
Reactive Oxygen Species as a Common Pathological Link Between Alcohol Use Disorder and Alzheimer’s Disease with Therapeutic Implications


Abstract
1. Introduction
2. Reactive Oxygen Species (ROS)
3. Alcohol, ROS, and Mitochondria
4. ROS and AUD
5. ROS and AD
6. Mechanisms of ROS Dysregulation in AD
7. ROS Play Distinct Roles in AUD and AD
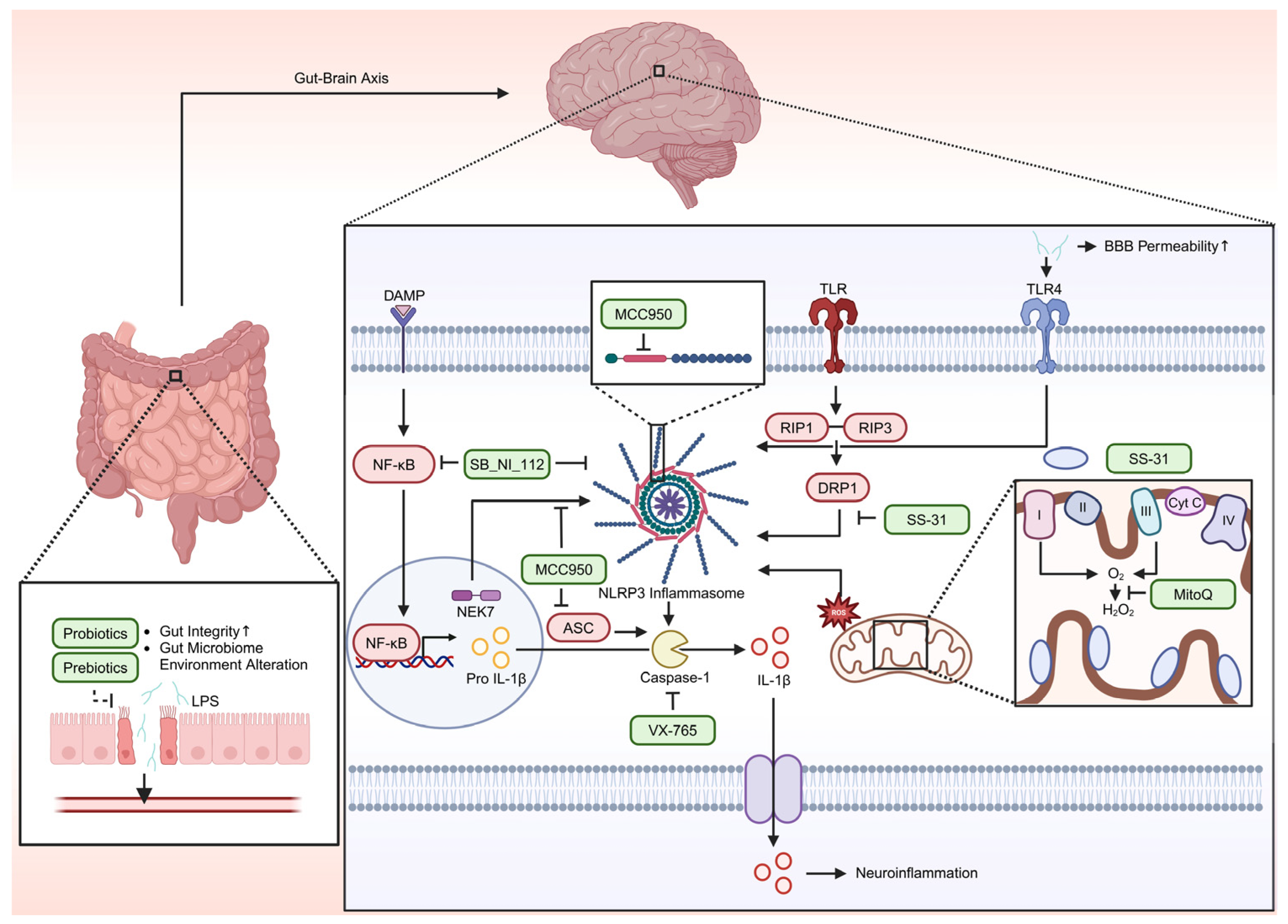
8. Therapeutic Target
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| AUD | Alcohol use disorder |
| AD | Alzheimer’s disease |
| Aβ | Amyloid-beta |
| TLR4 | Toll-like receptor 4 |
| LPS | Lipopolysaccharide |
| ETC | Electron transport chain |
| CYP2E1 | Cytochrome P450 2E1 |
| ADH | Alcohol dehydrogenase |
| MEOS | Microsomal ethanol oxidizing system |
| SOD | Superoxide dismutase |
| GPx | Glutathione peroxidase |
| CAT | Catalase |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| NOX | NADPH oxidase |
| Drp1 | Dynamin-related protein 1 |
| APP | Amyloid beta precursor protein |
| HFrEF | Heart failure with reduced ejection fraction |
| TEAEs | Treatment-emergent adverse events |
| mPTP | Mitochondrial permeability transition pore |
| MDA | Malondialdehyde |
| GLT-1 | Glutamate transporter-1 |
| UPR | Unfolded protein response |
| ABAD | Aβ-binding alcohol dehydrogenase |
| OXPHOS | Oxidative phosphorylation |
| NLRP3 | Pyrin domain-containing protein 3 |
| BBB | Blood–brain barrier |
| PI3K | Phosphoinositide 3-kinase |
| MAPK | Mitogen-activated protein kinase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| Keap1 | Kelch like-ECH-associated protein 1 |
| NF-κB | Nuclear factor kappa B |
| PAMP | Pathogen-associated molecular patterns |
| miRNAs | microRNAs |
| EAAT1 | Excitatory amino acid transporter 1 |
| EAAT2 | Excitatory amino acid transporter 2 |
References
- Bhatt, S.; Puli, L.; Patil, C.R. Role of reactive oxygen species in the progression of Alzheimer’s disease. Drug Discov. Today 2021, 26, 794–803. [Google Scholar] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar]
- Pritchard, K.A., Jr.; Ackerman, A.W.; Gross, E.R.; Stepp, D.W.; Shi, Y.; Fontana, J.T.; Baker, J.E.; Sessa, W.C. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J. Biol. Chem. 2001, 276, 17621–17624. [Google Scholar] [PubMed]
- Ye, Z.; Liu, Y.; Jin, X.; Wu, Y.; Zhao, H.; Gao, T.; Deng, Q.; Cheng, J.; Lin, J.; Tong, Z. Abeta-binding with alcohol dehydrogenase drives Alzheimer’s disease pathogenesis: A review. Int. J. Biol. Macromol. 2024, 264 Pt 2, 130580. [Google Scholar]
- Tamagno, E.; Parola, M.; Bardini, P.; Piccini, A.; Borghi, R.; Guglielmotto, M.; Santoro, G.; Davit, A.; Danni, O.; Smith, M.A.; et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 2005, 92, 628–636. [Google Scholar]
- Kany, S.; Janicova, A.; Relja, B. Innate Immunity and Alcohol. J. Clin. Med. 2019, 8, 1981. [Google Scholar] [CrossRef]
- Subramaniyan, V.; Chakravarthi, S.; Jegasothy, R.; Seng, W.Y.; Fuloria, N.K.; Fuloria, S.; Hazarika, I.; Das, A. Alcohol-associated liver disease: A review on its pathophysiology, diagnosis and drug therapy. Toxicol. Rep. 2021, 8, 376–385. [Google Scholar] [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef]
- Kong, E.Q.Z.; Subramaniyan, V.; Lubau, N.S.A. Uncovering the impact of alcohol on internal organs and reproductive health: Exploring TLR4/NF-kB and CYP2E1/ROS/Nrf2 pathways. Anim. Model. Exp. Med. 2024, 7, 444–459. [Google Scholar]
- Schwarzinger, M.; Pollock, B.G.; Hasan, O.S.M.; Dufouil, C.; Rehm, J.; QalyDays Study Group. Contribution of alcohol use disorders to the burden of dementia in France 2008-13: A nationwide retrospective cohort study. Lancet Public Health 2018, 3, e124–e132. [Google Scholar]
- Zhang, P.; Edenberg, H.J.; Nurnberger, J.; Lai, D.; Cheng, F.; Liu, Y. Alcohol use disorder is associated with higher risks of Alzheimer’s and Parkinson’s diseases: A study of US insurance claims data. Alzheimer’s Dement. 2022, 14, e12370. [Google Scholar]
- Kang, S.; Lee, J.; Ali, D.N.; Choi, S.; Nesbitt, J.; Min, P.H.; Trushina, E.; Choi, D.S. Low to moderate ethanol exposure reduces astrocyte-induced neuroinflammatory signaling and cognitive decline in presymptomatic APP/PS1 mice. Sci. Rep. 2024, 14, 23989. [Google Scholar]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [PubMed]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Balu, D.; Valencia-Olvera, A.C.; Nguyen, A.; Patnam, M.; York, J.; Peri, F.; Neumann, F.; LaDu, M.J.; Tai, L.M. A small-molecule TLR4 antagonist reduced neuroinflammation in female E4FAD mice. Alzheimer’s Res. Ther. 2023, 15, 181. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, R.; Hu, D.; Sun, X.; Fujioka, H.; Lundberg, K.; Chan, E.R.; Wang, Q.; Xu, R.; Flanagan, M.E.; et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci. Adv. 2020, 6, eabb8680. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar]
- Koopman, W.J.; Nijtmans, L.G.; Dieteren, C.E.; Roestenberg, P.; Valsecchi, F.; Smeitink, J.A.; Willems, P.H. Mammalian mitochondrial complex I: Biogenesis, regulation, and reactive oxygen species generation. Antioxid. Redox Signal 2010, 12, 1431–1470. [Google Scholar] [PubMed]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar]
- Kaminskyy, V.O.; Zhivotovsky, B. Free radicals in cross talk between autophagy and apoptosis. Antioxid. Redox Signal. 2014, 21, 86–102. [Google Scholar]
- Benhar, M. Oxidants, Antioxidants and Thiol Redox Switches in the Control of Regulated Cell Death Pathways. Antioxidants 2020, 9, 309. [Google Scholar] [CrossRef]
- Covarrubias, L.; Hernandez-Garcia, D.; Schnabel, D.; Salas-Vidal, E.; Castro-Obregon, S. Function of reactive oxygen species during animal development: Passive or active? Dev. Biol. 2008, 320, 1–11. [Google Scholar]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar]
- Poyton, R.O.; Castello, P.R.; Ball, K.A.; Woo, D.K.; Pan, N. Mitochondria and hypoxic signaling: A new view. Ann. N. Y. Acad. Sci. 2009, 1177, 48–56. [Google Scholar] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. 1998, 105, 855–870. [Google Scholar] [PubMed]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109 (Suppl. S1), 153–159. [Google Scholar]
- Koop, D.R.; Coon, M.J. Ethanol oxidation and toxicity: Role of alcohol P-450 oxygenase. Alcohol. Clin. Exp. Res. 1986, 10 (Suppl. S6), 44S–49S. [Google Scholar] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [PubMed]
- Koop, D.R. Alcohol metabolism’s damaging effects on the cell: A focus on reactive oxygen generation by the enzyme cytochrome P450 2E1. Alcohol Res. Health 2006, 29, 274–280. [Google Scholar]
- Okoye, C.N.; Koren, S.A.; Wojtovich, A.P. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. 2023, 67, 102926. [Google Scholar]
- Terada, T.; Therriault, J.; Kang, M.S.; Savard, M.; Pascoal, T.A.; Lussier, F.; Tissot, C.; Wang, Y.T.; Benedet, A.; Poltronetti, N.M.; et al. Mitochondrial complex I abnormalities underlie neurodegeneration and cognitive decline in Alzheimer’s disease. Eur. J. Neurol. 2022, 29, 1324–1334. [Google Scholar]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar]
- Brand, M.D. Riding the tiger—Physiological and pathological effects of superoxide and hydrogen peroxide generated in the mitochondrial matrix. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 592–661. [Google Scholar]
- Diaz, F.; Garcia, S.; Padgett, K.R.; Moraes, C.T. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum. Mol. Genet. 2012, 21, 5066–5077. [Google Scholar]
- Bayo Jimenez, M.T.; Frenis, K.; Hahad, O.; Steven, S.; Cohen, G.; Cuadrado, A.; Munzel, T.; Daiber, A. Protective actions of nuclear factor erythroid 2-related factor 2 (NRF2) and downstream pathways against environmental stressors. Free Radic. Biol. Med. 2022, 187, 72–91. [Google Scholar]
- Cross, C.E.; Halliwell, B.; Allen, A. Antioxidant protection: A function of tracheobronchial and gastrointestinal mucus. Lancet 1984, 1, 1328–1330. [Google Scholar]
- Wu, D.; Cederbaum, A.I. Oxidative stress and alcoholic liver disease. Semin. Liver Dis. 2009, 29, 141–154. [Google Scholar] [PubMed]
- Palmieri, V.O.; Grattagliano, I.; Palasciano, G. Ethanol induces secretion of oxidized proteins by pancreatic acinar cells. Cell Biol. Toxicol. 2007, 23, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, L.; Liu, T.; Wang, J.; Wen, A.; Ding, Y. Ellagic acid protects mice against sleep deprivation-induced memory impairment and anxiety by inhibiting TLR4 and activating Nrf2. Aging 2020, 12, 10457–10472. [Google Scholar] [PubMed]
- Lv, Y.N.; Cui, Y.; Zhang, B.; Huang, S.M. Sleep deficiency promotes Alzheimer’s disease development and progression. Front. Neurol. 2022, 13, 1053942. [Google Scholar] [CrossRef] [PubMed]
- Tavares, W.M.; Araujo de Franca, S.; Paiva, W.S.; Teixeira, M.J. Early tracheostomy versus late tracheostomy in severe traumatic brain injury or stroke: A systematic review and meta-analysis. Aust. Crit. Care 2023, 36, 1110–1116. [Google Scholar] [CrossRef]
- Leon, B.E.; Kang, S.; Franca-Solomon, G.; Shang, P.; Choi, D.S. Alcohol-Induced Neuroinflammatory Response and Mitochondrial Dysfunction on Aging and Alzheimer’s Disease. Front. Behav. Neurosci. 2021, 15, 778456. [Google Scholar] [CrossRef]
- Waddell, J.; McKenna, M.C.; Kristian, T. Brain ethanol metabolism and mitochondria. Curr. Top. Biochem. Res. 2022, 23, 1–13. [Google Scholar]
- Jin, M.; Ande, A.; Kumar, A.; Kumar, S. Regulation of cytochrome P450 2e1 expression by ethanol: Role of oxidative stress-mediated pkc/jnk/sp1 pathway. Cell Death Dis. 2013, 4, e554. [Google Scholar]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef]
- Feng, J.; Zheng, Y.; Guo, M.; Ares, I.; Martinez, M.; Lopez-Torres, B.; Martinez-Larranaga, M.R.; Wang, X.; Anadon, A.; Martinez, M.A. Oxidative stress, the blood-brain barrier and neurodegenerative diseases: The critical beneficial role of dietary antioxidants. Acta Pharm. Sin. B 2023, 13, 3988–4024. [Google Scholar] [CrossRef]
- Tsermpini, E.E.; Plemenitas Iljes, A.; Dolzan, V. Alcohol-Induced Oxidative Stress and the Role of Antioxidants in Alcohol Use Disorder: A Systematic Review. Antioxidants 2022, 11, 1374. [Google Scholar] [CrossRef] [PubMed]
- Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and mitochondrial effects of alcohol consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281–4304. [Google Scholar] [CrossRef]
- Mansouri, A.; Gaou, I.; De Kerguenec, C.; Amsellem, S.; Haouzi, D.; Berson, A.; Moreau, A.; Feldmann, G.; Letteron, P.; Pessayre, D.; et al. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology 1999, 117, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Loyola, R.; Canelo, F.; Aranguiz, A.; Tapia-Monsalves, C.; Osorio-Fuentealba, C.; Quintanilla, R.A. NADPH oxidase contributes to oxidative damage and mitochondrial impairment induced by acute ethanol treatment in rat hippocampal neurons. Neuropharmacology 2020, 171, 108100. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1alpha Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef]
- Chen, L.; Qin, Y.; Liu, B.; Gao, M.; Li, A.; Li, X.; Gong, G. PGC-1alpha-Mediated Mitochondrial Quality Control: Molecular Mechanisms and Implications for Heart Failure. Front. Cell Dev. Biol. 2022, 10, 871357. [Google Scholar]
- Rius-Perez, S.; Torres-Cuevas, I.; Millan, I.; Ortega, A.L.; Perez, S. PGC-1alpha, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Shang, P.; Lindberg, D.; Starski, P.; Peyton, L.; Hong, S.I.; Choi, S.; Choi, D.S. Chronic Alcohol Exposure Induces Aberrant Mitochondrial Morphology and Inhibits Respiratory Capacity in the Medial Prefrontal Cortex of Mice. Front. Neurosci. 2020, 14, 561173. [Google Scholar]
- Oberdoerster, J.; Rabin, R.A. Enhanced caspase activity during ethanol-induced apoptosis in rat cerebellar granule cells. Eur. J. Pharmacol. 1999, 385, 273–282. [Google Scholar] [PubMed]
- De Nicolo, B.; Cataldi-Stagetti, E.; Diquigiovanni, C.; Bonora, E. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants 2023, 12, 353. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Pan, C.H.; Chen, C.C.; Huang, M.C. Increased oxidative DNA damage in patients with alcohol dependence and its correlation with alcohol withdrawal severity. Alcohol. Clin. Exp. Res. 2011, 35, 338–344. [Google Scholar]
- Yang, M.; Zhou, X.; Tan, X.; Huang, X.; Yuan, L.; Zhang, Z.; Yang, Y.; Xu, M.; Wan, Y.; Li, Z. The Status of Oxidative Stress in Patients with Alcohol Dependence: A Meta-Analysis. Antioxidants 2022, 11, 1919. [Google Scholar] [CrossRef]
- Pemberton, P.W.; Smith, A.; Warnes, T.W. Non-invasive monitoring of oxidant stress in alcoholic liver disease. Scand. J. Gastroenterol. 2005, 40, 1102–1108. [Google Scholar] [CrossRef]
- Kitagaki, H.; Araki, Y.; Funato, K.; Shimoi, H. Ethanol-induced death in yeast exhibits features of apoptosis mediated by mitochondrial fission pathway. FEBS Lett. 2007, 581, 2935–2942. [Google Scholar]
- Breido, I.S. On the 120th anniversary of the antiseptic method of Joseph Lister (1867–1987). Vestn. Khir Im. I I Grek. 1988, 140, 123–125. [Google Scholar]
- Kieron, M.; Zekanowski, C.; Falk, A.; Wezyk, M. Oxidative DNA Damage Signalling in Neural Stem Cells in Alzheimer’s Disease. Oxid. Med. Cell Longev. 2019, 2019, 2149812. [Google Scholar] [CrossRef]
- Rehm, J.; Hasan, O.S.M.; Black, S.E.; Shield, K.D.; Schwarzinger, M. Alcohol use and dementia: A systematic scoping review. Alzheimer’s Res. Ther. 2019, 11, 1. [Google Scholar]
- Sullivan, E.V.; Pfefferbaum, A. Brain-behavior relations and effects of aging and common comorbidities in alcohol use disorder: A review. Neuropsychology 2019, 33, 760–780. [Google Scholar] [PubMed]
- Lopez-Valencia, L.; Moya, M.; Escudero, B.; Garcia-Bueno, B.; Orio, L. Bacterial lipopolysaccharide forms aggregates with apolipoproteins in male and female rat brains after ethanol binges. J. Lipid Res. 2024, 65, 100509. [Google Scholar]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Wang, Q.; Nagy, L.E. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol. 2006, 79, 1348–1356. [Google Scholar]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar]
- Vorhees, C.V.; Williams, M.T. Assessing spatial learning and memory in rodents. ILAR J. 2014, 55, 310–332. [Google Scholar]
- Di Lorenzo, F.; De Castro, C.; Silipo, A.; Molinaro, A. Lipopolysaccharide structures of Gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol. Rev. 2019, 43, 257–272. [Google Scholar] [PubMed]
- Crews, F.T.; Sarkar, D.K.; Qin, L.; Zou, J.; Boyadjieva, N.; Vetreno, R.P. Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Res. 2015, 37, 331–341, 344–351. [Google Scholar]
- McManus, R.M.; Latz, E. NLRP3 inflammasome signalling in Alzheimer’s disease. Neuropharmacology 2024, 252, 109941. [Google Scholar]
- Ramos, A.; Joshi, R.S.; Szabo, G. Innate immune activation: Parallels in alcohol use disorder and Alzheimer’s disease. Front. Mol. Neurosci. 2022, 15, 910298. [Google Scholar]
- Rasool, A.E.; Furlong, T.; Prasad, A.A. Microglia activity in the human basal ganglia is altered in alcohol use disorder and reversed with remission from alcohol. Addict. Biol. 2024, 29, e13374. [Google Scholar]
- Graves, A.R.; Moore, S.J.; Bloss, E.B.; Mensh, B.D.; Kath, W.L.; Spruston, N. Hippocampal pyramidal neurons comprise two distinct cell types that are countermodulated by metabotropic receptors. Neuron 2012, 76, 776–789. [Google Scholar] [PubMed]
- Fukui, K.; Takatsu, H.; Shinkai, T.; Suzuki, S.; Abe, K.; Urano, S. Appearance of amyloid beta-like substances and delayed-type apoptosis in rat hippocampus CA1 region through aging and oxidative stress. J. Alzheimer’s Dis. 2005, 8, 299–309. [Google Scholar]
- Igarashi, K.M. Entorhinal cortex dysfunction in Alzheimer’s disease. Trends Neurosci. 2023, 46, 124–136. [Google Scholar]
- Olajide, O.J.; Suvanto, M.E.; Chapman, C.A. Molecular mechanisms of neurodegeneration in the entorhinal cortex that underlie its selective vulnerability during the pathogenesis of Alzheimer’s disease. Biol. Open 2021, 10, bio056796. [Google Scholar] [CrossRef]
- de Flores, R.; Das, S.R.; Xie, L.; Wisse, L.E.M.; Lyu, X.; Shah, P.; Yushkevich, P.A.; Wolk, D.A. Medial Temporal Lobe Networks in Alzheimer’s Disease: Structural and Molecular Vulnerabilities. J. Neurosci. 2022, 42, 2131–2141. [Google Scholar] [PubMed]
- Du, A.T.; Schuff, N.; Kramer, J.H.; Rosen, H.J.; Gorno-Tempini, M.L.; Rankin, K.; Miller, B.L.; Weiner, M.W. Different regional patterns of cortical thinning in Alzheimer’s disease and frontotemporal dementia. Brain 2007, 130, 1159–1166. [Google Scholar]
- Gonzalez-Rodriguez, M.; Villar-Conde, S.; Astillero-Lopez, V.; Villanueva-Anguita, P.; Ubeda-Banon, I.; Flores-Cuadrado, A.; Martinez-Marcos, A.; Saiz-Sanchez, D. Human amygdala involvement in Alzheimer’s disease revealed by stereological and dia-PASEF analysis. Brain Pathol. 2023, 33, e13180. [Google Scholar] [PubMed]
- Sakato, Y.; Shima, A.; Terada, Y.; Takeda, K.; Sakamaki-Tsukita, H.; Nishida, A.; Yoshimura, K.; Wada, I.; Furukawa, K.; Kambe, D.; et al. Delineating three distinct spatiotemporal patterns of brain atrophy in Parkinson’s disease. Brain 2024, 147, 3702–3713. [Google Scholar]
- Choi, D.H.; Lee, K.H.; Kim, J.H.; Seo, J.H.; Kim, H.Y.; Shin, C.Y.; Han, J.S.; Han, S.H.; Kim, Y.S.; Lee, J. NADPH oxidase 1, a novel molecular source of ROS in hippocampal neuronal death in vascular dementia. Antioxid. Redox Signal. 2014, 21, 533–550. [Google Scholar]
- Yi, J.H.; Kim, D.H.; Piers, T.M.; Kim, S.C.; Whitcomb, D.J.; Regan, P.; Cho, K. Postsynaptic p47phox regulates long-term depression in the hippocampus. Cell Discov. 2018, 4, 44. [Google Scholar]
- Davolio, C.; Greenamyre, J.T. Selective vulnerability of the CA1 region of hippocampus to the indirect excitotoxic effects of malonic acid. Neurosci. Lett. 1995, 192, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Muthuraju, S.; Badal, S.; Wooden, J.; Leasure, J.L.; Roman, G.; Das, J. Differential Expression of Presynaptic Munc13-1 and Munc13-2 in Mouse Hippocampus Following Ethanol Drinking. Neuroscience 2022, 487, 166–183. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, K.S.; Adra, N.; Salz, D.M.; Kemppainen, M.I.; Ruiz, S.M.; Harris, G.J.; Oscar-Berman, M. Hippocampal subfield volumes in abstinent men and women with a history of alcohol use disorder. PLoS ONE 2020, 15, e0236641. [Google Scholar]
- Zahr, N.M.; Pohl, K.M.; Saranathan, M.; Sullivan, E.V.; Pfefferbaum, A. Hippocampal subfield CA2+3 exhibits accelerated aging in Alcohol Use Disorder: A preliminary study. Neuroimage Clin. 2019, 22, 101764. [Google Scholar] [PubMed]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar]
- Floyd, R.A.; Carney, J.M. Free radical damage to protein and DNA: Mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann. Neurol. 1992, 32, S22–S27. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Tabner, B.J.; El-Agnaf, O.M.; German, M.J.; Fullwood, N.J.; Allsop, D. Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem. Soc. Trans. 2005, 33 Pt 5, 1082–1086. [Google Scholar]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.C.; Stern, D.M.; Arancio, O.; Yan, S.S. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005, 19, 597–598. [Google Scholar]
- Hurley, T.D.; Edenberg, H.J. Genes encoding enzymes involved in ethanol metabolism. Alcohol Res. 2012, 34, 339–344. [Google Scholar]
- Smith, K.R.; Lank, K.M.; Dismukes, W.E.; Cobbs, C.G. In vitro comparison of cilofungin alone and in combination with other antifungal agents against clinical isolates of Candida species. Eur. J. Clin. Microbiol. Infect. Dis. 1991, 10, 588–592. [Google Scholar] [PubMed]
- Pellerin, L.; Magistretti, P.J. Ampakine CX546 bolsters energetic response of astrocytes: A novel target for cognitive-enhancing drugs acting as alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor modulators. J. Neurochem. 2005, 92, 668–677. [Google Scholar]
- Frausto, D.M.; Engen, P.A.; Naqib, A.; Jackson, A.; Tran, L.; Green, S.J.; Shaikh, M.; Forsyth, C.B.; Keshavarzian, A.; Voigt, R.M. Impact of alcohol-induced intestinal microbiota dysbiosis in a rodent model of Alzheimer’s disease. Front. Aging 2022, 3, 916336. [Google Scholar]
- Frausto, D.M.; Forsyth, C.B.; Keshavarzian, A.; Voigt, R.M. Dietary Regulation of Gut-Brain Axis in Alzheimer’s Disease: Importance of Microbiota Metabolites. Front. Neurosci. 2021, 15, 736814. [Google Scholar]
- Peng, B.; Yang, Q.; B Joshi, R.; Liu, Y.; Akbar, M.; Song, B.J.; Zhou, S.; Wang, X. Role of Alcohol Drinking in Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2316. [Google Scholar] [CrossRef]
- Rao, P.S.; Bell, R.L.; Engleman, E.A.; Sari, Y. Targeting glutamate uptake to treat alcohol use disorders. Front. Neurosci. 2015, 9, 144. [Google Scholar]
- Rao, P.S.; Sari, Y. Glutamate transporter 1: Target for the treatment of alcohol dependence. Curr. Med. Chem. 2012, 19, 5148–5156. [Google Scholar]
- Kim, J.; Yoo, I.D.; Lim, J.; Moon, J.S. Pathological phenotypes of astrocytes in Alzheimer’s disease. Exp. Mol. Med. 2024, 56, 95–99. [Google Scholar]
- Zoia, C.; Cogliati, T.; Tagliabue, E.; Cavaletti, G.; Sala, G.; Galimberti, G.; Rivolta, I.; Rossi, V.; Frattola, L.; Ferrarese, C. Glutamate transporters in platelets: EAAT1 decrease in aging and in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 149–157. [Google Scholar] [PubMed]
- Alotaibi, A.; Travaglianti, S.; Wong, W.; Abou-Gharbia, M.; Childers, W.; Sari, Y. Effects of MC-100093 on Ethanol Drinking and the Expression of Astrocytic Glutamate Transporters in the Mesocorticolimbic Brain Regions of Male and Female Alcohol-Preferring Rats. Neuroscience 2024, 552, 89–99. [Google Scholar] [PubMed]
- Sari, Y.; Sreemantula, S.N. Neuroimmunophilin GPI-1046 reduces ethanol consumption in part through activation of GLT1 in alcohol-preferring rats. Neuroscience 2012, 227, 327–335. [Google Scholar] [PubMed]
- Liu, Y.; Tan, Y.; Zhang, Z.; Yi, M.; Zhu, L.; Peng, W. The interaction between ageing and Alzheimer’s disease: Insights from the hallmarks of ageing. Transl. Neurodegener. 2024, 13, 7. [Google Scholar]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar]
- Marzec, J.M.; Christie, J.D.; Reddy, S.P.; Jedlicka, A.E.; Vuong, H.; Lanken, P.N.; Aplenc, R.; Yamamoto, T.; Yamamoto, M.; Cho, H.Y.; et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007, 21, 2237–2246. [Google Scholar]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar]
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2N neurons. Biochimie 2010, 92, 245–253. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Arauna, D.; Fuentes, F.; Araya-Maturana, R.; Palomo, I.; Alarcon, M.; Sebastian, D.; Zorzano, A.; Fuentes, E. Mitoquinone (MitoQ) Inhibits Platelet Activation Steps by Reducing ROS Levels. Int. J. Mol. Sci. 2020, 21, 6192. [Google Scholar] [CrossRef]
- Kirkman, D.L.; Robinson, A.T.; Rossman, M.J.; Seals, D.R.; Edwards, D.G. Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2080–H2100. [Google Scholar] [PubMed]
- Sang, W.; Chen, S.; Lin, L.; Wang, N.; Kong, X.; Ye, J. Antioxidant mitoquinone ameliorates EtOH-LPS induced lung injury by inhibiting mitophagy and NLRP3 inflammasome activation. Front. Immunol. 2022, 13, 973108. [Google Scholar]
- Jin, B.R.; Lim, C.Y.; Kim, H.J.; Lee, M.; An, H.J. Antioxidant mitoquinone suppresses benign prostatic hyperplasia by regulating the AR-NLRP3 pathway. Redox Biol. 2023, 65, 102816. [Google Scholar]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409. [Google Scholar] [PubMed]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimer’s Dis. 2010, 20 (Suppl. S2), S609–S631. [Google Scholar] [CrossRef]
- Ciocca, M.; Pizzamiglio, C. Clinical Benefits of Therapeutic Interventions Targeting Mitochondria in Parkinson’s Disease Patients. CNS Neurol. Disord. Drug Targets 2024, 23, 554–561. [Google Scholar]
- Linder, B.A.; Stute, N.L.; Hutchison, Z.J.; Barnett, A.M.; Tharpe, M.A.; Kavazis, A.N.; Kirkman, D.L.; Gutierrez, O.M.; Robinson, A.T. Acute high-dose MitoQ does not increase urinary kidney injury markers in healthy adults: A randomized crossover trial. Am. J. Physiol. Renal Physiol. 2024, 326, F135–F142. [Google Scholar] [CrossRef]
- Ganguly, E.; Aljunaidy, M.M.; Kirschenman, R.; Spaans, F.; Morton, J.S.; Phillips, T.E.J.; Case, C.P.; Cooke, C.M.; Davidge, S.T. Sex-Specific Effects of Nanoparticle-Encapsulated MitoQ (nMitoQ) Delivery to the Placenta in a Rat Model of Fetal Hypoxia. Front. Physiol. 2019, 10, 562. [Google Scholar] [CrossRef]
- Marquez, B.T.; Leung, T.C.S.; Hui, J.; Charron, F.; McKinney, R.A.; Watt, A.J. A mitochondrial-targeted antioxidant (MitoQ) improves motor coordination and reduces Purkinje cell death in a mouse model of ARSACS. Neurobiol. Dis. 2023, 183, 106157. [Google Scholar]
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial protein interaction landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373. [Google Scholar]
- Li, M.; Kong, D.; Meng, L.; Wang, Z.; Bai, Z.; Wu, G. Discovery of novel SS-31 (d-Arg-dimethylTyr-Lys-Phe-NH(2)) derivatives as potent agents to ameliorate inflammation and increase mitochondrial ATP synthesis. RSC Adv. 2024, 14, 29789–29799. [Google Scholar]
- Zhu, Y.; Luo, M.; Bai, X.; Li, J.; Nie, P.; Li, B.; Luo, P. SS-31, a Mitochondria-Targeting Peptide, Ameliorates Kidney Disease. Oxid. Med. Cell. Longev. 2022, 2022, 1295509. [Google Scholar] [PubMed]
- Zhong, L.; Ren, X.; Ai, Y.; Liu, Z. SS-31 Improves Cognitive Function in Sepsis-Associated Encephalopathy by Inhibiting the Drp1-NLRP3 Inflammasome Activation. Neuromol. Med. 2023, 25, 230–241. [Google Scholar]
- Saad, A.; Herrmann, S.M.S.; Eirin, A.; Ferguson, C.M.; Glockner, J.F.; Bjarnason, H.; McKusick, M.A.; Misra, S.; Lerman, L.O.; Textor, S.C. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients with Atherosclerotic Renal Artery Stenosis. Circ. Cardiovasc. Interv. 2017, 10, 944. [Google Scholar]
- Karaa, A.; Bertini, E.; Carelli, V.; Cohen, B.H.; Enns, G.M.; Falk, M.J.; Goldstein, A.; Gorman, G.S.; Haas, R.; Hirano, M.; et al. Efficacy and Safety of Elamipretide in Individuals with Primary Mitochondrial Myopathy: The MMPOWER-3 Randomized Clinical Trial. Neurology 2023, 101, e238–e252. [Google Scholar] [PubMed]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of Elamipretide on Left Ventricular Function in Patients with Heart Failure with Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail. 2020, 26, 429–437. [Google Scholar]
- Brinkschulte, R.; Fussholler, D.M.; Hoss, F.; Rodriguez-Alcazar, J.F.; Lauterbach, M.A.; Kolbe, C.C.; Rauen, M.; Ince, S.; Herrmann, C.; Latz, E.; et al. ATP-binding and hydrolysis of human NLRP3. Commun. Biol. 2022, 5, 1176. [Google Scholar]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar]
- Liu, S.Q.; Xie, S.Y.; Zhang, T.; Zhang, H.; Chen, M.Y.; Xing, Y.; Zhao, N.; Li, L.; Chen, S.; Wang, S.S.; et al. Impeding Nucleotide-Binding Oligomerization Domain-Like Receptor 3 Inflammasome Ameliorates Cardiac Remodeling and Dysfunction in Obesity-Associated Cardiomyopathy. J. Am. Heart Assoc. 2024, 13, e035234. [Google Scholar]
- Naeem, A.; Prakash, R.; Kumari, N.; Ali Khan, M.; Quaiyoom Khan, A.; Uddin, S.; Verma, S.; Ab Robertson, A.; Boltze, J.; Shadab Raza, S. MCC950 reduces autophagy and improves cognitive function by inhibiting NLRP3-dependent neuroinflammation in a rat model of Alzheimer’s disease. Brain Behav. Immun. 2024, 116, 70–84. [Google Scholar]
- Ostergaard, J.A.; Jha, J.C.; Sharma, A.; Dai, A.; Choi, J.S.Y.; de Haan, J.B.; Cooper, M.E.; Jandeleit-Dahm, K. Adverse renal effects of NLRP3 inflammasome inhibition by MCC950 in an interventional model of diabetic kidney disease. Clin. Sci. 2022, 136, 167–180. [Google Scholar]
- Li, H.; Guan, Y.; Liang, B.; Ding, P.; Hou, X.; Wei, W.; Ma, Y. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome. Eur. J. Pharmacol. 2022, 928, 175091. [Google Scholar]
- Yan, W.; Shen, Y.; Huang, J.; Lu, L.; Zhang, Q. MCC950 Ameliorates Acute Liver Injury Through Modulating Macrophage Polarization and Myeloid-Derived Suppressor Cells Function. Front. Med. 2021, 8, 752223. [Google Scholar]
- Risen, S.J.; Boland, S.W.; Sharma, S.; Weisman, G.M.; Shirley, P.M.; Latham, A.S.; Hay, A.J.D.; Gilberto, V.S.; Hines, A.D.; Brindley, S.; et al. Targeting Neuroinflammation by Pharmacologic Downregulation of Inflammatory Pathways Is Neuroprotective in Protein Misfolding Disorders. ACS Chem. Neurosci. 2024, 15, 1533–1547. [Google Scholar] [PubMed]
- Anton, P.E.; Nagpal, P.; Moreno, J.; Burchill, M.A.; Chatterjee, A.; Busquet, N.; Mesches, M.; Kovacs, E.J.; McCullough, R.L. NF-kappaB/NLRP3 Translational Inhibition by Nanoligomer Therapy Mitigates Ethanol and Advanced Age-Related Neuroinflammation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Risen, S.; Sharma, S.; Gilberto, V.S.; Brindley, S.; Aguilar, M.; Brown, J.M.; Chatterjee, A.; Moreno, J.A.; Nagpal, P. Large- and Small-Animal Studies of Safety, Pharmacokinetics, and Biodistribution of Inflammasome-Targeting Nanoligomer in the Brain and Other Target Organs. ACS Pharmacol. Transl. Sci. 2024, 7, 3439–3451. [Google Scholar] [PubMed]
- Liang, Y.B.; Luo, R.X.; Lu, Z.; Mao, Y.; Song, P.P.; Li, Q.W.; Peng, Z.Q.; Zhang, Y.S. VX-765 attenuates secondary damage and beta-amyloid accumulation in ipsilateral thalamus after experimental stroke in rats. Exp. Neurol. 2025, 385, 115097. [Google Scholar]
- Feng, X.; Chen, Z.; Cheng, W.; Liu, C.; Liu, Q. Role for NLRP3 inflammasome-mediated, Caspase1-dependent response in glaucomatous trabecular meshwork cell death and regulation of aqueous humor outflow. Heliyon 2024, 10, e38258. [Google Scholar]
- Mbareche, H.; Dumont-Leblond, N.; Bilodeau, G.J.; Duchaine, C. An Overview of Bioinformatics Tools for DNA Meta-Barcoding Analysis of Microbial Communities of Bioaerosols: Digest for Microbiologists. Life 2020, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Dhani, S.; Zhao, Y.; Zhivotovsky, B. A long way to go: Caspase inhibitors in clinical use. Cell Death Dis. 2021, 12, 949. [Google Scholar]
- Shandilya, S.; Kumar, S.; Kumar Jha, N.; Kumar Kesari, K.; Ruokolainen, J. Interplay of gut microbiota and oxidative stress: Perspective on neurodegeneration and neuroprotection. J. Adv. Res. 2022, 38, 223–244. [Google Scholar]
- Yang, D.; Wang, Z.; Chen, Y.; Guo, Q.; Dong, Y. Interactions between gut microbes and NLRP3 inflammasome in the gut-brain axis. Comput. Struct. Biotechnol. J. 2023, 21, 2215–2227. [Google Scholar] [PubMed]
- Koutromanos, I.; Legaki, E.; Gazouli, M.; Vasilopoulos, E.; Kouzoupis, A.; Tzavellas, E. Gut microbiome in alcohol use disorder: Implications for health outcomes and therapeutic strategies-a literature review. World J. Methodol. 2024, 14, 88519. [Google Scholar]
- Aleman, R.S.; Moncada, M.; Aryana, K.J. Leaky Gut and the Ingredients That Help Treat It: A Review. Molecules 2023, 28, 619. [Google Scholar] [CrossRef] [PubMed]
- Escudero, B.; Moya, M.; Lopez-Valencia, L.; Arias, F.; Orio, L. Reelin Plasma Levels Identify Cognitive Decline in Alcohol Use Disorder Patients During Early Abstinence: The Influence of APOE4 Expression. Int. J. Neuropsychopharmacol. 2023, 26, 545–556. [Google Scholar]
- Hammond, T.C.; Xing, X.; Yanckello, L.M.; Stromberg, A.; Chang, Y.H.; Nelson, P.T.; Lin, A.L. Human Gray and White Matter Metabolomics to Differentiate APOE and Stage Dependent Changes in Alzheimer’s Disease. J. Cell Immunol. 2021, 3, 397–412. [Google Scholar]


{kind=link}
{kind=link}
| Category | Alcohol Use Disorder (AUD) | Alzheimer’s Disease (AD) | References |
|---|---|---|---|
| Primary Causes | Chronic heavy drinking and alcohol dependence | Aging, heredity (APOE4), environmental factors | [114,157] |
| Key Pathological Mechanisms | -Increased ROS production via alcohol metabolism -Mitochondrial dysfunction and ATP depletion -Neuroinflammation (TLR4, NLRP3 activation) | -Aβ plaque and tau protein aggregation -Oxidative stress and mitochondrial dysfunction -Neuroinflammation (TLR4, NLRP3 activation) | [2,10,57,59,115] |
| Neuronal Damage Mechanisms | -Cytotoxic effects from alcohol metabolism -Mitochondrial dysfunction-induced neuronal loss -Activation of inflammasome pathways | -Aβ-mediated cytotoxicity -Mitochondrial dysfunction-induced neuronal loss -Chronic neuroinflammation and synaptic loss | [7,8,12,14] |
| Affected Brain Regions | Frontal cortex, Hippocampus, Basal ganglia | Frontal cortex, Hippocampus, Temporal lobe, Amygdala | [1,81,87] |
| Cognitive Impairments | Memory loss, impaired impulse control, reduced attention span | Memory loss, spatial disorientation, impaired judgment | [85,86] |
| Psychiatric Effects | Depression, anxiety, increased impulsivity | Depression, social withdrawal, personality changes | [88,158] |
| Therapeutic Strategies | -Antioxidants (MitoQ, SS-31) -Inflammation modulators (MCC950, SB_NI_112) -Neuroprotective treatments under investigation | -Antioxidants (MitoQ, SS-31) -Research on Aβ clearance therapies | [132,142,149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.; Lee, J.; Lee, Y.; Kim, S.; Kang, S. Reactive Oxygen Species as a Common Pathological Link Between Alcohol Use Disorder and Alzheimer’s Disease with Therapeutic Implications. Int. J. Mol. Sci. 2025, 26, 3272. https://doi.org/10.3390/ijms26073272
Song H, Lee J, Lee Y, Kim S, Kang S. Reactive Oxygen Species as a Common Pathological Link Between Alcohol Use Disorder and Alzheimer’s Disease with Therapeutic Implications. International Journal of Molecular Sciences. 2025; 26(7):3272. https://doi.org/10.3390/ijms26073272
Chicago/Turabian StyleSong, Hyein, Jiyong Lee, Yeeun Lee, Seungju Kim, and Shinwoo Kang. 2025. "Reactive Oxygen Species as a Common Pathological Link Between Alcohol Use Disorder and Alzheimer’s Disease with Therapeutic Implications" International Journal of Molecular Sciences 26, no. 7: 3272. https://doi.org/10.3390/ijms26073272
APA StyleSong, H., Lee, J., Lee, Y., Kim, S., & Kang, S. (2025). Reactive Oxygen Species as a Common Pathological Link Between Alcohol Use Disorder and Alzheimer’s Disease with Therapeutic Implications. International Journal of Molecular Sciences, 26(7), 3272. https://doi.org/10.3390/ijms26073272