Zammataro, L.; Nagarkatti, N.; Zaidi, S.; et al. A phase 2 evaluation of pembrolizumab for recurrent Lynch-like versus sporadic endometrial cancers with microsatellite instability. Cancer 2022, 128, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stinton, C.; Jordan, M.; Fraser, H.; Auguste, P.; Court, R.; Al-Khudairy, L.; Madan, J.; Grammatopoulos, D.; Taylor-Phillips, S. Testing Strategies for Lynch Syndrome in People with Endometrial Cancer: Systematic Reviews and Economic Evaluation; NIHR Journals Library: Southampton, UK, 2021; Health Technology Assessment, No. 25.42, Chapter 1, Introduction. Available online: https://www.ncbi.nlm.nih.gov/books/NBK571386/ (accessed on 31 January 2025).

{kind=link}
{kind=link}
| Study and Author | Country/Region | Study Period | Design | Sample Size (n) | Age Range | MMR Testing Method |
|---|---|---|---|---|---|---|
| Schofield et al. [21] | Australia (W. Australia) | 2000–2006 | Population-based | 1344 | <60 years | MSI (BAT-26), BRAF, IHC (MLH1/PMS2/MSH2/MSH6) |
| Buchanan et al. [22] | Australia (Victoria) | 1997–2009 | Two prospective cohorts | 1639 (total) | <60 primarily | MSI multi-marker, BRAF, IHC, germline |
| Schofield et al. [23] | Australia (W. Australia) | 2009–2010 | State-wide program | 270 | <60 years | MSI (pentaplex) + BRAF, IHC |
| Ward et al. [24] | Australia (Sydney) | 1994–2004 | Prospective single site | 786 individuals (836 tumors) | <60 subset included | MSI multi-marker, IHC (MLH1/MSH2) |
| Watson et al. [25] | Australia (W. Australia) | 2000–2004 | Population-based | 1003 | <60 years | MSI (BAT-26), BRAF, IHC (all 4 MMR) |
| Study | Total CRC < 60 (n) | MSI+ (%) | BRAF+ Among MSI+ | Germline Testing | Mutation Yield |
|---|---|---|---|---|---|
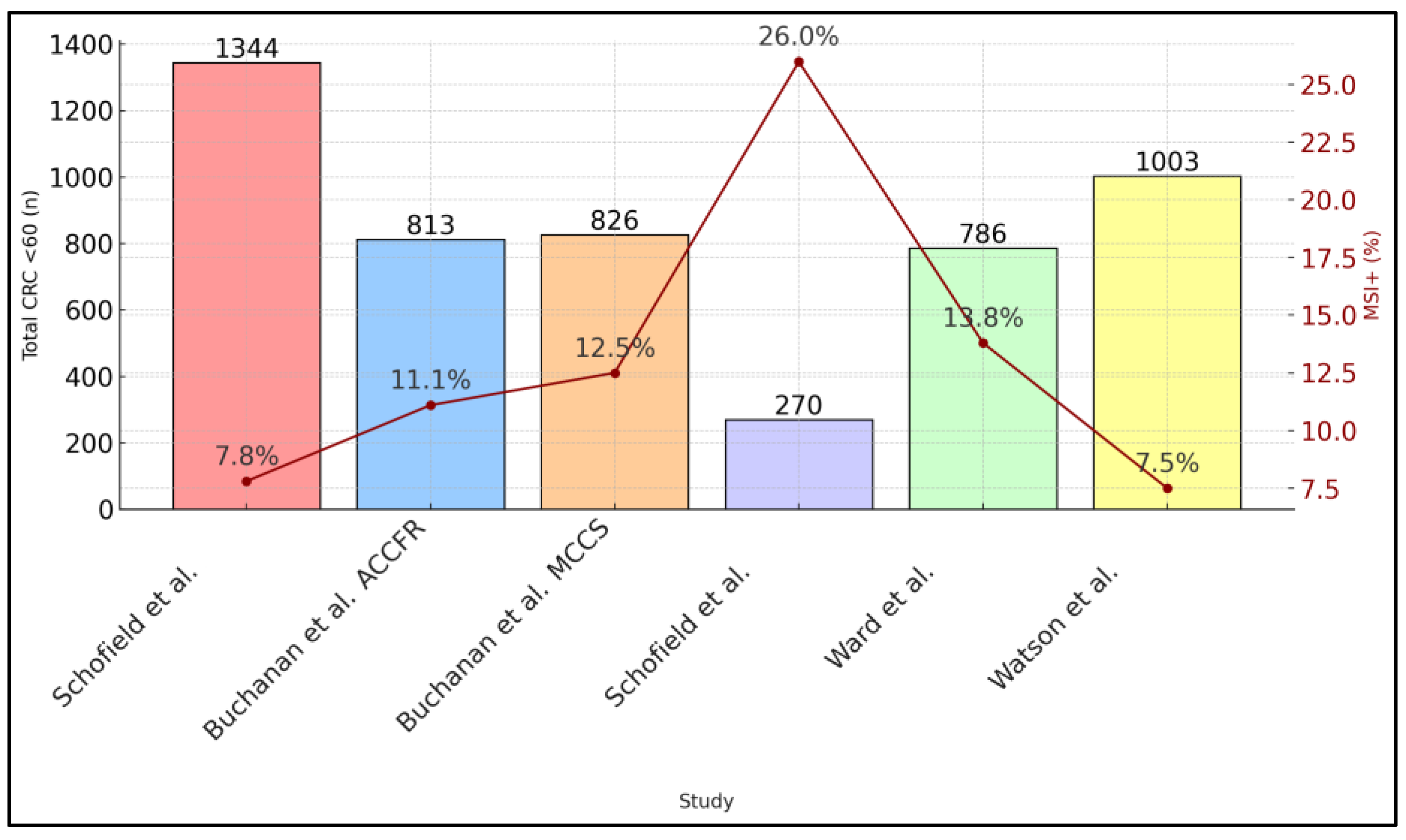
| Schofield et al. [21] | 1344 | 7.8 | 7/105 (6.7%) | 98 “red flag” → 35 tested | 11 newly discovered + 25 known = 36 total |
| Buchanan et al. [22] | ACCFR: 813; MCCS: 826 | 11.1 (ACCFR), 12.5 (MCCS) | NA | MMR-def tumors → germline | ACCFR: 5.2% carriers, MCCS: 0.8% carriers |
| Schofield et al. [23] | 270 | 70/270 (26) | 25/70 (35.7%) | 45 “red flag” → 31 tested | 15 carriers (48% tested) |
| Ward et al. [24] | 786 individuals | 115/836 (13.8) | Not stated | 108 with loss on IHC → subset tested | ∼0.8% for confirmed LS |
| Watson et al. [25] | 1003 | 7.5 | 6/75 (8%) | 69 MSI+/BRAF− → germline | 18 known carriers, 3 newly ID’d (≥2.1%) |
| Study | Major Genes Screened | Most Common Mutation(s) | IHC Loss Pattern | Comments |
|---|---|---|---|---|
| Schofield et al. [21] | MLH1, MSH2, MSH6, PMS2 | MSH2 and MLH1 predominantly | Often dual loss: MLH1/PMS2 or MSH2/MSH6 | Some cases of solitary PMS2 or MSH6 loss were noted. |
| Buchanan et al. [22] | MLH1, MSH2, MSH6, PMS2 | MLH1 or MSH2 in the majority | Standard patterns, plus occasional heterogeneity | MSH6 and PMS2 carriers were a smaller fraction. |
| Schofield et al. [23] | MLH1, MSH2, MSH6, PMS2 | 15 carriers found: distribution not fully specified | Distinct patterns of loss consistent with germline | 8/15 had single-protein losses (PMS2 or MSH6). |
| Ward et al. [24] | MLH1, MSH2 primarily | MLH1, MSH2 only | Rare solitary MSH6 or PMS2 not assessed widely | Study focused primarily on MLH1/MSH2 IHC. |
| Watson et al. [25] | MLH1, PMS2, MSH2, MSH6 | 18 known carriers, mostly MSH2, MLH1 | 51% had MLH1/PMS2 loss, 25% MSH2/MSH6 loss; 12% heterogeneous | Heterogeneous staining complicated interpretation. |
| Study | MMR-Def Patients | Stage Distribution | Survival/Prognosis | Notable Findings |
|---|---|---|---|---|
| Schofield et al. [21] | 7.8% MSI+ <60 years | More often Stage II or III but with fewer Stage IV | Improved overall survival among MMR-def. Some minimal 5-FU benefit reported. | Early detection critical; 36 total carriers identified. |
| Buchanan et al. [22] | 11.1% (ACCFR) and 12.5% (MCCS) MMR-def | Stage II overrepresented among MMR-def | MMR-def tumors had better 5-year survival, especially if node-negative. | Lynch patients often had synchronous/metachronous disease. |
| Schofield et al. [23] | 26% MSI+ (selective cohort) | Variable stage, but most “red flags” were earlier | MMR-def had a low mortality rate over short follow-up. | Subset analysis suggests no added benefit from 5-FU chemo. |
| Ward et al. [24] | 13.8% MSI+ <60 subset | MMR-def frequently high grade but fewer metastases | MMR-def was an independent good prognostic factor (p < 0.05). | Adjuvant therapy utility questioned in stage II MMR-def. |
| Watson et al. [25] | 7.5% MSI+ in <60 years | Predominantly right-sided, often Stage II | LS carriers had improved survival vs. sporadic MSI or MSS patients. | Heterogeneous IHC patterns complicated staging correlation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muntean, C.; Gaborean, V.; Vonica, R.C.; Faur, A.M.; Rus, V.I.; Faur, I.F.; Feier, C.V.I. The Clinical Outcomes Among Patients Under 60 Years Old with Lynch Syndrome: Variations Based on Different Mutation Patterns. Int. J. Mol. Sci. 2025, 26, 3383. https://doi.org/10.3390/ijms26073383
Muntean C, Gaborean V, Vonica RC, Faur AM, Rus VI, Faur IF, Feier CVI. The Clinical Outcomes Among Patients Under 60 Years Old with Lynch Syndrome: Variations Based on Different Mutation Patterns. International Journal of Molecular Sciences. 2025; 26(7):3383. https://doi.org/10.3390/ijms26073383
Chicago/Turabian StyleMuntean, Calin, Vasile Gaborean, Razvan Constantin Vonica, Alaviana Monique Faur, Vladut Iosif Rus, Ionut Flaviu Faur, and Catalin Vladut Ionut Feier. 2025. "The Clinical Outcomes Among Patients Under 60 Years Old with Lynch Syndrome: Variations Based on Different Mutation Patterns" International Journal of Molecular Sciences 26, no. 7: 3383. https://doi.org/10.3390/ijms26073383
APA StyleMuntean, C., Gaborean, V., Vonica, R. C., Faur, A. M., Rus, V. I., Faur, I. F., & Feier, C. V. I. (2025). The Clinical Outcomes Among Patients Under 60 Years Old with Lynch Syndrome: Variations Based on Different Mutation Patterns. International Journal of Molecular Sciences, 26(7), 3383. https://doi.org/10.3390/ijms26073383