1. Introduction
Typically, cytomegalovirus (CMV)-seropositive individuals harbor a high percentage of CMV-specific CD8
pos T cells directed against immunodominant viral peptides, which may increase with age up to 20% [
1]. These so-called ‘inflationary’ anti-CMV CD8
pos T cells possess an effector–memory phenotype, maintain a ready-to-go functional status, and exhibit the capacity to migrate into virtually all tissues, including tumor tissues [
2]. In recent years, several strategies that aim to redirect the unique cytotoxic potential of inflationary anti-CMV CD8
pos T cells to selectively eliminate cancer cells have been developed (reviewed in [
3]).
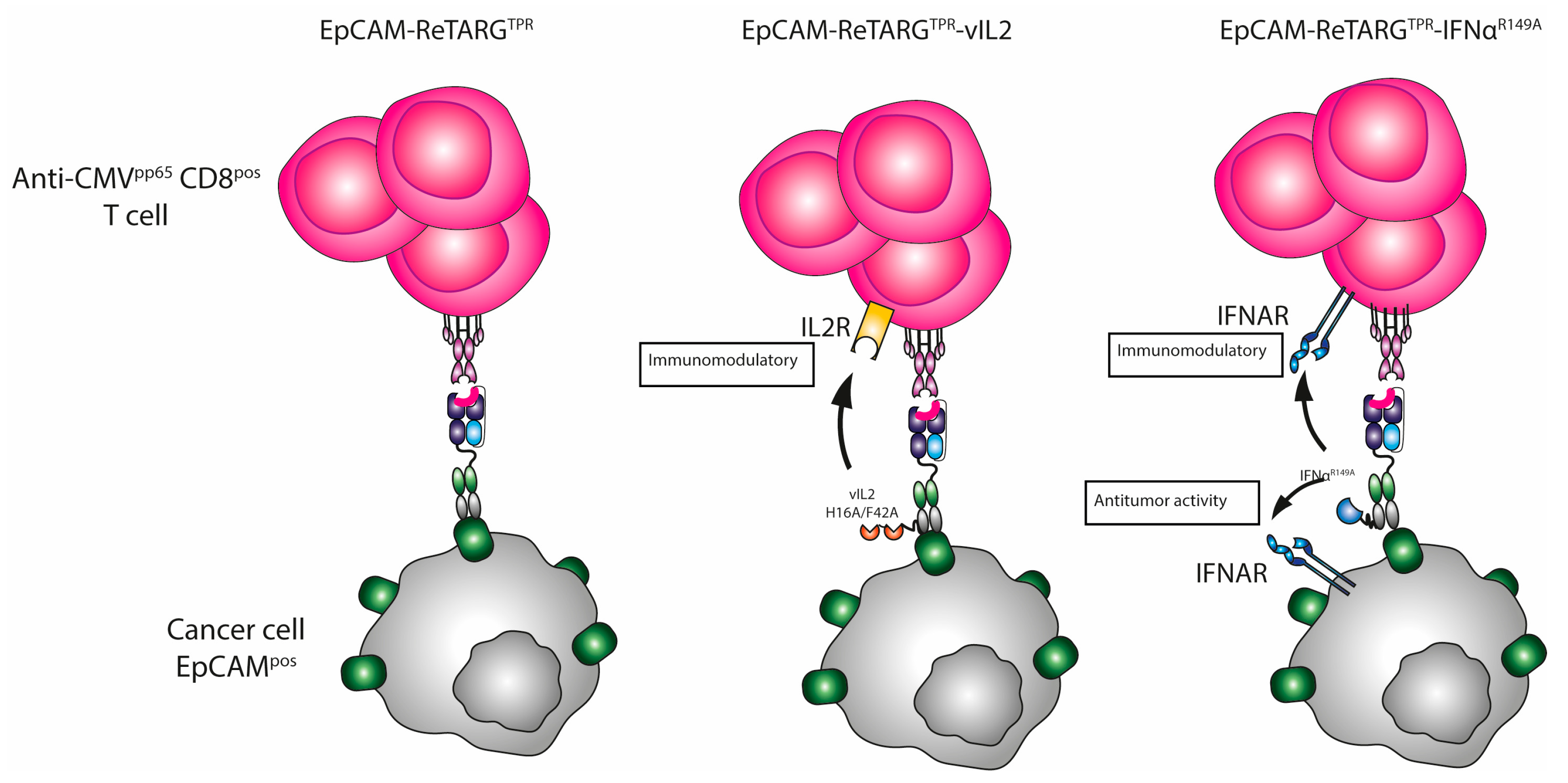
Previously, we reported on a novel Fab–peptide–HLA-I fusion protein, designated EpCAM-ReTARG
TPR, that consists of an EpCAM-directed Fab antibody fragment fused to HLA-B*07:02-β2-microglobulin (β2M) and genetically equipped with the CMV pp65-derived peptide TPRVTGGGAM (TPR). EpCAM-ReTARG
TPR showed potent in vitro capacity to redirect TPR-specific CD8
pos T cells derived from CMV-seropositive/HLA-B*07:02
pos individuals to eliminate various types of EpCAM-expressing carcinomas [
4]. The ReTARG approach selectively engages the TCR-CD3 signaling complex of cognate CMV-specific CD8
+ T cells in a physiologically normal manner, effectively eliminating EpCAM
pos cancer cells without excessive cytokine release. In contrast, CD3-based T-cell engagers like the BiTE solitomab activate all CD3
pos T cells, including inhibitory CD4
pos Tregs, by stimulating the CD3ε chain regardless of T-cell specificity. This non-physiological hyperactivation is strongly associated with massive cytokine release and immune-related side effects.
Previously, we compared EpCAM-ReTARGTPR and solitomab in terms of cancer cell elimination and T-cell-secreted cytokines. Our results confirmed that EpCAM-ReTARGTPR effectively engaged CMV-specific CD8pos T cells to eliminate EpCAMpos cancer cells with potency comparable to solitomab, without inducing excessive cytokine release.
Building upon our findings, in the present work, we aimed to further enhance the anticancer activity of the EpCAM-ReTARGTPR approach by genetically equipping this fusion protein with the immune-potentiating cytokine muteins interleukin-2 (IL2) and IFNα-2b, respectively.
IL2 is a pleiotropic cytokine which acts via low-, intermediate-, and high-affinity IL2 receptors (IL2Rs), which are differentially expressed on various types of immune cells. CD25 (also known as IL2Rα) acts as the low-affinity IL2R. The intermediate-affinity IL2R is composed of CD122 (IL2Rβ) and CD132, which are typically expressed on resting immune cells. However, upon TCR-mediated activation, CD25 is rapidly co-expressed and binds with low affinity to IL2, which results in the formation of the heterotrimeric high-affinity IL2R by engaging with CD122 and CD132 (2R) [
5]. In contrast, highly immunosuppressive intratumoral regulatory T cells (T
regs) constitutively express high levels of CD25, which leads to enhanced cell surface levels of the high-affinity IL2R, thereby depleting IL2 that is essential for the expansion and activity of neighboring anticancer T cells [
6,
7]. Therefore, engineered variant forms of IL2 have been developed with attenuated binding affinity for CD25. In particular, IL2, containing the amino acid mutations H16A and F42A (hereafter referred to as vIL2), has shown to have >100-fold reduced affinity for CD25, which reduces the bias towards T
reg activation and selectively promotes the expansion of CD8
pos effector T cells [
8,
9,
10]. We therefore reasoned that arming EpCAM-ReTARG
TPR with vIL2 may enhance its anticancer capacity in an EpCAM-directed manner. To this end, we constructed EpCAM-ReTARG
TPRvIL2 in which two linker-interspersed copies of mutein vIL2 were genetically fused to the constant domain of the light chain (CL) of the anti-EpCAM Fab fragment.
Analogously, we constructed EpCAM-ReTARG
TPRIFNα
R149A in which mutein IFNα
R149A is genetically fused to the CL domain of the anti-EpCAM Fab fragment. The pleiotropic activity of IFNα appears to be of particular promise given its potent dual anticancer and immune-potentiating activities. Like all type 1 IFN family members, IFNα binds to the interferon-α/β receptor (IFNAR), which is composed of two subunits, namely IFNAR1 and IFNAR2. Various recombinant IFNα formulations have been employed for a wide range of indications, including cancer, although their efficacy is hampered by dose-limiting side effects as a result of IFNAR expression on essentially all nucleated cells. It was previously shown that the fusion of IFNα mutein IFNα
R149A, with a 200-fold reduced affinity for the IFNAR2, to a cancer-directed antibody is less likely to bind to IFNAR expressed on normal cells, thus potentially reducing deleterious off-cancer side effects while ‘en route’ to cancer cells [
11,
12]. Importantly, antibody-mediated binding to the cancer cell surface locally increases the concentration of IFNα
R149A such that this compensates for its reduced capacity to bind and activate IFNAR1/2. Since IFNAR1/2 molecules are present on both cancer cells and neighboring tumor-infiltrating immune cells, dual anticancer and immune-potentiating activities are potentially achieved in a tumor-directed manner. Therefore, we reasoned that it may be feasible to exploit these favorable characteristics by genetically equipping EpCAM-ReTARG
TPR with the IFNα
R149A domain.
Here, we investigated the effects of arming peptide–HLA-I fusion proteins with potentiating cytokines and assessed whether their antiproliferative and immune-potentiating activities resulted in overall enhanced anticancer capacity.
3. Discussion
Previously, we reported on a novel Fab–peptide–HLA-I fusion protein, designated EpCAM-ReTARG
TPR, that consists of an EpCAM-directed Fab antibody fragment fused to HLA-B*07:02-β2M that is genetically equipped with the CMV pp65-derived peptide TPR. EpCAM-ReTARG
TPR showed potent in vitro capacity to redirect anti-CMV
pp65 CD8
pos T cells from CMV-seropositive/HLA-B*07:02
pos donors to selectively eliminate various EpCAM-expressing carcinoma cell lines and primary patient-derived cancer cells [
4]. Building upon these results, we aimed to further enhance and broaden its anticancer activities by fusing it to the immune-potentiating cytokines vIL2 and IFNα
R149A, respectively. EpCAM-ReTARG
TPRvIL2 was equipped with two IL2
H16A, F42A mutein (vIL2) molecules; we hypothesize that this would selectively promote the cytotoxic activity of redirected TPR-specific CD8
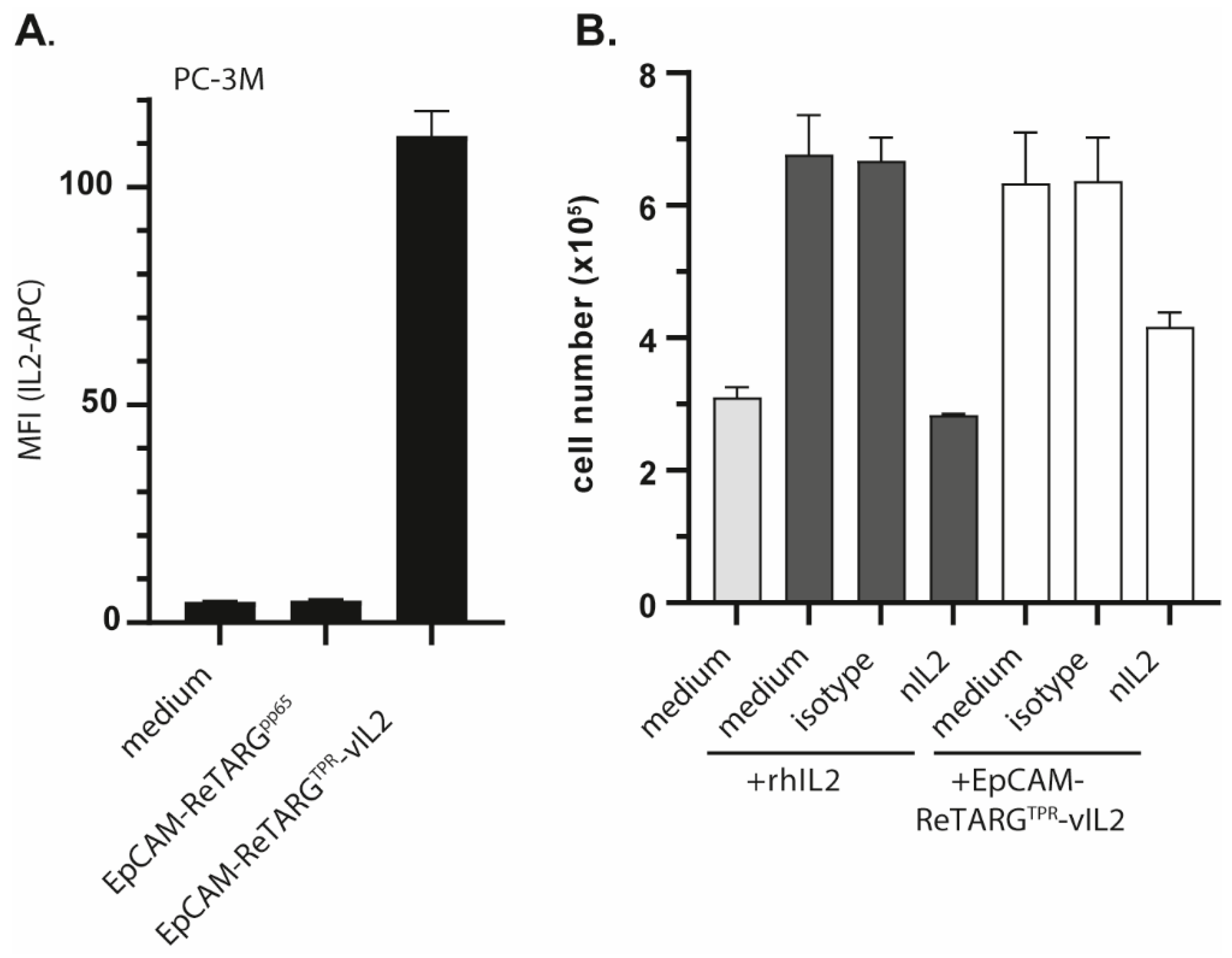
pos T cells. However, compared to EpCAM-ReTARG
TPR, this showed only marginal enhanced capacity to induce cancer cell lysis. Moreover, EpCAM-selective anticancer activity of EpCAM-ReTARG
TPRvIL2 appeared to be strongly compromised. Consequently, we decided to exclude EpCAM-ReTARG
TPRvIL2 from our study.
Our results appear to contradict with those of Schardt et al., who reported increased cytotoxicity and the expansion of anti-CMV
pp65 CD8
pos T cells directed to EGFR
pos tumor cells when using an analogous guided-pMHC-staging (GPS) molecule that was equipped with the same IL2 mutein (H16A and F42A) [
16]. However, their experimental set-up of cytotoxicity assays may be incomplete since the authors did not include control experiments using cancer cells in which EGFR expression was knocked down/out.
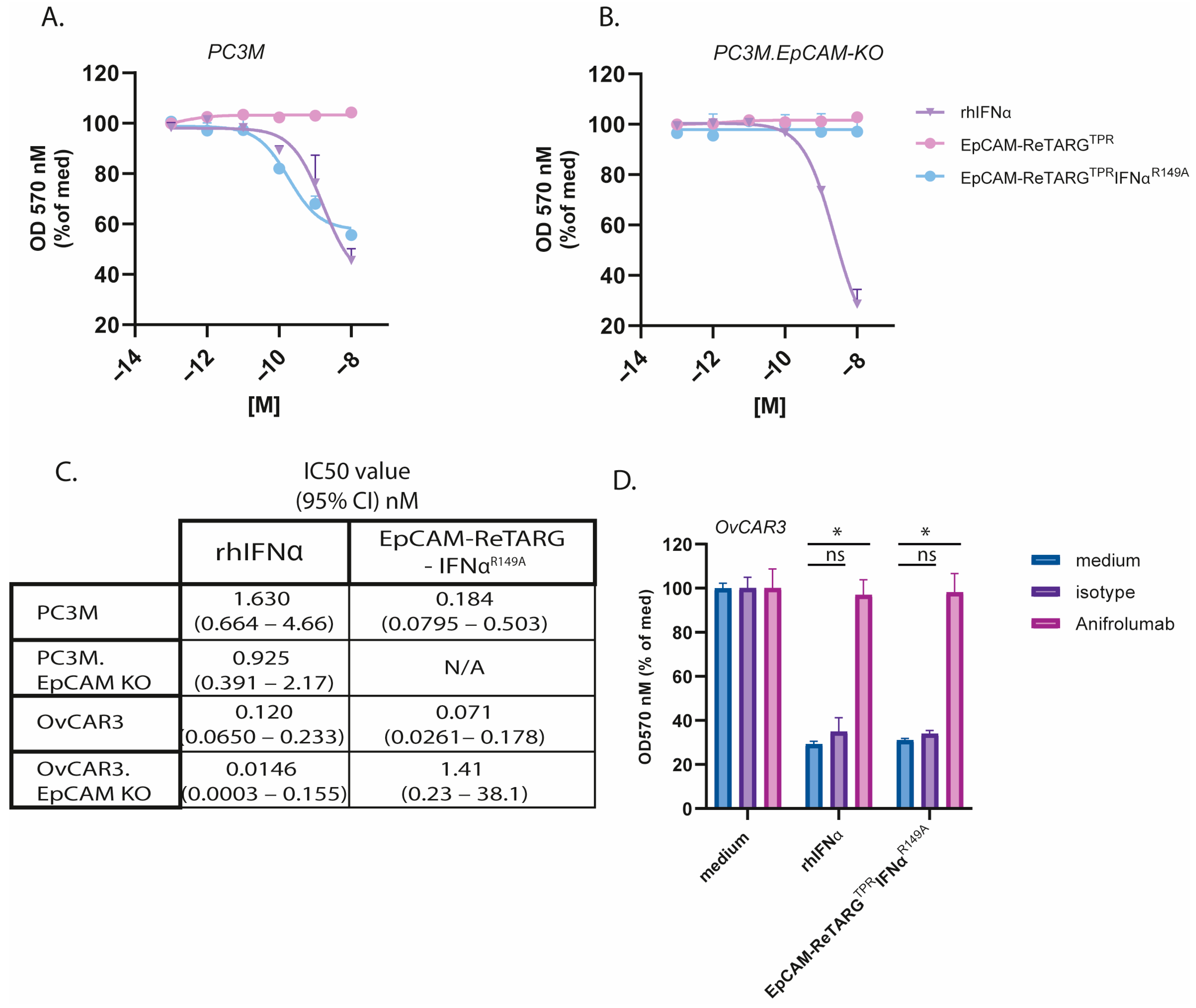
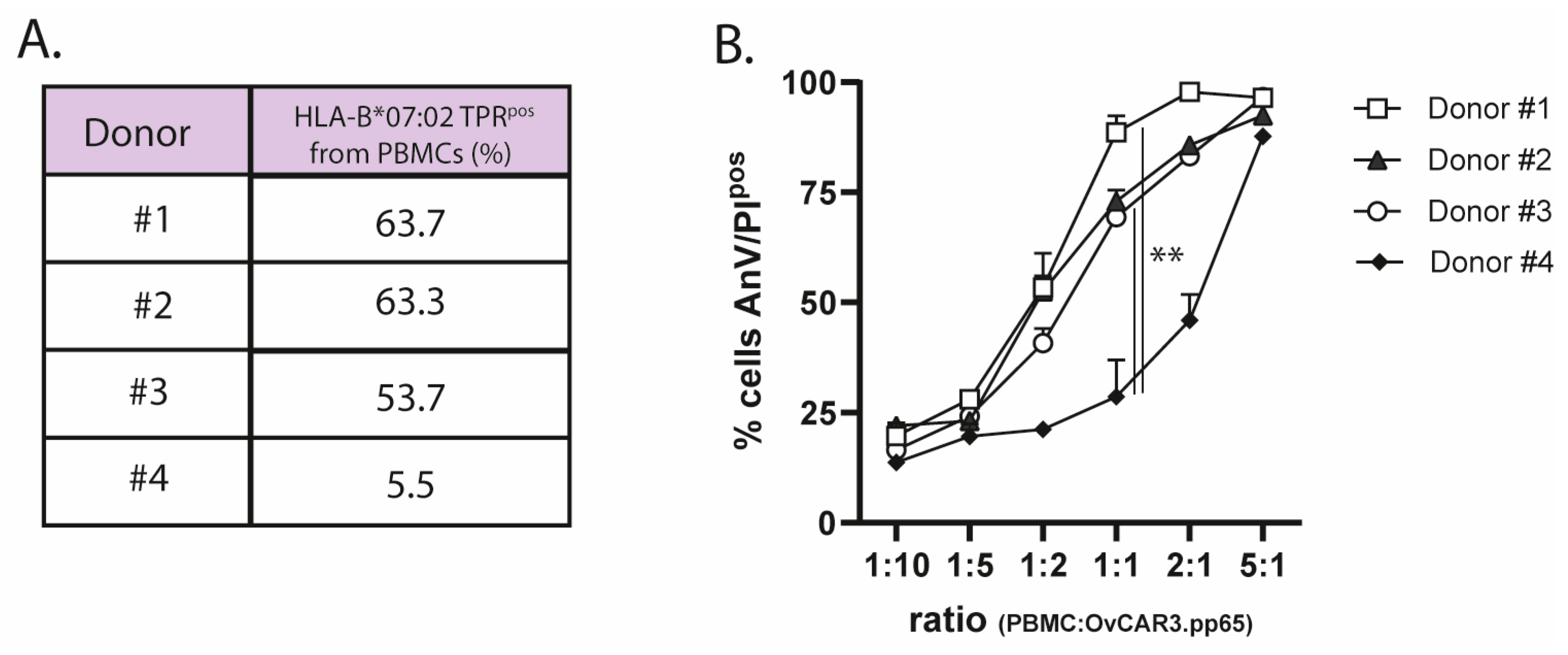
We equipped EpCAM-ReTARGTPRIFNαR149A with one IFNαR149A mutein unit, which was reported to have a 200-fold reduced binding affinity for IFNAR2. Intriguingly, EpCAM-ReTARGTPRIFNαR149A potently enhanced the cytotoxic capacity of TPR/IL2-expanded PBMCs. Notably, unlike EpCAM-ReTARGTPRvIL2, EpCAM-ReTARGTPRIFNαR149A-enhanced cytotoxicity remained fully EpCAM-restricted. Interestingly, EpCAM-ReTARG TPRIFNαR149A enhanced cancer cell lysis when PBMCs derived from donor #4 were used, who had a low percentage of TPR CD8pos T cells compared to donor #1. This indicates that EpCAM-ReTARGTPRIFNαR149A may augment the cytotoxic abilities of not only specific TPR CD8pos T cells but also other immune cells within the PBMC population, which could be particularly advantageous for individuals with a low proportion of TPR-specific cells. In 120 h target/effector cell co-culture experiments, EpCAM-ReTARGTPRIFNαR149A significantly reduced the viable cell number of EpCAMpos cancer cells compared to EpCAM-ReTARGTPR.
It appears reasonable to assume that the unique and multifold anticancer activities we observed in our co-culture experiments with EpCAM-ReTARGTPRIFNαR149A are in part attributable to the pleiotropic and possibly mutually reinforcing biological activities of its IFNαR149A domain towards the targeted cancer cells as well as the engaged TPR-specific CD8pos T cells.
In this respect, in the absence of TPR-specific CD8
pos T cells, the treatment of OvCAR3 and PC3M cancer cells with EpCAM-ReTARG
TPRIFNα
R149A inhibits proliferation and stimulates cancer cell-secreted CXCL10 (IP-10). CXCL10 is a chemoattractant for activated T cells and may enhance the homing of T cells to the tumor site(s) [
13]. Additionally, in co-culture experiments, treatment with EpCAM-ReTARG
TPRIFNα
R149A boosted T-cell-secreted IFNγ, and we observed that treatment with EpCAM-ReTARG
TPRIFNα
R149A prolonged the survival of TPR/IL2-expanded PBMCs. Together, these characteristics enhance the anticancer activity of EpCAM-ReTARG
TPRIFNα
R149A, leading to improved cancer cell lysis, which corroborates findings by Hervas-Stubbs et al., who showed that treatment with IFNα during the in vitro expansion of anti-CMV CD8
pos T cells increases IFNγ production and enhances their cytolytic capacity [
17].
Pogue et al. demonstrated that an anti-CD38 antibody genetically equipped with an attenuated version of human IFNα (designated anti-CD38-IFNα(att)) exhibits 10,000-fold increased specificity for CD38
pos cells in vitro compared to non-targeted IFNα. As a result, anti-CD38-IFNα(att) is approximately 6000-fold less toxic to normal bone marrow cells in vitro than IFNα [
18]. Although anti-CD38-IFNα(att) provided potent anti-tumor activity in various MM cell lines and in human xenograft MM tumor models, it is not addressed whether it induces T cell/immune cell activation. In a study by Daneels et al., the in vivo antitumor potential of the huCD20-IFNαR149A fusion protein (huCD20-Fc-AFN) was explored using tumor-bearing human immune system (HIS) mice. huCD20-Fc-AFN not only directly affected cancer cell growth but also significantly enhanced immune cell-mediated tumor cell elimination. Upon huCD8
pos T-cell depletion, tumor growth was significantly increased, suggesting a crucial role of T cells in cancer cell elimination [
19]. Future studies aimed to evaluate the in vivo anti-tumor activity of EpCAM-ReTARG
TPRIFNα
R149A should use tumor-bearing mice engrafted with peripheral blood mononuclear cells (PBMCs) from CMV-seropositive/HLA-B*07:02-matched donors.
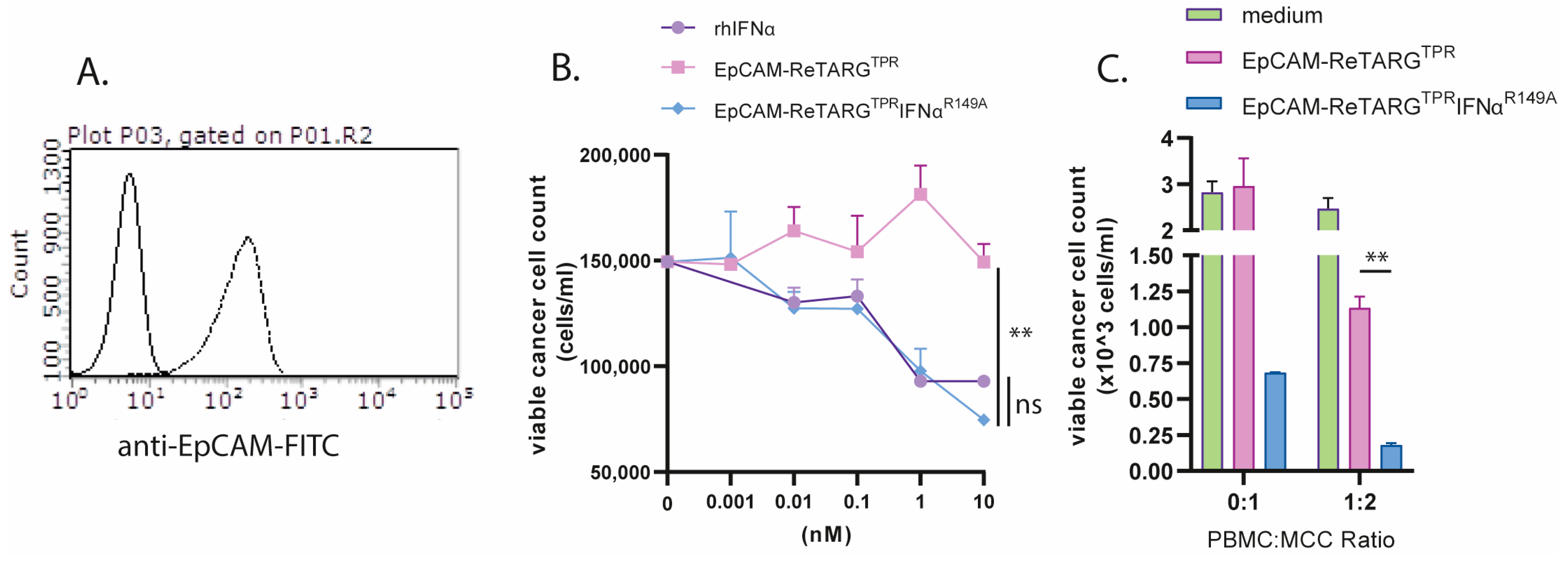
Merkel cell carcinoma (MCC) is a rare EpCAM-expressing refractory skin cancer. It has been reported that MCC is sensitive to treatment with IFNα [
15]. Therefore, we reasoned that EpCAM-ReTARG
TPRIFNα
R149A could be an attractive treatment option for this cancer type. Indeed, our results show that MCC cells were sensitive to EpCAM-ReTARG
TPRIFNα
R149A-induced growth inhibition, and they were more effectively eliminated by TPR/IL2-expanded PBMCs compared to EpCAM-ReTARG
TPR. Recent studies have shown that Merkel cell polyomavirus (MCPyV) plays a vital role in the development of MCC by encoding oncoproteins. Interestingly, T cells targeting MCPyV oncogenes, specifically large T (LTA) and small (STA) antigens, are found exclusively in MCC patients, and these LTA- and STA-specific T cells can effectively kill HLA-matched MCPyV-positive MCC cells [
20]. Given the sensitivity of MCC cells to IFNα, developing IFNα-equipped Fab–peptide–HLA-I fusion proteins equipped with MCPyV-derived epitopes may represent a promising new immunotherapy approach for MCPyV-associated MCC.
Recombinant untargeted IFNα is currently used in the treatment of several cancers, including melanoma, hairy cell leukemia, and renal cell carcinoma (RCC) [
21]. Notably, a study by Rosato et al. [
2] found that anti-CMV T cells infiltrate RCC tumors and can be activated by viral peptides, suggesting that EpCAM-ReTARG
TPRIFNα
R149A could be particularly effective for RCC treatment. Additionally, other IFNα-sensitive EpCAM
pos cancer types may also be promising targets for EpCAM-ReTARG
TPRIFNα
R149A therapy.
Decoration of of EpCAMpos cancer cells with EpCAM-ReTARGTPR enables physiological engagement of cognate anti-CMV CD8pos T cells and subsequent effective target cell elimination in the absence of excessive cytokine release. Compared to an analogous BiTE, we hypothesize that EpCAM-ReTARGTPRIFNαR149A is less likely to induce severe side effects such as cytokine release syndrome. However, future in vivo studies should be conducted to directly compare the respective anticancer efficacies and cytokine secretion profiles. It is important to note that EpCAM is expressed at low levels on the basolateral side of epithelial cells, which may result in off-target effects. Truly cancer-selective targets are rare, but if identified, the ReTARG fusion protein can be readily adapted to target them.
In conclusion, the armoring of the carcinoma-directed peptide–HLA-I fusion protein EpCAM-ReTARGTPR with IFNαR149A potently enhanced the efficacy of pre-existing anti-CMV CD8pos T-cell immunity to selectively eliminate EpCAMpos cancer cells.
4. Materials and Methods
4.1. Antibodies and Reagents
The following primary (fluorescent)-labeled monoclonal antibodies directed against human antigens were used: FITC-labeled anti-EpCAM (clone VU-1D9, STEMCELL Technologies Germany GmbH, Köln, Germany, APC-labeled anti-HLA-B7 (clone BB7.1), BioLegend Europe B.V., Amsterdam, The Netherlands, APC-labeled anti-IL2 (clone MQI-17H12, and CaptureSelect™ Biotin Anti-IgG-CH1 conjugate (were from Thermo Fisher Scientific, Waltham, MA, USA). Anti-human IFNAR1 (anifrolumab) #SIM0022 was from BioXCell, Lebanon, NH, USA. The neutralizing IL2 antibody (clone 5334) was from R&D Systems, Inc., Minneapolis, NE, USA. The following reagents were used: trypan blue (Sigma Aldrich, Zwijndrecht, The Netherlands), Streptavidin-AlexaFluor™647 (Thermo Fisher Scientific, Waltham, MA USA), FITC-labeled Annexin-V (ImmunoTools GmbH, Friesoythe, Germany), and propidium iodide (PI) (Thermo Fisher Scientific, Waltham, MA USA). The following recombinant proteins were used: human IFNα-2b and human IL2. They were from ImmunoTools GmbH, Friesoythe, Germany. Biotinylated MHC I-Strep HLA-B*0702, CMV pp65 (TPRVTGGGAM), and APC-labeled strep-Tactin were from IBA Lifesciences GmbH, Göttingen, Germany.
4.2. Cell Lines and Transfectants
The cell lines Jurkat (T-ALL), CTLL2 (mouse cytotoxic T-cell clone), PC3M (prostate cancer), and OvCAR3 (ovarian cancer) were obtained from the ATCC (Manassas, VA, USA). PC3M.EpCAM-KO and OvCAR3.EpCAM-KO cells were generated using CRISPR-Cas9 gene editing technology via transfection with the plasmid pSpCas9 BB-2A-GFP (PX458) containing sgRNA 5′-TAATGTTATCACTATTGATC-3′ [
13]. Subsequently, EpCAM-KO cancer cells were obtained through limited dilution. OvCAR3.pp65 cells were generated via lipofection (Fugene-HD, Promega BNL, Leiden, The Netherlands) of the plasmid pCMV6-pp65 (OriGene Technologies GmbH, Herford, Germany), and OvCAR3.pp65 cells stably expressing the CMV pp65 protein were obtained after limited dilution. Cells were cultured in RPMI-1640 or DMEM (Lonza, Geleen, The Netherlands) supplemented with 10% FCS at 37° C in a humidified 5% CO
2 atmosphere.
4.3. Merkel Cell Carcinoma Patient Sample
Merkel cell carcinoma tissues was obtained from surgical resection waste materials. Tumor tissue was minced and short-term cultured in RPMI/10% fetal calf serum. Cell phenotype was analyzed through flow cytometry (Guava Easycyte, Merck millipore, Amsterdam, The Netherlands) using fluorescently labeled antibodies. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the medical ethical committee of the UMCG, under approval number EUCTR2012-000507-33-EN on 26 November 2012.
4.4. Assessment of Cytokine Activity of EpCAM-ReTARGTPRvIL2
The presence of the vIL2 domains in EpCAM-ReTARGTPRvIL2 was confirmed using flow cytometry. In short, EpCAMpos cancer cells were incubated with EpCAM-ReTARGTPR or EpCAM-ReTARGTPRvIL2. Subsequently, cancer cell surface-bounded vIL2 was assessed using an APC-labeled anti-IL2 antibody and an IL2-neutralizing antibody clone 5334 (R&D Systems, Inc., Minneapolis, NE, USA) with a secondary anti-mouse-647 antibody (Thermo Fisher Scientific, Waltham, MA USA). The T-cell proliferative activity of EpCAM-ReTARGTPRvIL2 was assessed using the IL2-dependent mouse cell line CTLL2. In short, CTLL2 cells were cultured in the presence of increasing concentrations of EpCAM-ReTARGTPRvIL2 or EpCAM-ReTARGTPR (0.078–20 nM). IL2-mediated proliferation of cells was determined after 72 h using flow cytometry. Moreover, EpCAM-ReTARGTPRvIL2-induced CTLL2 proliferation was assessed in the presence of an IL2-neutralizing antibody.
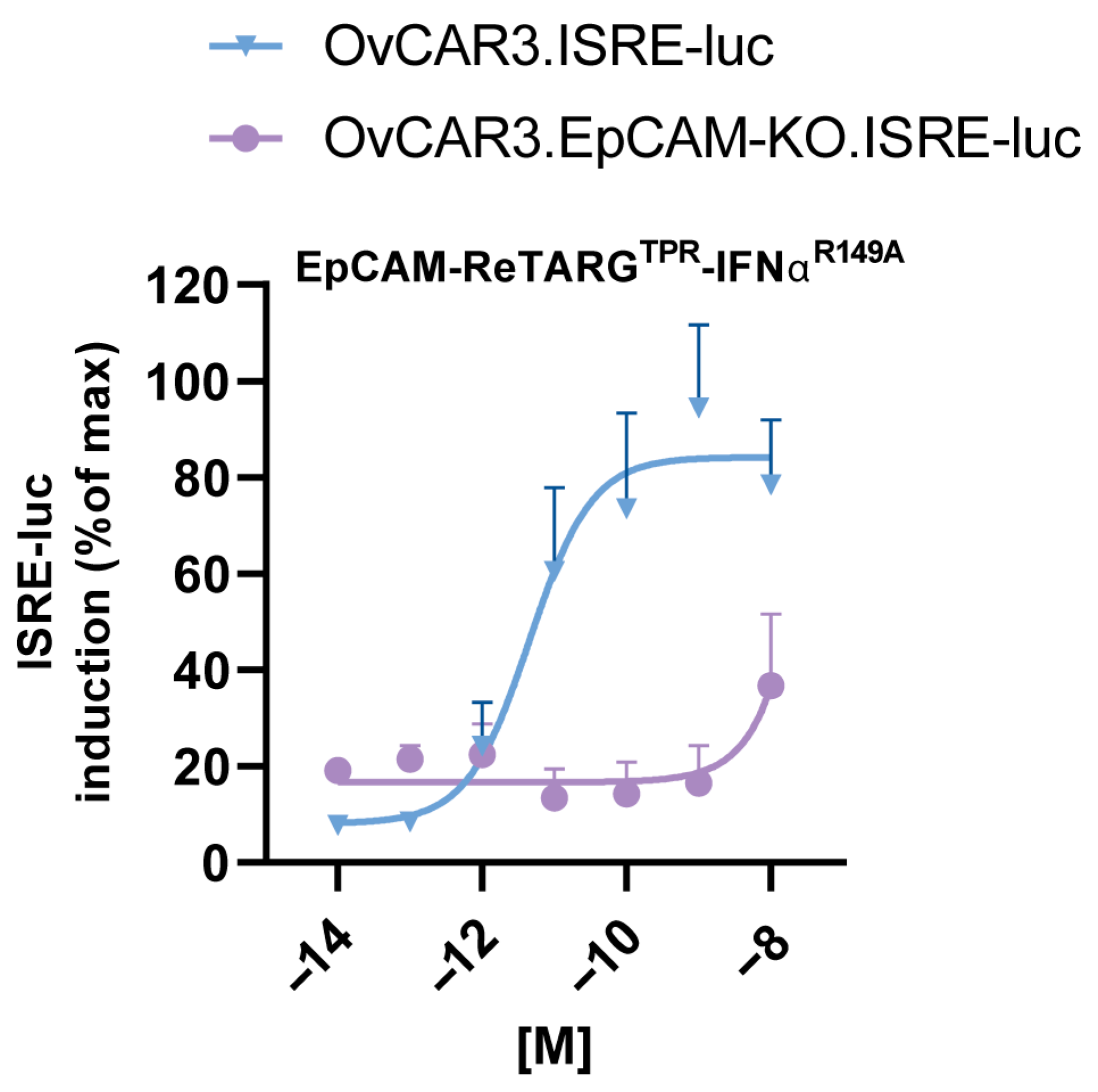
4.5. Assessment of Cytokine Activity of EpCAM-ReTARGTPRIFNαR149A
OvCAR3, OvCAR3.EpCAM-KO, and Jurkat cells were transduced with the interferon-stimulated response element (ISRE) luciferase reporter lentivirus (BPS Bioscience, San Diego, CA, USA #79824) at MOIs of 100, 100, 10, respectively. After 72 h, transduced cells were selected from the culture medium supplemented with 1.0 µg/mL puromycin. Subsequently, ISRE luc induction by IFNα was assessed. In short, OvCAR3 cells and OvCAR3.EpCAM-KO ISRE reporter cells were seeded (each 8000 cells/well) using 96-well black/clear bottom plates (ThermoFisher) and were left to adhere overnight. Cells were treated with increasing concentrations (0.0001–10 nM) of IFNα-2b, EpCAM-ReTARGTPR, and EpCAM-ReTARGTPRIFNαR149A for 6 h. After treatment, the Bio-Glo™ luciferase reagent (Promega BNL, Leiden, The Netherlands)) was added to each well, and luminescence was measured using a SpectraMaxi3X molecular device. Analogously, a suspension of 50,000 Jurkat.ISRE-luc reporter cells was added to 96-wells black/clear bottom plates, and cells were treated and evaluated as described above.
4.6. Ex Vivo Expansion of PBMCs from HLA-B*07:02pos/CMV-Seropositive Individuals
Peripheral blood mononuclear cells (PBMCs) from HLA-B*07:02pos/CMV-seropositive individuals were purchased from CTL - Europe GmbH, Rutesheim Germany PBMCs were harvested, washed, and cultured in 6-well plates (a final concentration of 4 × 106 cells/mL, with 2 mL per well) in the RPMI medium. PBMCs were stimulated with 0.5 µg/mL CMV pp65-derived peptide TPR for 4 d. Next, TPR-stimulated PBMCs were harvested, resuspended in a fresh X-VIVO15 medium (Lonza, Geleen, The Netherlands) supplemented with 50 IU/mL IL2, and cultured for an additional 7 d. The percentage of TPR-specific HLA-B*07:02pos CD8pos T cells was determined using streptamer staining through flow cytometry.
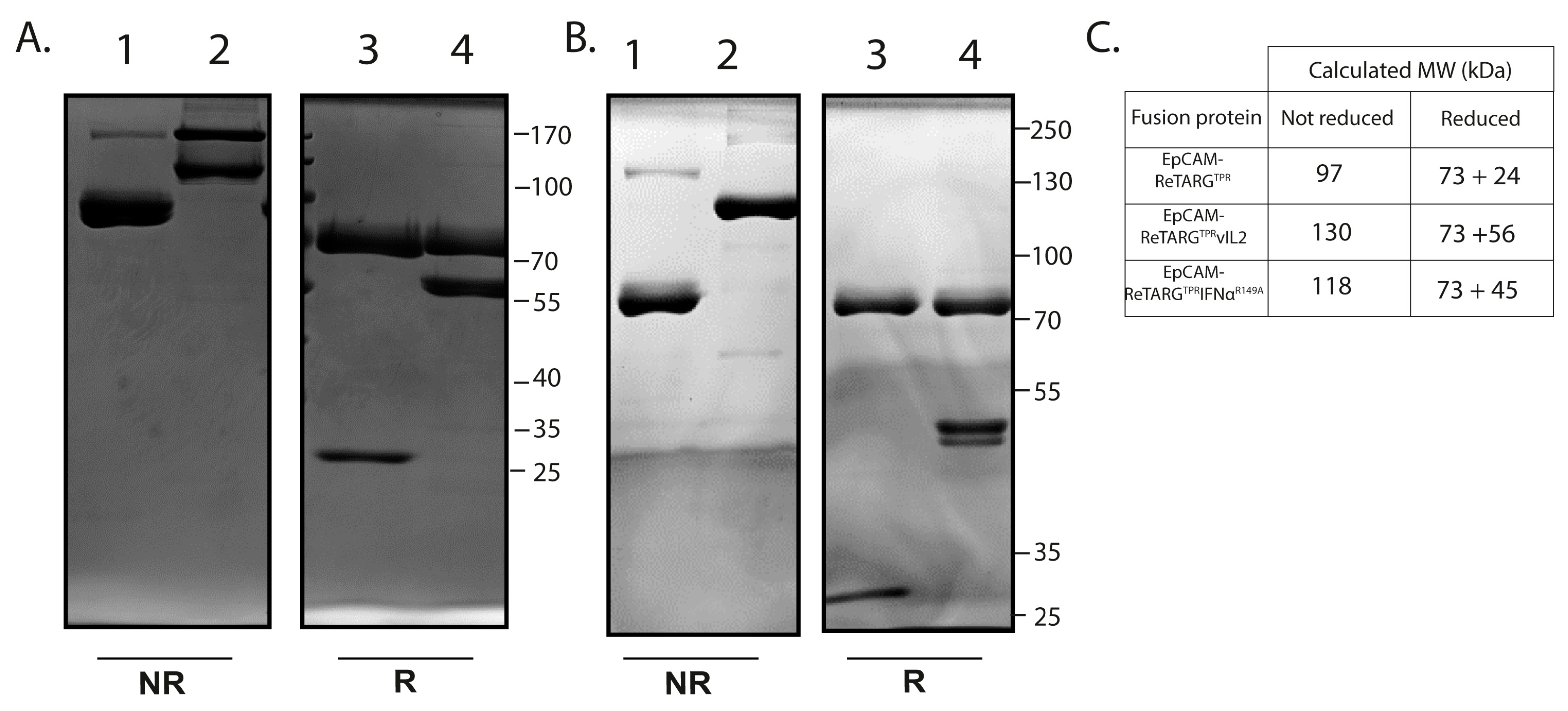
4.7. Construction, Production, and Purification of EpCAM-ReTARGTPR, EpCAM-ReTARGTPRvIL2, and EpCAM-ReTARGTPRIFNαR149A
EpCAM-ReTARG
TPR was designed as a monomeric recombinant fusion protein consisting of the antigenic CMV pp65 peptide TPR, β2M, and a truncated HLA-B*07:02 heavy chain lacking the transmembrane and intracellular domains. To enhance the stability of EpCAM-ReTARG
TPR, The C-terminus of TPR was fused to the flexible linker sequence G
CGGSGGGGSGGGGS, which was engineered to contain a cysteine residue (in bold) that promotes the formation of a stabilizing intramolecular disulfide bridge with a cysteine residue inserted in the α1 domain HLA-B*07:02 heavy chain. Using a flexible linker, the HLA-I α chain was genetically fused to a high-affinity anti-EpCAM Fab antibody fragment containing the VH-VL gene segments of the humanized scFv 4D5 MOC-B [
14]. In this Fab, we employed human kappa CL and CH1 domains based on UniProt accession numbers P01834 and P01857, respectively. To construct EpCAM-ReTARG
TPRvIL2, two mutein IL2 (vIL2) molecules containing the point mutations, F42A and H16A, were genetically fused to the C-terminal of the constant domain of the light chain (CL) of the anti-EpCAM Fab antibody of EpCAM-ReTARG
TPR. To construct EpCAM-ReTARG
TPRIFNα
R149A, an IFNα-2b-encoding DNA fragment with the R149A point mutation was genetically fused to the CL domain of the anti-EpCAM Fab antibody of EpCAM-ReTARG
TPR. The amino acid sequence of mutein IL2
H16A/F42A was obtained from a study by Quayle et al. [
8], and the amino acid sequence of mutein IFNα
R149A was obtained from Patent number US20180028616A1. The cDNAs encoding the respective fusion proteins were synthesized and then cloned into the eukaryotic expression plasmid pcDNA3.1-hygro by Genscript (Rijswijk, The Netherlands) and then transfected (Fugene-HD, Promega) into Hek293AD production cells. After 7 d, conditioned cell culture supernatants were harvested and cleared via centrifugation (4000×
g, 30 min). EpCAM-ReTARG
TPR, EpCAM-ReTARG
TPRvIL2, and EpCAM-ReTARG
TPRIFNα
R149A were purified using a Capture Select™ CH1-XL column (Thermo Fisher Scientific, Waltham, MA, USA) connected to an ÄKTA Start chromatography system (GE Healthcare Life Sciences, Eindhoven, The Netherlands), diluted in PBS to 1 mg/mL, and stored at −20 °C until use.
4.8. SDS-PAGE Analysis
Purified EpCAM-ReTARGTPR, EpCAM-ReTARGTPRvIL2, and EpCAM-ReTARG TPRIFNαR149A (2 μg protein per lane) were separated using SDS-PAGE (10% acrylamide) under reducing or non-reducing conditions and stained using Coomassie brilliant blue.
4.9. Assessment of EpCAM-Binding Activity of EpCAM-ReTARGTPR, EpCAM-ReTARGTPRvIL2, and EpCAM-ReTARGTPRIFNαR149A
The binding of EpCAM-ReTARGTPR, EpCAM-ReTARGTPRvIL2, and EpCAM-ReTARGTPRIFNαR149A to cell surface-expressed EpCAM was assessed through flow cytometry. In short, EpCAMpos- and EpCAM-KO-derived PC3M cancer cells were incubated with 10 nM of either EpCAM-ReTARGTPR, EpCAM-ReTARGTPRvIL2, or EpCAM-ReTARG TPRIFNαR149A at 4° C for 45 min, after which binding was evaluated using CaptureSelect™ Biotin Anti-IgG-CH1 Conjugate plus Streptavidin-AlexaFluor™647 (Thermo Fisher Scientific, Waltham, MA USA) or the fluorescently labeled anti-HLA-B7 antibody.
4.10. Cancer Cell Proliferation Assay
To evaluate the antiproliferative activity of EpCAM-ReTARGTPRIFNαR149A, EpCAM-expressing cancer cells (or EpCAM-KO derivatives) were seeded in 48-well plates at 3500 cells/well. Cells were treated with 0.001–10 nM of IFNα-2b, EpCAM-ReTARGTPR, or EpCAM-ReTARGTPRIFNαR149A for 8 d. Cancer cell proliferation was quantified via the OD of crystal violet staining measured at 570 nm using a microplate reader (SpectraMaxi3X, Molecular Devices, San Jose, CA, USA). Additionally, the antiproliferative activity of EpCAM-ReTARGTPRIFNαR149A towards cancer cells was assessed in the presence of the IFNAR1-blocking antibody anifrolumab. The IC50 was determined using a colorimetric crystal violet assay, a standard assay to evaluate cell viability, cell attachment, and cell proliferation. OD values from this assay were normalized to the percentage of the medium control (set to 100%). The concentrations of the EpCAM-ReTARGTPR fusion protein variants were transformed to the logarithm of the dose and plotted using the GraphPad Prism software, version 10.2.3. IC50 values, along with 95% confidence intervals, were calculated by performing a non-linear regression analysis using the “log(inhibitor) vs. response (three parameters)” equation. This method follows the guidelines outlined in the GraphPad Prism 10 Curve Fitting Guide for determining absolute IC50 values. A reference to this procedure is included in the revised manuscript.
4.11. Viability Assay
Ex vivo TPR/IL2-expanded PBMCs were seeded in a 48-well plate (400,000 cells/well) and cultured in the presence of IFNα-2b (referred to as rhIFNα), EpCAM-ReTARGTPR, or EpCAM-ReTARGTPRIFNαR149A, respectively, for 120 h. The number of viable PBMCs was evaluated using flow cytometry (PI staining).
4.12. In Vitro Cytotoxicity Assays
EpCAMpos cancer cells (or EpCAM-KO derivatives) were treated with either EpCAM-ReTARGTPR, EpCAM-ReTARGTPRvIL2, or EpCAM-ReTARGTPRIFNαR149A in the presence (or absence) of ex vivo-expanded PBMCs at the indicated PBMC-to-target cell ratios. Apoptotic cancer cell death was evaluated using flow cytometry at indicated timepoints (Annexin V/PI staining). In addition, the anticancer activity of EpCAM-ReTARGTPRIFNαR149A was assessed through live-cell imaging using a Phi HoloMonitor™ system (Phase Holographic Imaging PHI AB, Lund, Sweden) as the mean of cell confluency after 120 h.
4.13. Assessment of Cytokine Secretion
The activation of TPR/IL2-expanded PBMCs in response to treatment with EpCAM-ReTARGTPR and EpCAM-ReTARGTPR-IFNαR149A (10 nM each), respectively, was determined by co-culturing PBMCs with cancer cells at the indicated PBMC-to-target cell ratios. The conditioned culture media was collected after treatment for 72 h, after which the levels of human IFNγ were determined via ELISA (ThermoFisher). In addition, we assessed CXCL10 secretion by cancer cells after treatment for 72 h with rhIFNα, EpCAM-ReTARGTPR, and EpCAM-ReTARGTPRIFNαR149A (10 nM each), respectively, and assessed conditioned supernatants for CXCL10 secretion using a corresponding ELISA (ThermoFisher).
4.14. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software, version 10.2.3). Means of differences were calculated using one- and two-way ANOVA, respectively, followed by a multiple comparison test where appropriate. p-values considered significant are indicated by asterisks as follows: * p < 0.05; ** p < 0.01; *** p < 0.001.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}