

In Vivo Engineered CAR-T Cell Therapy: Lessons Built from COVID-19 mRNA Vaccines

 , , , , and
, , , , and
Abstract
1. Introduction
2. Current Status and Challenges of CAR-T Cell Therapy
2.1. Hematological Malignancies
2.2. Solid Tumors
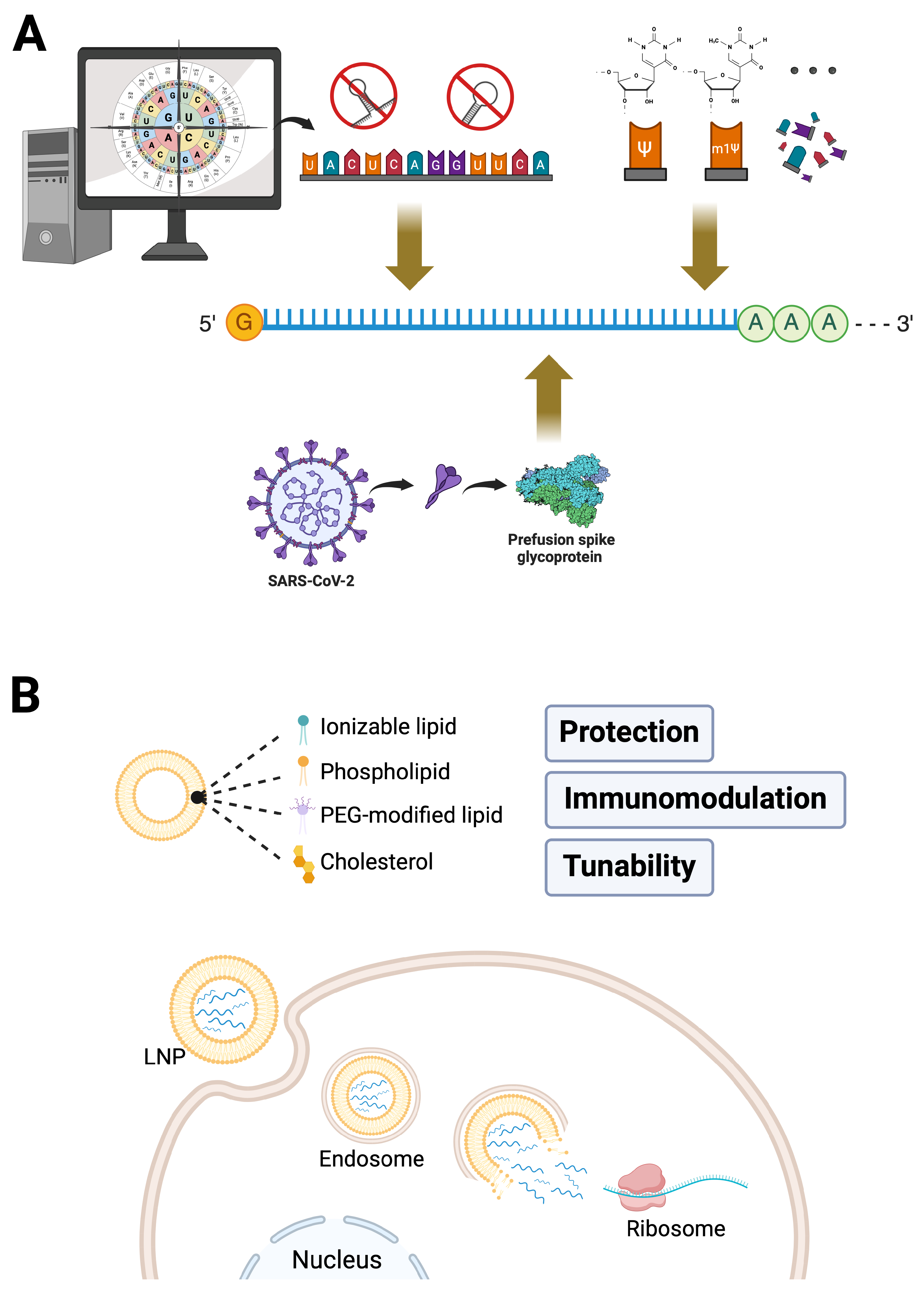
3. Critical Elements Behind the Rapid Development of mRNA Vaccines
3.1. Breakthroughs in mRNA Technology
3.1.1. Precise Antigen Design
3.1.2. Optimization of mRNA Sequences
3.1.3. Nucleoside Modifications
3.2. Advances in Delivery Systems
3.2.1. Protecting mRNA Integrity and Ensuring Efficient Delivery
3.2.2. Balancing Immunogenicity: Relatively Low Risk with Enhanced Efficacy
3.2.3. Tunability and Industrial Scalability
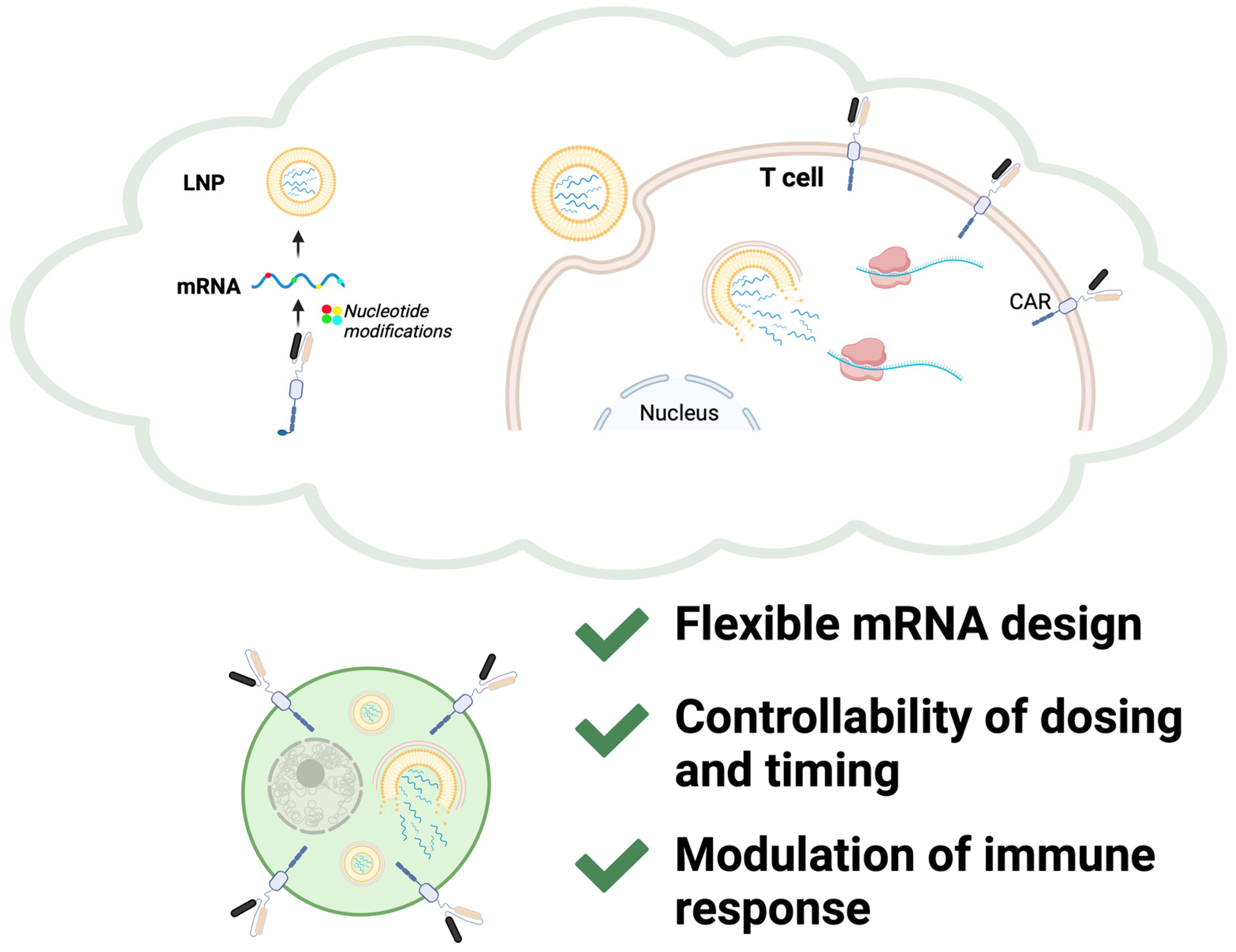
4. From Vaccines to CAR-T Therapy: Bridging Challenges with Technological Insights
4.1. Perspectives from mRNA Technology
4.2. Perspectives from LNP Delivery Systems
5. Future Directions
5.1. Refining Nucleotide Modifications in mRNA Technology
5.1.1. Methylation-Based Modifications
5.1.2. Non-Methylation-Based Modifications
5.2. Optimizing Targeting Strategies in LNP Systems
5.2.1. Passive Targeting and Physicochemical Properties
5.2.2. Active Targeting via Surface Modifications
5.2.3. Stimuli-Responsive Targeting
5.3. Balancing Immune Response
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR-T | Chimeric antigen receptor T-cell |
| LNP | Lipid nanoparticle |
| ALL | Acute lymphoblastic leukemia |
| CR rate | complete remission rate |
| CRS | Cytokine release syndrome |
| ICANS | Immune effector cell-associated neurotoxicity syndrome |
| TME | Tumor microenvironment |
| OTOT | On-target/off-tumor (toxicity) |
| Tregs | Regulatory T cells |
| RBD | Receptor-binding domain |
| UTRs | Untranslated regions |
| CDO | Codon deoptimization |
| TLR | Toll-like receptor |
| PEG | Polyethylene glycol |
| APCs | Antigen-presenting cells |
| m1Ψ | N1-methylpseudouridine |
| m6A | N6-Methyladenosine |
| m5C | 5-Methylcytosine |
| m5U | 5-methyluridine |
| m1A | N1-methyladenosine |
| s2U | 2-Thiouridine |
| mcm5 | 5-carboxymethylaminomethyl |
References
- Savoldo, B.; Grover, N.; Dotti, G. CAR T cells for hematological malignancies. J. Clin. Investig. 2024, 134, e177160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Amorós-Pérez, B.; Rivas-Pardo, B.; Gómez Del Moral, M.; Subiza, J.L.; Martínez-Naves, E. State of the Art in CAR-T Cell Therapy for Solid Tumors: Is There a Sweeter Future? Cells 2024, 13, 725. [Google Scholar] [CrossRef]
- Parhiz, H.; Atochina-Vasserman, E.N.; Weissman, D. mRNA-based therapeutics: Looking beyond COVID-19 vaccines. Lancet 2024, 403, 1192–1204. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Hernani, R.; Benzaquén, A.; Solano, C. Toxicities following CAR-T therapy for hematological malignancies. Cancer Treat. Rev. 2022, 111, 102479. [Google Scholar] [CrossRef]
- Rabilloud, T.; Potier, D.; Pankaew, S.; Nozais, M.; Loosveld, M.; Payet-Bornet, D. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat. Commun. 2021, 12, 865. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, G. The journey of CAR-T therapy in hematological malignancies. Mol. Cancer 2022, 21, 194. [Google Scholar] [CrossRef] [PubMed]
- Albelda, S.M. CAR T cell therapy for patients with solid tumours: Key lessons to learn and unlearn. Nat. Rev. Clin. Oncol. 2024, 21, 47–66. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef]
- D’Souza, R.R.; Dimou, P.; Bughda, R.; Hawkins, E.; Leboreiro Babe, C.; Klampatsa, A. Overcoming tumor antigen heterogeneity in CAR-T cell therapy for malignant mesothelioma (MM). J. Cancer Metastasis Treat. 2022, 8, 28. [Google Scholar]
- Chen, N.; Li, X.; Chintala, N.K.; Tano, Z.E.; Adusumilli, P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 2018, 51, 103–110. [Google Scholar] [CrossRef]
- Chhabra, Y.; Weeraratna, A.T. Fibroblasts in cancer: Unity in heterogeneity. Cell 2023, 186, 1580–1609. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.N.; Rathmell, J.C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 2023, 41, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Barkley, D.; Moncada, R.; Pour, M.; Liberman, D.A.; Dryg, I.; Werba, G.; Wang, W.; Baron, M.; Rao, A.; Xia, B.; et al. Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment. Nat. Genet. 2022, 54, 1192–1201. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, R.; Hogan, M.J.; Loré, K.; Pardi, N. Innate immune mechanisms of mRNA vaccines. Immunity 2022, 55, 1993–2005. [Google Scholar] [CrossRef]
- Jacob, F.; Perrin, D.; Sánchez, C.; Monod, J.; Edelstein, S. The operon: A group of genes with expression coordinated by an operator. [C.R.Acad. Sci. Paris 250 (1960) 1727–1729]. C. R. Biol. 2005, 328, 514–520. [Google Scholar] [CrossRef]
- Pardee, A.B. Nucleic acid precursors and protein synthesis. Proc. Natl. Acad. Sci. USA 1954, 40, 263–270. [Google Scholar] [CrossRef]
- Lockard, R.E.; Lingrel, J.B. The synthesis of mouse hemoglobin beta-chains in a rabbit reticulocyte cell-free system programmed with mouse reticulocyte 9S RNA. Biochem. Biophys. Res. Commun. 1969, 37, 204–212. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef]
- Karikó, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. BNT162b2 mRNA COVID-19 Vaccine: First Approval. Drugs 2021, 81, 495–501. [Google Scholar] [CrossRef]
- Naficy, A.; Kuxhausen, A.; Seifert, H.; Hastie, A.; Leav, B.; Miller, J.; Anteyi, K.; Mwakingwe-Omari, A. No immunological interference or concerns about safety when seasonal quadrivalent influenza vaccine is co-administered with a COVID-19 mRNA-1273 booster vaccine in adults: A randomized trial. Hum. Vaccines Immunother. 2024, 20, 2327736. [Google Scholar] [CrossRef]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e13. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.C.; Kepl, E.; Navarro, M.J.; Chen, C.; Johnson, M.; Sprouse, K.R.; Stewart, C.; Palser, A.; Valdez, A.; Pettie, D.; et al. Potent neutralization of SARS-CoV-2 variants by RBD nanoparticle and prefusion-stabilized spike immunogens. NPJ Vaccines 2024, 9, 184. [Google Scholar] [CrossRef]
- Presnyak, V.; Alhusaini, N.; Chen, Y.H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ng, W.H.; Zusinaite, E.; Freitas, J.; Taylor, A.; Yerragunta, V.; Aavula, S.M.; Gorriparthi, S.; Ponsekaran, S.; Bonda, R.L.; et al. A single-dose intranasal live-attenuated codon deoptimized vaccine provides broad protection against SARS-CoV-2 and its variants. Nat. Commun. 2024, 15, 7225. [Google Scholar] [CrossRef]
- Qin, Q.; Yan, H.; Gao, W.; Cao, R.; Liu, G.; Zhang, X.; Wang, N.; Zuo, W.; Yuan, L.; Gao, P.; et al. Engineered mRNAs With Stable Structures Minimize Double-stranded RNA Formation and Increase Protein Expression. J. Mol. Biol. 2024, 436, 168822. [Google Scholar] [CrossRef]
- Jin, L.; Zhou, Y.; Zhang, S.; Chen, S.J. mRNA Vaccine Sequence and Structure Design and Optimization: Advances and Challenges. J. Biol. Chem. 2024, 301, 108015. [Google Scholar] [CrossRef]
- Jia, L.; Mao, Y.; Ji, Q.; Dersh, D.; Yewdell, J.W.; Qian, S.B. Decoding mRNA translatability and stability from the 5′ UTR. Nat. Struct. Mol. Biol. 2020, 27, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Sample, P.J.; Wang, B.; Reid, D.W.; Presnyak, V.; McFadyen, I.J.; Morris, D.R.; Seelig, G. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 2019, 37, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef]
- Passmore, L.A.; Coller, J. Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat. Rev. Mol. Cell Biol. 2022, 23, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Biziaev, N.; Shuvalov, A.; Salman, A.; Egorova, T.; Shuvalova, E.; Alkalaeva, E. The impact of mRNA poly(A) tail length on eukaryotic translation stages. Nucleic Acids Res. 2024, 52, 7792–7808. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, H.; Shao, F.; Zhang, Y.; Nie, H.; Zhang, J.; Li, C.; Hou, Z.; Chen, Z.J.; Wang, J.; et al. Remodeling of maternal mRNA through poly(A) tail orchestrates human oocyte-to-embryo transition. Nat. Struct. Mol. Biol. 2023, 30, 200–215. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, Q.; Feng, X.H.; Liu, J. Synthetic modified messenger RNA for therapeutic applications. Acta Biomater. 2021, 131, 1–15. [Google Scholar] [CrossRef]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef]
- Dolgin, E. CureVac COVID vaccine let-down spotlights mRNA design challenges. Nature 2021, 594, 483. [Google Scholar] [CrossRef]
- Horne, R.W.; Bangham, A.D.; Whittaker, V.P. Negatively stained lipoprotein membranes. Nature 1963, 200, 1340. [Google Scholar] [CrossRef]
- Bangham, A.D.; Horne, R.W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, F.; Kamali, H.; Gholami, L.; McCloskey, A.P.; Kesharwani, P.; Sahebkar, A. Solid lipid nanoparticles: A promising tool for insulin delivery. Expert Opin. Drug Deliv. 2022, 19, 1577–1595. [Google Scholar] [CrossRef]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles—From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef] [PubMed]
- Eygeris, Y.; Gupta, M.; Kim, J.; Sahay, G. Chemistry of Lipid Nanoparticles for RNA Delivery. Acc. Chem. Res. 2022, 55, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Fang, E.; Liu, X.; Li, M.; Zhang, Z.; Song, L.; Zhu, B.; Wu, X.; Liu, J.; Zhao, D.; Li, Y. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target. Ther. 2022, 7, 94. [Google Scholar] [CrossRef]
- Hald Albertsen, C.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef]
- Xiao, Y.; Tang, Z.; Huang, X.; Chen, W.; Zhou, J.; Liu, H.; Liu, C.; Kong, N.; Tao, W. Emerging mRNA technologies: Delivery strategies and biomedical applications. Chem. Soc. Rev. 2022, 51, 3828–3845. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Alameh, M.G.; Tombácz, I.; Bettini, E.; Lederer, K.; Sittplangkoon, C.; Wilmore, J.R.; Gaudette, B.T.; Soliman, O.Y.; Pine, M.; Hicks, P.; et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 2021, 54, 2877–2892.e7. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lee, A.; Grigoryan, L.; Arunachalam, P.S.; Scott, M.K.D.; Trisal, M.; Wimmers, F.; Sanyal, M.; Weidenbacher, P.A.; Feng, Y.; et al. Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine. Nat. Immunol. 2022, 23, 543–555. [Google Scholar] [CrossRef]
- Tahtinen, S.; Tong, A.J.; Himmels, P.; Oh, J.; Paler-Martinez, A.; Kim, L.; Wichner, S.; Oei, Y.; McCarron, M.J.; Freund, E.C.; et al. IL-1 and IL-1ra are key regulators of the inflammatory response to RNA vaccines. Nat. Immunol. 2022, 23, 532–542. [Google Scholar] [CrossRef]
- Ndeupen, S.; Qin, Z.; Jacobsen, S.; Bouteau, A.; Estanbouli, H.; Igyártó, B.Z. The mRNA-LNP platform’s lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. iScience 2021, 24, 103479. [Google Scholar] [CrossRef]
- Ju, Y.; Lee, W.S.; Pilkington, E.H.; Kelly, H.G.; Li, S.; Selva, K.J.; Wragg, K.M.; Subbarao, K.; Nguyen, T.H.O.; Rowntree, L.C.; et al. Anti-PEG Antibodies Boosted in Humans by SARS-CoV-2 Lipid Nanoparticle mRNA Vaccine. ACS Nano 2022, 16, 11769–11780. [Google Scholar] [CrossRef]
- Szebeni, J.; Storm, G.; Ljubimova, J.Y.; Castells, M.; Phillips, E.J.; Turjeman, K.; Barenholz, Y.; Crommelin, D.J.A.; Dobrovolskaia, M.A. Applying lessons learned from nanomedicines to understand rare hypersensitivity reactions to mRNA-based SARS-CoV-2 vaccines. Nat. Nanotechnol. 2022, 17, 337–346. [Google Scholar] [CrossRef]
- Sato, Y.; Nakamura, T.; Yamada, Y.; Harashima, H. The impact of, and expectations for, lipid nanoparticle technology: From cellular targeting to organelle targeting. J. Control. Release 2024, 370, 516–527. [Google Scholar] [CrossRef]
- Hardianto, A.; Muscifa, Z.S.; Widayat, W.; Yusuf, M.; Subroto, T. The Effect of Ethanol on Lipid Nanoparticle Stabilization from a Molecular Dynamics Simulation Perspective. Molecules 2023, 28, 4836. [Google Scholar] [CrossRef]
- Short, L.; Holt, R.A.; Cullis, P.R.; Evgin, L. Direct in vivo CAR T cell engineering. Trends Pharmacol. Sci. 2024, 45, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.A.; Mei, H.; Sang, R.; Ortega, D.G.; Deng, W. Advancements and challenges in developing in vivo CAR T cell therapies for cancer treatment. EBioMedicine 2024, 106, 105266. [Google Scholar] [CrossRef] [PubMed]
- Parayath, N.N.; Stephan, S.B.; Koehne, A.L.; Nelson, P.S.; Stephan, M.T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 2020, 11, 6080. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Méndez Fernández, P.O.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Billingsley, M.M.; Singh, N.; Ravikumar, P.; Zhang, R.; June, C.H.; Mitchell, M.J. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20, 1578–1589. [Google Scholar] [CrossRef]
- Álvarez-Benedicto, E.; Tian, Z.; Chatterjee, S.; Orlando, D.; Kim, M.; Guerrero, E.D.; Wang, X.; Siegwart, D.J. Spleen SORT LNP Generated in situ CAR T Cells Extend Survival in a Mouse Model of Lymphoreplete B Cell Lymphoma. Angew. Chem. Int. Ed. Engl. 2023, 62, e202310395. [Google Scholar] [CrossRef]
- Hamilton, A.G.; Swingle, K.L.; Joseph, R.A.; Mai, D.; Gong, N.; Billingsley, M.M.; Alameh, M.G.; Weissman, D.; Sheppard, N.C.; June, C.H.; et al. Ionizable Lipid Nanoparticles with Integrated Immune Checkpoint Inhibition for mRNA CAR T Cell Engineering. Adv. Healthc. Mater. 2023, 12, e2301515. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Y.; Wang, P.; Li, S.; Dong, Y.; Zhou, M.; Shi, B.; Jiang, H.; Sun, R.; Li, Z. FAP-targeted CAR-T suppresses MDSCs recruitment to improve the antitumor efficacy of claudin18.2-targeted CAR-T against pancreatic cancer. J. Transl. Med. 2023, 21, 255. [Google Scholar] [CrossRef]
- Billingsley, M.M.; Hamilton, A.G.; Mai, D.; Patel, S.K.; Swingle, K.L.; Sheppard, N.C.; June, C.H.; Mitchell, M.J. Orthogonal Design of Experiments for Optimization of Lipid Nanoparticles for mRNA Engineering of CAR T Cells. Nano Lett. 2022, 22, 533–542. [Google Scholar] [CrossRef]
- Kobiyama, K.; Ishii, K.J. Making innate sense of mRNA vaccine adjuvanticity. Nat. Immunol. 2022, 23, 474–476. [Google Scholar] [CrossRef]
- Bevers, S.; Kooijmans, S.A.A.; Van de Velde, E.; Evers, M.J.W.; Seghers, S.; Gitz-Francois, J.; van Kronenburg, N.C.H.; Fens, M.; Mastrobattista, E.; Hassler, L.; et al. mRNA-LNP vaccines tuned for systemic immunization induce strong antitumor immunity by engaging splenic immune cells. Mol. Ther. 2022, 30, 3078–3094. [Google Scholar] [CrossRef] [PubMed]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Ranjan, P.; Suleiman, Z.G.; Goswami, S.K.; Li, J.; Prasad, R.; Verma, S.K. mRNA modifications in cardiovascular biology and disease: With a focus on m6A modification. Cardiovasc. Res. 2022, 118, 1680–1692. [Google Scholar] [CrossRef]
- Flamand, M.N.; Tegowski, M.; Meyer, K.D. The Proteins of mRNA Modification: Writers, Readers, and Erasers. Annu. Rev. Biochem. 2023, 92, 145–173. [Google Scholar] [CrossRef]
- Hara, T.; Meng, S.; Sato, H.; Tatekawa, S.; Sasaki, K.; Takeda, Y.; Tsuji, Y.; Arao, Y.; Ofusa, K.; Kitagawa, T.; et al. High N6-methyladenosine-activated TCEAL8 mRNA is a novel pancreatic cancer marker. Cancer Sci. 2024, 115, 2360–2370. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, S.; Pascual, G.; Feng, B.; Klann, K.; Behm, M.; Hotz-Wagenblatt, A.; Richter, K.; Zaoui, K.; Herpel, E.; Münch, C.; et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature 2022, 607, 593–603. [Google Scholar] [CrossRef]
- Witzenberger, M.; Burczyk, S.; Settele, D.; Mayer, W.; Welp, L.M.; Heiss, M.; Wagner, M.; Monecke, T.; Janowski, R.; Carell, T.; et al. Human TRMT2A methylates tRNA and contributes to translation fidelity. Nucleic Acids Res. 2023, 51, 8691–8710. [Google Scholar] [CrossRef]
- Gu, X.; Zhuang, A.; Yu, J.; Yang, L.; Ge, S.; Ruan, J.; Jia, R.; Fan, X.; Chai, P. Histone lactylation-boosted ALKBH3 potentiates tumor progression and diminished promyelocytic leukemia protein nuclear condensates by m1A demethylation of SP100A. Nucleic Acids Res. 2024, 52, 2273–2289. [Google Scholar] [CrossRef]
- Freund, I.; Buhl, D.K.; Boutin, S.; Kotter, A.; Pichot, F.; Marchand, V.; Vierbuchen, T.; Heine, H.; Motorin, Y.; Helm, M.; et al. 2′-O-methylation within prokaryotic and eukaryotic tRNA inhibits innate immune activation by endosomal Toll-like receptors but does not affect recognition of whole organisms. RNA 2019, 25, 869–880. [Google Scholar] [CrossRef]
- Keller, P.; Freund, I.; Marchand, V.; Bec, G.; Huang, R.; Motorin, Y.; Eigenbrod, T.; Dalpke, A.; Helm, M. Double methylation of tRNA-U54 to 2′-O-methylthymidine (Tm) synergistically decreases immune response by Toll-like receptor 7. Nucleic Acids Res. 2018, 46, 9764–9775. [Google Scholar] [CrossRef] [PubMed]
- Small-Saunders, J.L.; Sinha, A.; Bloxham, T.S.; Hagenah, L.M.; Sun, G.; Preiser, P.R.; Dedon, P.C.; Fidock, D.A. tRNA modification reprogramming contributes to artemisinin resistance in Plasmodium falciparum. Nat. Microbiol. 2024, 9, 1483–1498. [Google Scholar] [CrossRef]
- Mulroney, T.E.; Pöyry, T.; Yam-Puc, J.C.; Rust, M.; Harvey, R.F.; Kalmar, L.; Horner, E.; Booth, L.; Ferreira, A.P.; Stoneley, M.; et al. N(1)-methylpseudouridylation of mRNA causes +1 ribosomal frameshifting. Nature 2024, 625, 189–194. [Google Scholar] [CrossRef]
- Maeda, H. Macromolecular therapeutics in cancer treatment: The EPR effect and beyond. J. Control. Release 2012, 164, 138–144. [Google Scholar] [CrossRef]
- Chen, J.; Ye, Z.; Huang, C.; Qiu, M.; Song, D.; Li, Y.; Xu, Q. Lipid nanoparticle-mediated lymph node-targeting delivery of mRNA cancer vaccine elicits robust CD8(+) T cell response. Proc. Natl. Acad. Sci. USA 2022, 119, e2207841119. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.K.; Billingsley, M.M.; Frazee, C.; Han, X.; Swingle, K.L.; Qin, J.; Alameh, M.G.; Wang, K.; Weissman, D.; Mitchell, M.J. Hydroxycholesterol substitution in ionizable lipid nanoparticles for mRNA delivery to T cells. J. Control. Release 2022, 347, 521–532. [Google Scholar] [CrossRef]
- Li, B.; Raji, I.O.; Gordon, A.G.R.; Sun, L.; Raimondo, T.M.; Oladimeji, F.A.; Jiang, A.Y.; Varley, A.; Langer, R.S.; Anderson, D.G. Accelerating ionizable lipid discovery for mRNA delivery using machine learning and combinatorial chemistry. Nat. Mater. 2024, 23, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, M.M.; Gong, N.; Mukalel, A.J.; Thatte, A.S.; El-Mayta, R.; Patel, S.K.; Metzloff, A.E.; Swingle, K.L.; Han, X.; Xue, L.; et al. In Vivo mRNA CAR T Cell Engineering via Targeted Ionizable Lipid Nanoparticles with Extrahepatic Tropism. Small 2024, 20, e2304378. [Google Scholar] [CrossRef]
- Herrera-Barrera, M.; Ryals, R.C.; Gautam, M.; Jozic, A.; Landry, M.; Korzun, T.; Gupta, M.; Acosta, C.; Stoddard, J.; Reynaga, R.; et al. Peptide-guided lipid nanoparticles deliver mRNA to the neural retina of rodents and nonhuman primates. Sci. Adv. 2023, 9, eadd4623. [Google Scholar] [CrossRef]
- Ye, S.; Cheng, Y.; Guo, Z.; Wang, X.; Wei, W. A lipid toolbox of sugar alcohol fatty acid monoesters for single-component lipid nanoparticles with temperature-controlled release. Colloids Surf. B Biointerfaces 2023, 228, 113426. [Google Scholar] [CrossRef]
- Qu, M.; Mehrmohammadi, M.; Truby, R.; Graf, I.; Homan, K.; Emelianov, S. Contrast-enhanced magneto-photo-acoustic imaging in vivo using dual-contrast nanoparticles. Photoacoustics 2014, 2, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Kato, N.; Yoshida, M.; Hiu, T.; Matsuo, T.; Mizukami, S.; Omata, D.; Suzuki, R.; Maruyama, K.; Mukai, H.; et al. Focused ultrasound/microbubbles-assisted BBB opening enhances LNP-mediated mRNA delivery to brain. J. Control. Release 2022, 348, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, R.; Wang, Y.; Liu, M.; Hu, D.; Wang, Y.; Yang, L. A blood-brain barrier- and blood-brain tumor barrier-penetrating siRNA delivery system targeting gliomas for brain tumor immunotherapy. J. Control. Release 2024, 369, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Han, E.L.; Tang, S.; Kim, D.; Murray, A.M.; Swingle, K.L.; Hamilton, A.G.; Mrksich, K.; Padilla, M.S.; Palanki, R.; Li, J.J.; et al. Peptide-Functionalized Lipid Nanoparticles for Targeted Systemic mRNA Delivery to the Brain. Nano Lett. 2025, 25, 800–810. [Google Scholar] [CrossRef]
- Johnson, L.R.; Lee, D.Y.; Eacret, J.S.; Ye, D.; June, C.H.; Minn, A.J. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell 2021, 184, 4981–4995.e14. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Study/Company | mRNA-LNP System | Key Findings | Reference |
|---|---|---|---|
| Billingsley et al., 2020 | Ionizable LNPs (C14-4 LNPs) optimized for T cell transfection |
| [75] |
| Rurik et al., 2022 | CD5-targeted LNPs delivering nucleoside-modified mRNA encoding FAP CAR |
| [74] |
| Álvarez-Benedicto et al., 2023 | Spleen SORT LNPs for antibody-free T cell targeting |
| [76] |
| Hamilton et al., 2023 | mRNA/siRNA Co-Delivery LNPs for CAR expression and PD-1 knockdown |
| [77] |
| Capstan Therapeutics | CD8-targeted LNPs delivering sequence-enhanced mRNA encoding anti-CD19 CAR |
| |
| Orna Therapeutics | Circular RNA (oRNA)-LNPs with optimized internal ribosome entry sites (IRESs) for enhanced CAR expression |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, S.; Hara, T.; Miura, Y.; Arao, Y.; Saito, Y.; Inoue, K.; Hirotsu, T.; Vecchione, A.; Satoh, T.; Ishii, H. In Vivo Engineered CAR-T Cell Therapy: Lessons Built from COVID-19 mRNA Vaccines. Int. J. Mol. Sci. 2025, 26, 3119. https://doi.org/10.3390/ijms26073119
Meng S, Hara T, Miura Y, Arao Y, Saito Y, Inoue K, Hirotsu T, Vecchione A, Satoh T, Ishii H. In Vivo Engineered CAR-T Cell Therapy: Lessons Built from COVID-19 mRNA Vaccines. International Journal of Molecular Sciences. 2025; 26(7):3119. https://doi.org/10.3390/ijms26073119
Chicago/Turabian StyleMeng, Sikun, Tomoaki Hara, Yutaka Miura, Yasuko Arao, Yoshiko Saito, Kana Inoue, Takaaki Hirotsu, Andrea Vecchione, Taroh Satoh, and Hideshi Ishii. 2025. "In Vivo Engineered CAR-T Cell Therapy: Lessons Built from COVID-19 mRNA Vaccines" International Journal of Molecular Sciences 26, no. 7: 3119. https://doi.org/10.3390/ijms26073119
APA StyleMeng, S., Hara, T., Miura, Y., Arao, Y., Saito, Y., Inoue, K., Hirotsu, T., Vecchione, A., Satoh, T., & Ishii, H. (2025). In Vivo Engineered CAR-T Cell Therapy: Lessons Built from COVID-19 mRNA Vaccines. International Journal of Molecular Sciences, 26(7), 3119. https://doi.org/10.3390/ijms26073119