
Parkinson’s Disease: The Neurodegenerative Enigma Under the “Undercurrent” of Endoplasmic Reticulum Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Basic Concepts and Activation Mechanisms of ERS
2.1. Structure and Functions of the ER
2.2. Concept and Triggers of ERS
2.3. Signaling Pathways of ERS
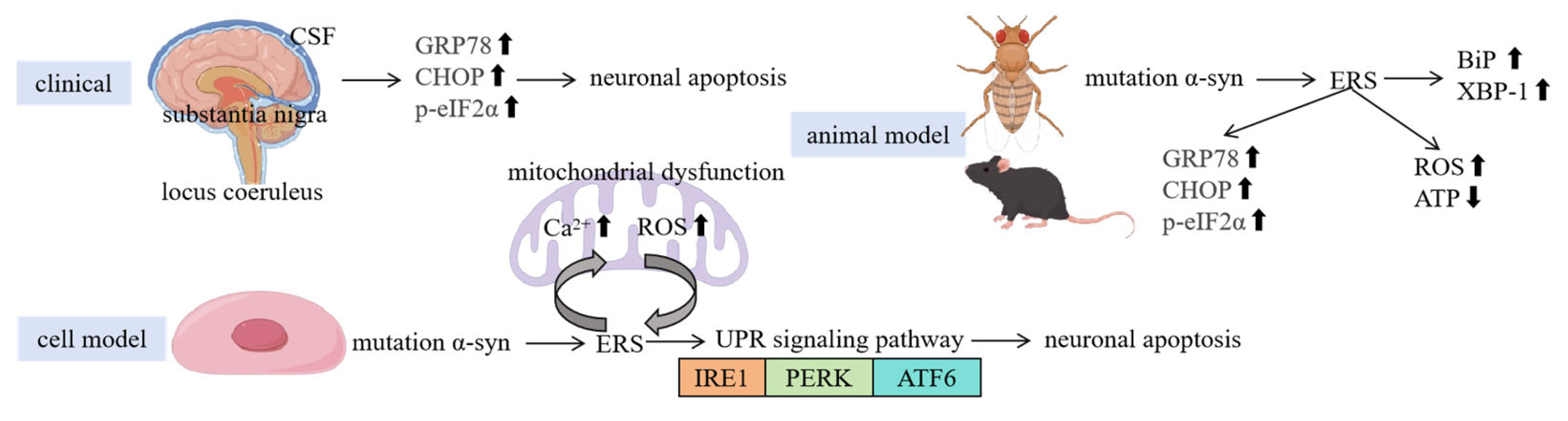
3. Relationship Between ERS and PD Pathological Features
3.1. Clinical Evidence
3.2. Animal Model Evidence
3.3. Cellular Experimental Evidence
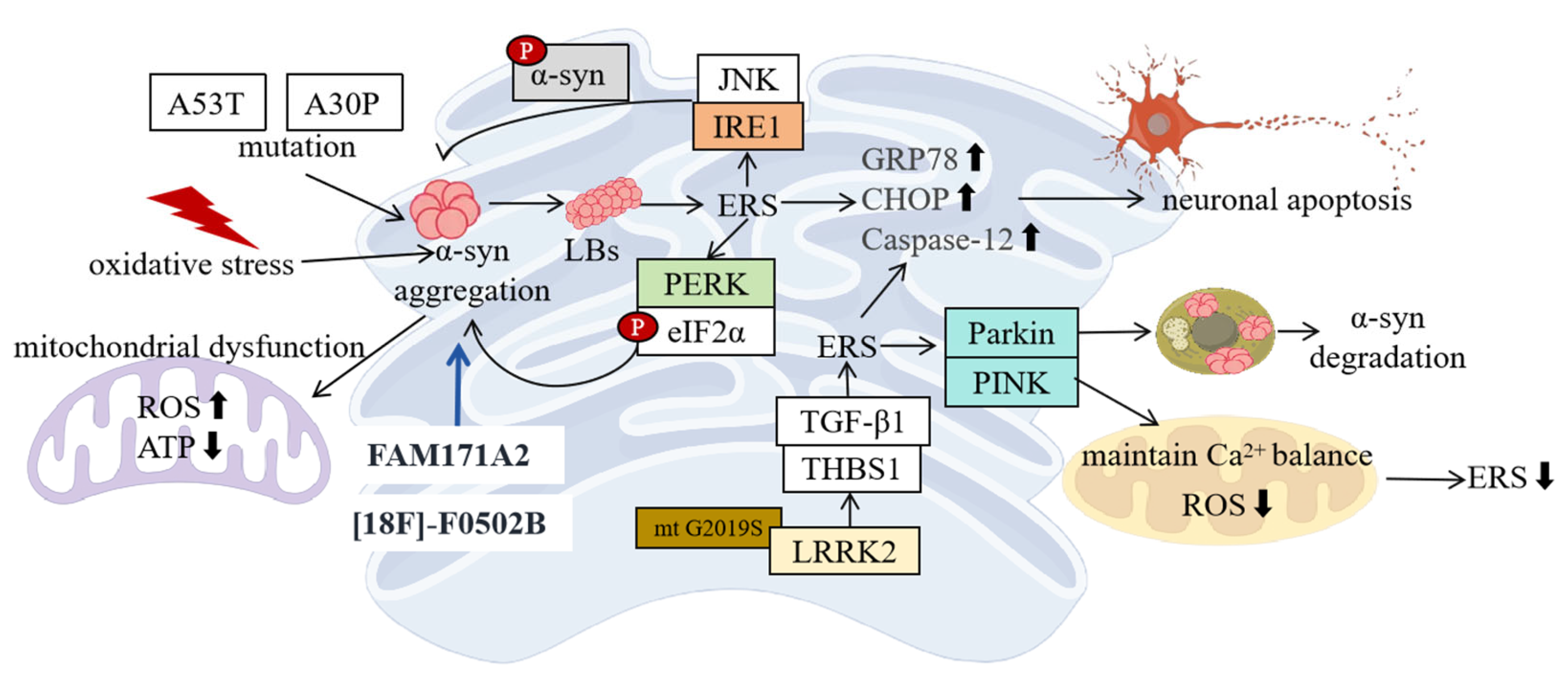
4. Molecular Mechanisms of ERS in PD
4.1. ERS and α-Syn
4.2. LRRK2 and ERS
4.3. Other Related Molecules and ERS
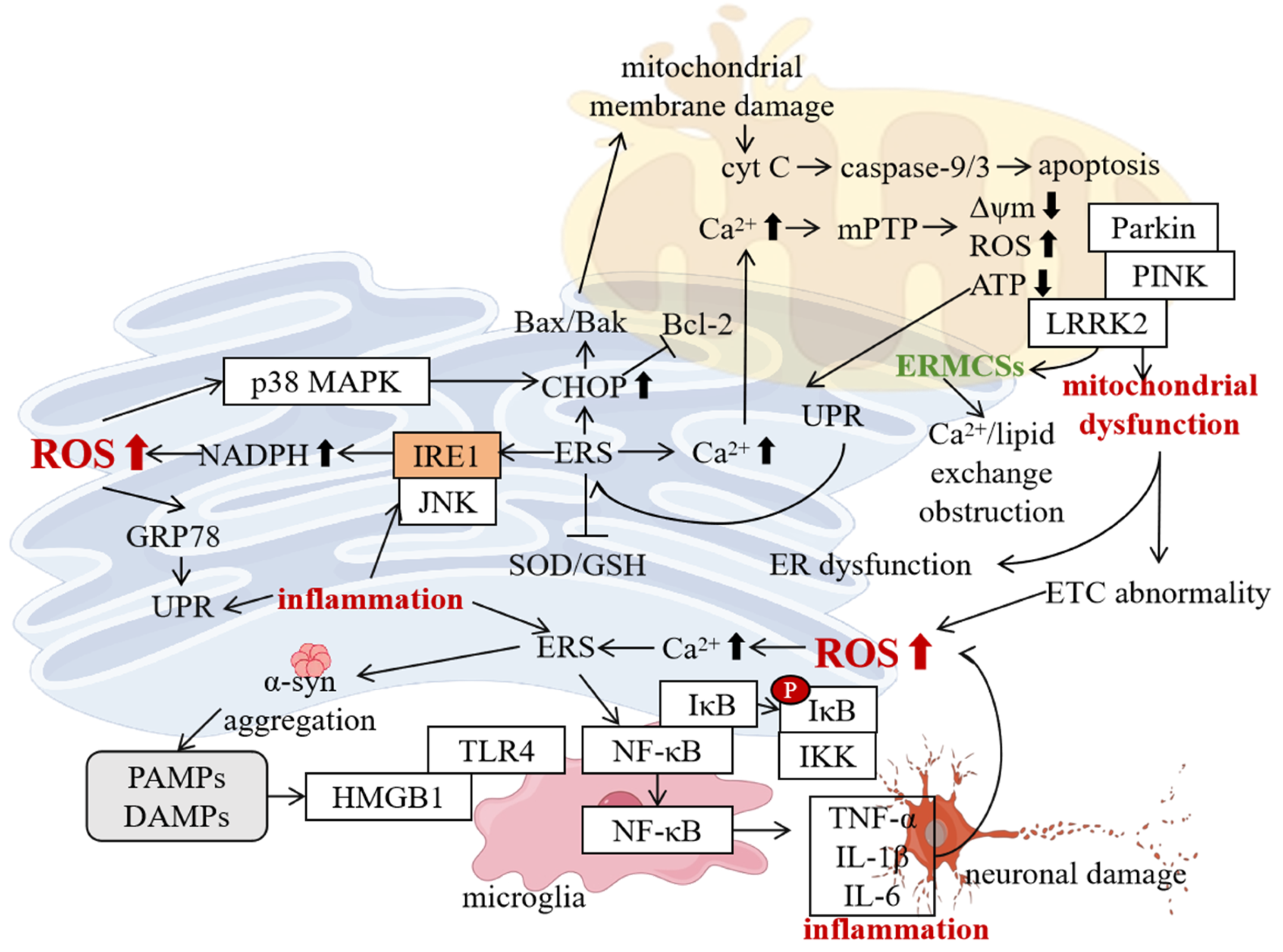
5. Interaction Between ERS and Other Pathological Mechanisms of PD
5.1. Interaction with Mitochondrial Dysfunction
5.2. Interaction with Oxidative Stress
5.3. Interaction with Neuroinflammation
6. Exploration of PD Treatment Strategies Based on ERS
6.1. Drug Treatment Strategies
6.2. Non-Drug Treatment Strategies
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, J.; Yang, N.; Huang, Y.; Hu, T.; Rao, C. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 2022, 13, 1051. [Google Scholar] [CrossRef]
- Wang, D.; Qu, S.; Zhang, Z.; Tan, L.; Chen, X.; Zhong, H.-J.; Chong, C.-M. Strategies targeting endoplasmic reticulum stress to improve Parkinson’s disease. Front. Pharmacol. 2023, 14, 1288894. [Google Scholar] [CrossRef]
- Jiang, M.; Yun, Q.; Shi, F.; Niu, G.; Gao, Y.; Xie, S.; Yu, S. Downregulation of miR-384-5p attenuates rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through inhibiting endoplasmic reticulum stress. Am. J. Physiol. Cell Physiol. 2016, 310, C755–C763. [Google Scholar] [CrossRef]
- Zeng, H.; Liu, Y.; Liu, X.; Li, J.; Lu, L.; Xue, C.; Wu, X.; Zhang, X.; Zheng, Z.; Lu, G. Interplay of α-Synuclein Oligomers and Endoplasmic Reticulum Stress in Parkinson’s Disease: Insights into Cellular Dysfunctions. Inflammation 2024, 1–17. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Cherepanova, N.; Shrimal, S.; Gilmore, R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 2016, 41, 57–65. [Google Scholar] [CrossRef]
- Zirkin, B.R.; Papadopoulos, V. Leydig cells: Formation, function, and regulation. Biol. Reprod. 2018, 99, 101–111. [Google Scholar] [CrossRef]
- Zhou, S.; Cheng, K.; Peng, Y.; Liu, Y.; Hu, Q.; Zeng, S.; Qi, X.; Yu, L. Regulation mechanism of endoplasmic reticulum stress on metabolic enzymes in liver diseases. Pharmacol. Res. 2024, 207, 107332. [Google Scholar] [CrossRef]
- Rayavarapu, S.; Coley, W.; Nagaraju, K. Endoplasmic reticulum stress in skeletal muscle homeostasis and disease. Curr. Rheumatol. Rep. 2012, 14, 238–243. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Merighi, A.; Lossi, L. Endoplasmic Reticulum Stress Signaling and Neuronal Cell Death. Int. J. Mol. Sci. 2022, 23, 15186. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Guan, L.; Ge, R.; Ma, S. Newsights of endoplasmic reticulum in hypoxia. Biomed. Pharmacother. 2024, 175, 116812. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Y.; Zhang, J.; Zhang, R.; Wang, Y.; Liu, F. ABCA1 deficiency-mediated glomerular cholesterol accumulation exacerbates glomerular endothelial injury and dysfunction in diabetic kidney disease. Metabolism 2023, 139, 155377. [Google Scholar] [CrossRef]
- Taylor, R.C.; Hetz, C. Mastering organismal aging through the endoplasmic reticulum proteostasis network. Aging Cell 2020, 19, e13265. [Google Scholar] [CrossRef]
- Siwecka, N.; Saramowicz, K.; Galita, G.; Rozpędek-Kamińska, W.; Majsterek, I. Inhibition of Protein Aggregation and Endoplasmic Reticulum Stress as a Targeted Therapy for α-Synucleinopathy. Pharmaceutics 2023, 15, 2051. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic β-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 4843. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Wiseman, R.L.; Mesgarzadeh, J.S.; Hendershot, L.M. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol. Cell 2022, 82, 1477–1491. [Google Scholar] [CrossRef]
- Dawes, S.; Hurst, N.; Grey, G.; Wieteska, L.; Wright, N.V.; Manfield, I.W.; Hussain, M.H.; Kalverda, A.P.; Lewandowski, J.R.; Chen, B.; et al. Chaperone BiP controls ER stress sensor Ire1 through interactions with its oligomers. Life Sci. Alliance 2024, 7, e202402702. [Google Scholar] [CrossRef]
- Brooks, N.C.; Marshall, A.H.; Qa’aty, N.; Hiyama, Y.; Boehning, D.; Jeschke, M.G. XBP-1s is linked to suppressed gluconeogenesis in the Ebb phase of burn injury. Mol. Med. 2013, 19, 72–78. [Google Scholar] [CrossRef]
- Arasi, F.P.; Shahrestanaki, M.K.; Aghaei, M. A2a adenosine receptor agonist improves endoplasmic reticulum stress in MIN6 cell line through protein kinase A/ protein kinase B/ Cyclic adenosine monophosphate response element-binding protein/ and Growth Arrest And DNA-Damage-Inducible 34/ eukaryotic Initiation Factor 2α pathways. J. Cell. Physiol. 2019, 234, 10500–10511. [Google Scholar] [CrossRef]
- Madhavan, A.; Kok, B.P.; Rius, B.; Grandjean, J.M.D.; Alabi, A.; Albert, V.; Sukiasyan, A.; Powers, E.T.; Galmozzi, A.; Saez, E.; et al. Pharmacologic IRE1/XBP1s activation promotes systemic adaptive remodeling in obesity. Nat. Commun. 2022, 13, 608. [Google Scholar] [CrossRef]
- She, C.; Wu, C.; Guo, W.; Xie, Y.; Li, S.; Liu, W.; Xu, C.; Li, H.; Cao, P.; Yang, Y.; et al. Combination of RUNX1 inhibitor and gemcitabine mitigates chemo-resistance in pancreatic ductal adenocarcinoma by modulating BiP/PERK/eIF2α-axis-mediated endoplasmic reticulum stress. J. Exp. Clin. Cancer Res. 2023, 42, 238. [Google Scholar] [CrossRef]
- Hao, L.; Zhong, W.; Dong, H.; Guo, W.; Sun, X.; Zhang, W.; Yue, R.; Li, T.; Griffiths, A.; Ahmadi, A.R.; et al. ATF4 activation promotes hepatic mitochondrial dysfunction by repressing NRF1-TFAM signalling in alcoholic steatohepatitis. Gut 2021, 70, 1933–1945. [Google Scholar] [CrossRef]
- Bahamondes Lorca, V.A.; Bastidas Mayorga, B.D.; Tong, L.; Wu, S. UVB-induced eIF2α phosphorylation in keratinocytes depends on decreased ATF4, GADD34 and CReP expression levels. Life Sci. 2021, 286, 120044. [Google Scholar] [CrossRef]
- Lei, Y.; Yu, H.; Ding, S.; Liu, H.; Liu, C.; Fu, R. Molecular mechanism of ATF6 in unfolded protein response and its role in disease. Heliyon 2024, 10, e25937. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Mnich, K.; Moghaddam, S.; Browne, P.; Counihan, T.; Fitzgerald, S.P.; Martin, K.; Richardson, C.; Samali, A.; Gorman, A.M. Endoplasmic Reticulum Stress-Regulated Chaperones as a Serum Biomarker Panel for Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 1476–1485. [Google Scholar] [CrossRef]
- Motawi, T.K.; Al-Kady, R.H.; Abdelraouf, S.M.; Senousy, M.A. Empagliflozin alleviates endoplasmic reticulum stress and augments autophagy in rotenone-induced Parkinson’s disease in rats: Targeting the GRP78/PERK/eIF2α/CHOP pathway and miR-211-5p. Chem. Biol. Interact. 2022, 362, 110002. [Google Scholar] [CrossRef]
- Baek, J.H.; Mamula, D.; Tingstam, B.; Pereira, M.; He, Y.; Svenningsson, P. GRP78 Level Is Altered in the Brain, but Not in Plasma or Cerebrospinal Fluid in Parkinson’s Disease Patients. Front. Neurosci. 2019, 13, 697. [Google Scholar] [CrossRef]
- Sunderhaus, E.R.; Law, A.D.; Kretzschmar, D. ER responses play a key role in Swiss-Cheese/Neuropathy Target Esterase-associated neurodegeneration. Neurobiol. Dis. 2019, 130, 104520. [Google Scholar] [CrossRef]
- Jiao, P.; Fan, W.; Ma, X.; Lin, R.; Zhao, Y.; Li, Y.; Zhang, H.; Jia, X.; Bi, Y.; Feng, X.; et al. SARS-CoV-2 nonstructural protein 6 triggers endoplasmic reticulum stress-induced autophagy to degrade STING1. Autophagy 2023, 19, 3113–3131. [Google Scholar] [CrossRef]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef]
- Kang, S.; Piao, Y.; Kang, Y.C.; Lim, S.; Pak, Y.K. DA-9805 protects dopaminergic neurons from endoplasmic reticulum stress and inflammation. Biomed. Pharmacother. 2022, 145, 112389. [Google Scholar] [CrossRef]
- Zhu, J.; Dou, S.; Jiang, Y.; Chen, J.; Wang, C.; Cheng, B. Apelin-13 protects dopaminergic neurons in MPTP-induced Parkinson’s disease model mice through inhibiting endoplasmic reticulum stress and promoting autophagy. Brain Res. 2019, 1715, 203–212. [Google Scholar] [CrossRef]
- Bulteau, A.L.; Mena, N.P.; Auchère, F.; Lee, I.; Prigent, A.; Lobsiger, C.S.; Camadro, J.M.; Hirsch, E.C. Dysfunction of mitochondrial Lon protease and identification of oxidized protein in mouse brain following exposure to MPTP: Implications for Parkinson disease. Free Radic. Biol. Med. 2017, 108, 236–246. [Google Scholar] [CrossRef]
- Davis, C.K.; Bathula, S.; Jeong, S.; Arruri, V.; Choi, J.; Subramanian, S.; Ostrom, C.M.; Vemuganti, R. An antioxidant and anti-ER stress combination therapy elevates phosphorylation of α-Syn at serine 129 and alleviates post-TBI PD-like pathology in a sex-specific manner in mice. Exp. Neurol. 2024, 377, 114795. [Google Scholar] [CrossRef]
- Zhang, G.F.; Zhang, Y.; Zhao, G. Crocin protects PC12 cells against MPP(+)-induced injury through inhibition of mitochondrial dysfunction and ER stress. Neurochem. Int. 2015, 89, 101–110. [Google Scholar] [CrossRef]
- Ge, B.; Li, S.L.; Li, F.R. Astragaloside-IV regulates endoplasmic reticulum stress-mediated neuronal apoptosis in a murine model of Parkinson’s disease via the lincRNA-p21/CHOP pathway. Exp. Mol. Pathol. 2020, 115, 104478. [Google Scholar] [CrossRef]
- Xia, N.; Zhang, Q.; Wang, S.T.; Gu, L.; Yang, H.M.; Liu, L.; Bakshi, R.; Yang, H.; Zhang, H. Blockade of metabotropic glutamate receptor 5 protects against DNA damage in a rotenone-induced Parkinson’s disease model. Free Radic. Biol. Med. 2015, 89, 567–580. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Liu, A.; Chen, Z.; Li, X.; Xie, C.; Chen, Y.; Su, X.; Chen, Y.; Zhang, M.; Chen, J.; Yang, T.; et al. C5a-C5aR1 induces endoplasmic reticulum stress to accelerate vascular calcification via PERK-eIF2α-ATF4-CREB3L1 pathway. Cardiovasc. Res. 2023, 119, 2563–2578. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.M. Endoplasmic Reticulum-Associated Biomarkers for Molecular Phenotyping of Rare Kidney Disease. Int. J. Mol. Sci. 2021, 22, 2161. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug Discov. 2022, 21, 115–140. [Google Scholar] [CrossRef]
- Groenendyk, J.; Agellon, L.B.; Michalak, M. Calcium signaling and endoplasmic reticulum stress. Int. Rev. Cell Mol. Biol. 2021, 363, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Mehra, S.; Sahay, S.; Singh, P.K.; Maji, S.K. α-synuclein aggregation and its modulation. Int. J. Biol. Macromol. 2017, 100, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Du, X.Y.; Xie, X.X.; Liu, R.T. The Role of α-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kou, M.; Deng, Q.; Yu, H.; Mei, J.; Gao, J.; Fu, W.; Ning, B. Acupuncture activates IRE1/XBP1 endoplasmic reticulum stress pathway in Parkinson’s disease model rats. Behav. Brain Res. 2024, 462, 114871. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.M.; Xu, Q.H.; Liu, Y.Q.; Feng, Y.W.; Han, S.D.; Zhang, Y.R.; Chen, S.D.; Guo, Y.; Wu, B.S.; Ma, L.Z.; et al. Neuronal FAM171A2 mediates α-synuclein fibril uptake and drives Parkinson’s disease. Science 2025, 387, 892–900. [Google Scholar] [CrossRef]
- Xu, W.; Han, S.D.; Zhang, C.; Li, J.Q.; Wang, Y.J.; Tan, C.C.; Li, H.Q.; Dong, Q.; Mei, C.; Tan, L.; et al. The FAM171A2 gene is a key regulator of progranulin expression and modifies the risk of multiple neurodegenerative diseases. Sci. Adv. 2020, 6, eabb3063. [Google Scholar] [CrossRef]
- Xiang, J.; Tao, Y.; Xia, Y.; Luo, S.; Zhao, Q.; Li, B.; Zhang, X.; Sun, Y.; Xia, W.; Zhang, M.; et al. Development of an α-synuclein positron emission tomography tracer for imaging synucleinopathies. Cell 2023, 186, 3350–3367.e19. [Google Scholar] [CrossRef]
- Pavia-Collado, R.; Cóppola-Segovia, V.; Miquel-Rio, L.; Alarcón-Aris, D.; Rodríguez-Aller, R.; Torres-López, M.; Paz, V.; Ruiz-Bronchal, E.; Campa, L.; Artigas, F.; et al. Intracerebral Administration of a Ligand-ASO Conjugate Selectively Reduces α-Synuclein Accumulation in Monoamine Neurons of Double Mutant Human A30P*A53T*α-Synuclein Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 2939. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Qu, L.; Liu, Z.; Liang, L.; Wang, Y.; Quan, S.; Wang, Y.; Tang, L. The IRE1/JNK signaling pathway regulates inflammation cytokines and production of glomerular extracellular matrix in the acute kidney injury to chronic kidney disease transition. Mol. Biol. Rep. 2022, 49, 7709–7718. [Google Scholar] [CrossRef]
- Suzuki, H.; Egawa, N.; Imamura, K.; Kondo, T.; Enami, T.; Tsukita, K.; Suga, M.; Yada, Y.; Shibukawa, R.; Takahashi, R.; et al. Mutant α-synuclein causes death of human cortical neurons via ERK1/2 and JNK activation. Mol. Brain 2024, 17, 14. [Google Scholar] [CrossRef]
- Li, H.Y.; Wang, X.C.; Xu, Y.M.; Luo, N.C.; Luo, S.; Hao, X.Y.; Cheng, S.Y.; Fang, J.S.; Wang, Q.; Zhang, S.J.; et al. Berberine Improves Diabetic Encephalopathy Through the SIRT1/ER Stress Pathway in db/db Mice. Rejuvenation Res. 2018, 21, 200–209. [Google Scholar] [CrossRef]
- Chakraborty, J.; Pakrashi, S.; Sarbajna, A.; Dutta, M.; Bandyopadhyay, J. Quercetin Attenuates Copper-Induced Apoptotic Cell Death and Endoplasmic Reticulum Stress in SH-SY5Y Cells by Autophagic Modulation. Biol. Trace Elem. Res. 2022, 200, 5022–5041. [Google Scholar] [CrossRef]
- Wu, H.; Guo, H.; Liu, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Copper sulfate-induced endoplasmic reticulum stress promotes hepatic apoptosis by activating CHOP, JNK and caspase-12 signaling pathways. Ecotoxicol. Environ. Saf. 2020, 191, 110236. [Google Scholar] [CrossRef]
- Kabiraj, P.; Valenzuela, C.A.; Marin, J.E.; Ramirez, D.A.; Mendez, L.; Hwang, M.S.; Varela-Ramirez, A.; Fenelon, K.; Narayan, M.; Skouta, R. The neuroprotective role of ferrostatin-1 under rotenone-induced oxidative stress in dopaminergic neuroblastoma cells. Protein J. 2015, 34, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, T.; Mirzaei-Behbahani, B.; Zadali, R.; Pirhaghi, M.; Morozova-Roche, L.A.; Meratan, A.A. Common Mechanisms Underlying α-Synuclein-Induced Mitochondrial Dysfunction in Parkinson’s Disease. J. Mol. Biol. 2023, 435, 167992. [Google Scholar] [CrossRef] [PubMed]
- Sosero, Y.L.; Gan-Or, Z. LRRK2 and Parkinson’s disease: From genetics to targeted therapy. Ann. Clin. Transl. Neurol. 2023, 10, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Lu, F.; Koc, S.; Zheng, Z.; Wang, B.; Zhang, S.; Skutella, T.; Lu, G. LRRK2 Gly2019Ser Mutation Promotes ER Stress via Interacting with THBS1/TGF-β1 in Parkinson’s Disease. Adv. Sci. 2023, 10, e2303711. [Google Scholar] [CrossRef]
- Singh, F.; Prescott, A.R.; Rosewell, P.; Ball, G.; Reith, A.D.; Ganley, I.G. Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. Elife 2021, 10, e67604. [Google Scholar] [CrossRef]
- Xiong, Y.; Yu, J. LRRK2 in Parkinson’s disease: Upstream regulation and therapeutic targeting. Trends Mol. Med. 2024, 30, 982–996. [Google Scholar] [CrossRef]
- Ohtonen, S.; Giudice, L.; Jäntti, H.; Fazaludeen, M.F.; Shakirzyanova, A.; Gómez-Budia, M.; Välimäki, N.N.; Niskanen, J.; Korvenlaita, N.; Fagerlund, I.; et al. Human iPSC-derived microglia carrying the LRRK2-G2019S mutation show a Parkinson’s disease related transcriptional profile and function. Sci. Rep. 2023, 13, 22118. [Google Scholar] [CrossRef]
- Pena, N.; Richbourg, T.; Gonzalez-Hunt, C.P.; Qi, R.; Wren, P.; Barlow, C.; Shanks, N.F.; Carlisle, H.J.; Sanders, L.H. G2019S selective LRRK2 kinase inhibitor abrogates mitochondrial DNA damage. NPJ Park. Dis. 2024, 10, 49. [Google Scholar] [CrossRef]
- Ramalingam, M.; Jang, S.; Hwang, J.; Kim, B.; Cho, H.H.; Kim, E.; Jeong, H.S. Neuroprotective Effects of the Neural-Induced Adipose-Derived Stem Cell Secretome against Rotenone-Induced Mitochondrial and Endoplasmic Reticulum Dysfunction. Int. J. Mol. Sci. 2023, 24, 5622. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, D.; Schips, T.G.; Vo, A.; Grimes, K.M.; Baldwin, T.A.; Brody, M.J.; Accornero, F.; Sargent, M.A.; Molkentin, J.D. Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nat Commun. 2021, 12, 3928. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Yeganeh, B.; Zeki, A.A.; Shojaei, S.; Kenyon, N.J.; Ott, S.; Samali, A.; Patterson, J.; Alizadeh, J.; Moghadam, A.R.; et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-β1 in human lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L493–L504. [Google Scholar] [CrossRef]
- Deng, R.; Li, C.; Wang, X.; Chang, L.; Ni, S.; Zhang, W.; Xue, P.; Pan, D.; Wan, M.; Deng, L.; et al. Periosteal CD68+ F4/80+ Macrophages Are Mechanosensitive for Cortical Bone Formation by Secretion and Activation of TGF-β1. Adv. Sci. 2022, 9, e2103343. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Feng, W.; Lv, C.; Cheng, L.; Song, X.; Li, X.; Xie, H.; Chen, S.; Wang, X.; Xue, L.; Zhang, C.; et al. Targeting ERS-mitophagy in hippocampal neurons to explore the improvement of memory by tea polyphenols in aged type 2 diabetic rats. Free Radic. Biol. Med. 2024, 213, 293–308. [Google Scholar] [CrossRef]
- Clausen, L.; Okarmus, J.; Voutsinos, V.; Meyer, M.; Lindorff-Larsen, K.; Hartmann-Petersen, R. PRKN-linked familial Parkinson’s disease: Cellular and molecular mechanisms of disease-linked variants. Cell. Mol. Life Sci. 2024, 81, 223. [Google Scholar] [CrossRef]
- Uoselis, L.; Lindblom, R.; Lam, W.K.; Küng, C.J.; Skulsuppaisarn, M.; Khuu, G.; Nguyen, T.N.; Rudler, D.L.; Filipovska, A.; Schittenhelm, R.B.; et al. Temporal landscape of mitochondrial proteostasis governed by the UPRmt. Sci. Adv. 2023, 9, eadh8228. [Google Scholar] [CrossRef]
- El Manaa, W.; Duplan, E.; Goiran, T.; Lauritzen, I.; Vaillant Beuchot, L.; Lacas-Gervais, S.; Morais, V.A.; You, H.; Qi, L.; Salazar, M.; et al. Transcription- and phosphorylation-dependent control of a functional interplay between XBP1s and PINK1 governs mitophagy and potentially impacts Parkinson disease pathophysiology. Autophagy 2021, 17, 4363–4385. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, S.; Song, R.; Lai, X.; Shen, M.; Wu, J.; Yan, J. A novel mechanism of PHB2-mediated mitophagy participating in the development of Parkinson’s disease. Neural Regen. Res. 2024, 19, 1828–1834. [Google Scholar] [CrossRef]
- Quarato, G.; Mari, L.; Barrows, N.J.; Yang, M.; Ruehl, S.; Chen, M.J.; Guy, C.S.; Low, J.; Chen, T.; Green, D.R. Mitophagy restricts BAX/BAK-independent, Parkin-mediated apoptosis. Sci. Adv. 2023, 9, eadg8156. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hussain, R.; Mehmood, K.; Tang, Z.; Zhang, H.; Li, Y. Mitochondrial-Endoplasmic Reticulum Communication-Mediated Oxidative Stress and Autophagy. Biomed. Res. Int. 2022, 2022, 6459585. [Google Scholar] [CrossRef]
- Mao, H.; Chen, W.; Chen, L.; Li, L. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochem. Pharmacol. 2022, 199, 115011. [Google Scholar] [CrossRef]
- Gan, Z.Y.; Callegari, S.; Cobbold, S.A.; Cotton, T.R.; Mlodzianoski, M.J.; Schubert, A.F.; Geoghegan, N.D.; Rogers, K.L.; Leis, A.; Dewson, G.; et al. Activation mechanism of PINK1. Nature 2022, 602, 328–335, Erratum in Nature 2022, 603, E33. [Google Scholar] [CrossRef]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER-Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef]
- Barodia, S.K.; Prabhakaran, K.; Karunakaran, S.; Mishra, V.; Tapias, V. Editorial: Mitochondria and Endoplasmic Reticulum Dysfunction in Parkinson’s Disease. Front. Neurosci. 2019, 13, 1171. [Google Scholar] [CrossRef]
- Chen, J.; Li, L.; Bai, X.; Xiao, L.; Shangguan, J.; Zhang, W.; Zhang, X.; Wang, S.; Liu, G. Inhibition of Autophagy Prevents Panax Notoginseng Saponins (PNS) Protection on Cardiac Myocytes Against Endoplasmic Reticulum (ER) Stress-Induced Mitochondrial Injury, Ca2+ Homeostasis and Associated Apoptosis. Front. Pharmacol. 2021, 12, 620812. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Blasiak, J.; Liton, P.; Boulton, M.; Klionsky, D.J.; Sinha, D. Autophagy in age-related macular degeneration. Autophagy 2023, 19, 388–400. [Google Scholar] [CrossRef]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 2021, 17, 385–401. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2023, 77, 619–639. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Qu, M.; Shen, T.; Su, J.; Xu, Y.; Xu, C.; Barkat, M.Q.; Cai, J.; Zhu, H.; Zeng, L.H.; et al. Control of mitochondria-associated endoplasmic reticulum membranes by protein S-palmitoylation: Novel therapeutic targets for neurodegenerative diseases. Ageing Res. Rev. 2023, 87, 101920. [Google Scholar] [CrossRef]
- Liu, N.; Bai, L.; Lu, Z.; Gu, R.; Zhao, D.; Yan, F.; Bai, J. TRPV4 contributes to ER stress and inflammation: Implications for Parkinson’s disease. J. Neuroinflammation 2022, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Kamarehei, M.; Kabudanian Ardestani, S.; Firouzi, M.; Zahednasab, H.; Keyvani, H.; Harirchian, M.H. Increased expression of endoplasmic reticulum stress-related caspase-12 and CHOP in the hippocampus of EAE mice. Brain Res. Bull. 2019, 147, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Wang, X.L.; Rao, J.Y.; Zhu, H.F.; Qi, H.Y.; Tian, Z. Periplaneta americana L. extract exerts neuroprotective effects by inhibiting endoplasmic reticulum stress via AKT-dependent pathway in experimental models of Parkinson’s disease. Chin. Med. 2024, 19, 157. [Google Scholar] [CrossRef]
- Monzel, A.S.; Enríquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef]
- Baker, Z.N.; Forny, P.; Pagliarini, D.J. Mitochondrial proteome research: The road ahead. Nat. Rev. Mol. Cell Biol. 2024, 25, 65–82. [Google Scholar] [CrossRef]
- Wang, H.F.; Wang, Z.Q.; Ding, Y.; Piao, M.H.; Feng, C.S.; Chi, G.F.; Luo, Y.N.; Ge, P.F. Endoplasmic reticulum stress regulates oxygen-glucose deprivation-induced parthanatos in human SH-SY5Y cells via improvement of intracellular ROS. CNS Neurosci. Ther. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Duan, J.; Matute, J.D.; Unger, L.W.; Hanley, T.; Schnell, A.; Lin, X.; Krupka, N.; Griebel, P.; Lambden, C.; Sit, B.; et al. Endoplasmic reticulum stress in the intestinal epithelium initiates purine metabolite synthesis and promotes Th17 cell differentiation in the gut. Immunity 2023, 56, 1115–1131.e9. [Google Scholar] [CrossRef]
- Fernández, A.; Ordóñez, R.; Reiter, R.J.; González-Gallego, J.; Mauriz, J.L. Melatonin and endoplasmic reticulum stress: Relation to autophagy and apoptosis. J. Pineal Res. 2015, 59, 292–307. [Google Scholar] [CrossRef]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Dadarao Chakole, R.; Nemade, L.S.; Kishor Kale, N.; Borah, S.; Shrikant Deokar, S.; et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis -An updated review. Mitochondrion 2023, 71, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, H.; Gu, W.; Wei, Y.; Mou, S.; Wang, Y.; Zhang, J.; Zhong, Q. CGI1746 targets σ1R to modulate ferroptosis through mitochondria-associated membranes. Nat. Chem. Biol. 2024, 20, 699–709. [Google Scholar] [CrossRef]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef]
- Weindel, C.G.; Martinez, E.L.; Zhao, X.; Mabry, C.J.; Bell, S.L.; Vail, K.J.; Coleman, A.K.; VanPortfliet, J.J.; Zhao, B.; Wagner, A.R.; et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 2022, 185, 3214–3231.e23. [Google Scholar] [CrossRef]
- Markovinovic, A.; Greig, J.; Martín-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic reticulum-mitochondria signaling in neurons and neurodegenerative diseases. J. Cell Sci. 2022, 135, jcs248534. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Li, Z.; Zhao, M.; Wang, D.; Sun, Z.; Wen, P.; Dai, Y.; Gou, F.; Ji, Y.; et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases. Ageing Res. Rev. 2023, 84, 101817. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, H.; Li, A.M.; Liu, Y.T.; Liu, Y.; Zhang, W.; Yang, C.; Song, N.; Zhan, M.; Yang, S. VDR regulates mitochondrial function as a protective mechanism against renal tubular cell injury in diabetic rats. Redox Biol. 2024, 70, 103062. [Google Scholar] [CrossRef]
- Yi, J.; Wang, H.L.; Lu, G.; Zhang, H.; Wang, L.; Li, Z.Y.; Wang, L.; Wu, Y.; Xia, D.; Fang, E.F.; et al. Spautin-1 promotes PINK1-PRKN-dependent mitophagy and improves associative learning capability in an alzheimer disease animal model. Autophagy 2024, 20, 2655–2676. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 2020, 39, e100875, Erratum in EMBO J. 2020, 39, e105826. [Google Scholar] [CrossRef]
- Grossmann, D.; Malburg, N.; Glaß, H.; Weeren, V.; Sondermann, V.; Pfeiffer, J.F.; Petters, J.; Lukas, J.; Seibler, P.; Klein, C.; et al. Mitochondria-Endoplasmic Reticulum Contact Sites Dynamics and Calcium Homeostasis Are Differentially Disrupted in PINK1-PD or PRKN-PD Neurons. Mov. Disord. 2023, 38, 1822–1836. [Google Scholar] [CrossRef]
- Erpapazoglou, Z.; Corti, O. The endoplasmic reticulum/mitochondria interface: A subcellular platform for the orchestration of the functions of the PINK1-Parkin pathway? Biochem. Soc. Trans. 2015, 43, 297–301. [Google Scholar] [CrossRef]
- Zhang, S.; Lv, Y.; Luo, X.; Weng, X.; Qi, J.; Bai, X.; Zhao, C.; Zeng, M.; Bao, X.; Dai, X.; et al. Homocysteine promotes atherosclerosis through macrophage pyroptosis via endoplasmic reticulum stress and calcium disorder. Mol. Med. 2023, 29, 73. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, C.J.; Ho, T.C.; Brumwell, A.M.; Kathiriya, J.J.; Wei, Y.; Hughes, J.B.; Garakani, K.; Atabai, K.; Auyeung, V.C.; Papa, F.R.; et al. BCL-2 Modulates IRE1α Activation to Attenuate Endoplasmic Reticulum Stress and Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2024, 70, 247–258. [Google Scholar] [CrossRef]
- Zhou, Z.; Torres, M.; Sha, H.; Halbrook, C.J.; Van den Bergh, F.; Reinert, R.B.; Yamada, T.; Wang, S.; Luo, Y.; Hunter, A.H.; et al. Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science 2020, 368, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef]
- Chang, C.Y.; Pan, P.H.; Wu, C.C.; Liao, S.L.; Chen, W.Y.; Kuan, Y.H.; Wang, W.Y.; Chen, C.J. Endoplasmic Reticulum Stress Contributes to Gefitinib-Induced Apoptosis in Glioma. Int. J. Mol. Sci. 2021, 22, 3934. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Zhou, F.; Gao, H.; Shang, L.; Li, J.; Zhang, M.; Wang, S.; Li, R.; Ye, L.; Yang, S. Oridonin promotes endoplasmic reticulum stress via TP53-repressed TCF4 transactivation in colorectalcancer. J. Exp. Clin. Cancer Res. 2023, 42, 150. [Google Scholar] [CrossRef]
- Xie, X.; Deng, T.; Duan, J.; Xie, J.; Yuan, J.; Chen, M. Exposure to polystyrene microplastics causes reproductive toxicity through oxidative stress and activation of the p38 MAPK signaling pathway. Ecotoxicol. Environ. Saf. 2020, 190, 110133. [Google Scholar] [CrossRef]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative stress and regulated cell death inParkinson’s disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef] [PubMed]
- Fujii, C.; Zorumski, C.F.; Izumi, Y. Endoplasmic reticulum stress, autophagy, neuroinflammation, and sigma 1 receptors as contributors to depression and its treatment. Neural Regen. Res. 2024, 19, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Akhter, N.; Wilson, A.; Arefanian, H.; Thomas, R.; Kochumon, S.; Al-Rashed, F.; Abu-Farha, M.; Al-Madhoun, A.; Al-Mulla, F.; Ahmad, R.; et al. Endoplasmic Reticulum Stress Promotes the Expression of TNF-α in THP-1 Cells by Mechanisms Involving ROS/CHOP/HIF-1α and MAPK/NF-κB Pathways. Int. J. Mol. Sci. 2023, 24, 15186. [Google Scholar] [CrossRef]
- Jo, S.L.; Yang, H.; Lee, H.W.; Hong, E.J. Curcumae radix Reduces Endoplasmic Reticulum Stress in Mice with Chronic Neuroinflammation. Biomedicines 2023, 11, 2107. [Google Scholar] [CrossRef]
- Shields, D.C.; Haque, A.; Banik, N.L. Neuroinflammatory responses of microglia in central nervous system trauma. J. Cereb Blood Flow. Metab. 2020, 40 (Suppl. 1), S25–S33. [Google Scholar] [CrossRef]
- Hasel, P.; Rose, I.V.L.; Sadick, J.S.; Kim, R.D.; Liddelow, S.A. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat. Neurosci. 2021, 24, 1475–1487. [Google Scholar] [CrossRef]
- Xiao, P.; Hu, Z.; Lang, J.; Pan, T.; Mertens, R.T.; Zhang, H.; Guo, K.; Shen, M.; Cheng, H.; Zhang, X.; et al. Mannose metabolism normalizes gut homeostasis by blocking the TNF-α-mediated proinflammatory circuit. Cell. Mol. Immunol. 2023, 20, 119–130. [Google Scholar] [CrossRef]
- Krupkova, O.; Sadowska, A.; Kameda, T.; Hitzl, W.; Hausmann, O.N.; Klasen, J.; Wuertz-Kozak, K. p38 MAPK Facilitates Crosstalk Between Endoplasmic Reticulum Stress and IL-6 Release in the Intervertebral Disc. Front. Immunol. 2018, 9, 1706. [Google Scholar] [CrossRef]
- Yoo, Y.M.; Joo, S.S. Melatonin Can Modulate Neurodegenerative Diseases by RegulatingEndoplasmic Reticulum Stress. Int. J. Mol. Sci. 2023, 24, 2381. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.W.; Wang, Z.M.; Sun, S.M.; Su, Y.; Li, Z.H.; Shao, J.J.; Tan, S.Z.; Chen, A.P.; Wang, S.J.; Zhang, Z.L.; et al. Endoplasmic reticulum stress and protein degradation in chronic liver disease. Pharmacol. Res. 2020, 161, 105218. [Google Scholar] [CrossRef]
- Chen, W.; Hu, Y.; Ju, D. Gene therapy for neurodegenerative disorders: Advances, insights and prospects. Acta Pharm. Sin. B 2020, 10, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Sharmin, M.M.; Hayashi, S.; Miyaji, M.; Ishizaki, H.; Matsuyama, H.; Haga, S.; Yonekura, S. Insulin-like growth factor-1 induces IRE1-XBP1-dependent endoplasmic reticulum biogenesis in bovine mammary epithelial cells. J. Dairy Sci. 2021, 104, 12094–12104. [Google Scholar] [CrossRef]
- Guo, J.; Ren, R.; Sun, K.; Yao, X.; Lin, J.; Wang, G.; Guo, Z.; Xu, T.; Guo, F. PERK controls bone homeostasis through the regulation of osteoclast differentiation and function. Cell Death Dis. 2020, 11, 847. [Google Scholar] [CrossRef]
- Almeida, L.M.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. The PERKs of mitochondria protection during stress: Insights for PERK modulation in neurodegenerative and metabolic diseases. Biol. Rev. Camb. Philos. Soc. 2022, 97, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Muqit, M.M.K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 2019, 705, 7–13. [Google Scholar] [CrossRef]
- Sun, C.P.; Zhou, J.J.; Yu, Z.L.; Huo, X.K.; Zhang, J.; Morisseau, C.; Hammock, B.D.; Ma, X.C. Kurarinone alleviated Parkinson’s disease via stabilization of epoxyeicosatrienoic acids in animal model. Proc. Natl. Acad. Sci. USA 2022, 119, e2118818119. [Google Scholar] [CrossRef]
- Hu, H.; Cao, B.; Huang, D.; Lin, Y.; Zhou, B.; Ying, J.; Huang, L.; Zhang, L. Withaferin a modulation of microglia autophagy mitigates neuroinflammation and enhances cognitive function in POCD. Sci. Rep. 2024, 14, 26112. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Zheng, Y.; Zhang, Y.; Zhang, X.J.; Wang, H.; Du, Y.; Guan, J.; Wang, X.; Fu, J. NAD+ improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J. Neuroinflammation 2021, 18, 207. [Google Scholar] [CrossRef]
- Han, X.; Xu, T.; Fang, Q.; Zhang, H.; Yue, L.; Hu, G.; Sun, L. Quercetin hinders microglialactivation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol. 2021, 44, 102010. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, X.; Liu, T.; Wei, J. Parkinson’s Disease: The Neurodegenerative Enigma Under the “Undercurrent” of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2025, 26, 3367. https://doi.org/10.3390/ijms26073367
Kong X, Liu T, Wei J. Parkinson’s Disease: The Neurodegenerative Enigma Under the “Undercurrent” of Endoplasmic Reticulum Stress. International Journal of Molecular Sciences. 2025; 26(7):3367. https://doi.org/10.3390/ijms26073367
Chicago/Turabian StyleKong, Xiangrui, Tingting Liu, and Jianshe Wei. 2025. "Parkinson’s Disease: The Neurodegenerative Enigma Under the “Undercurrent” of Endoplasmic Reticulum Stress" International Journal of Molecular Sciences 26, no. 7: 3367. https://doi.org/10.3390/ijms26073367
APA StyleKong, X., Liu, T., & Wei, J. (2025). Parkinson’s Disease: The Neurodegenerative Enigma Under the “Undercurrent” of Endoplasmic Reticulum Stress. International Journal of Molecular Sciences, 26(7), 3367. https://doi.org/10.3390/ijms26073367