
Dressed in Collagen: 2D and 3D Cardiac Fibrosis Models


Abstract
1. Introduction
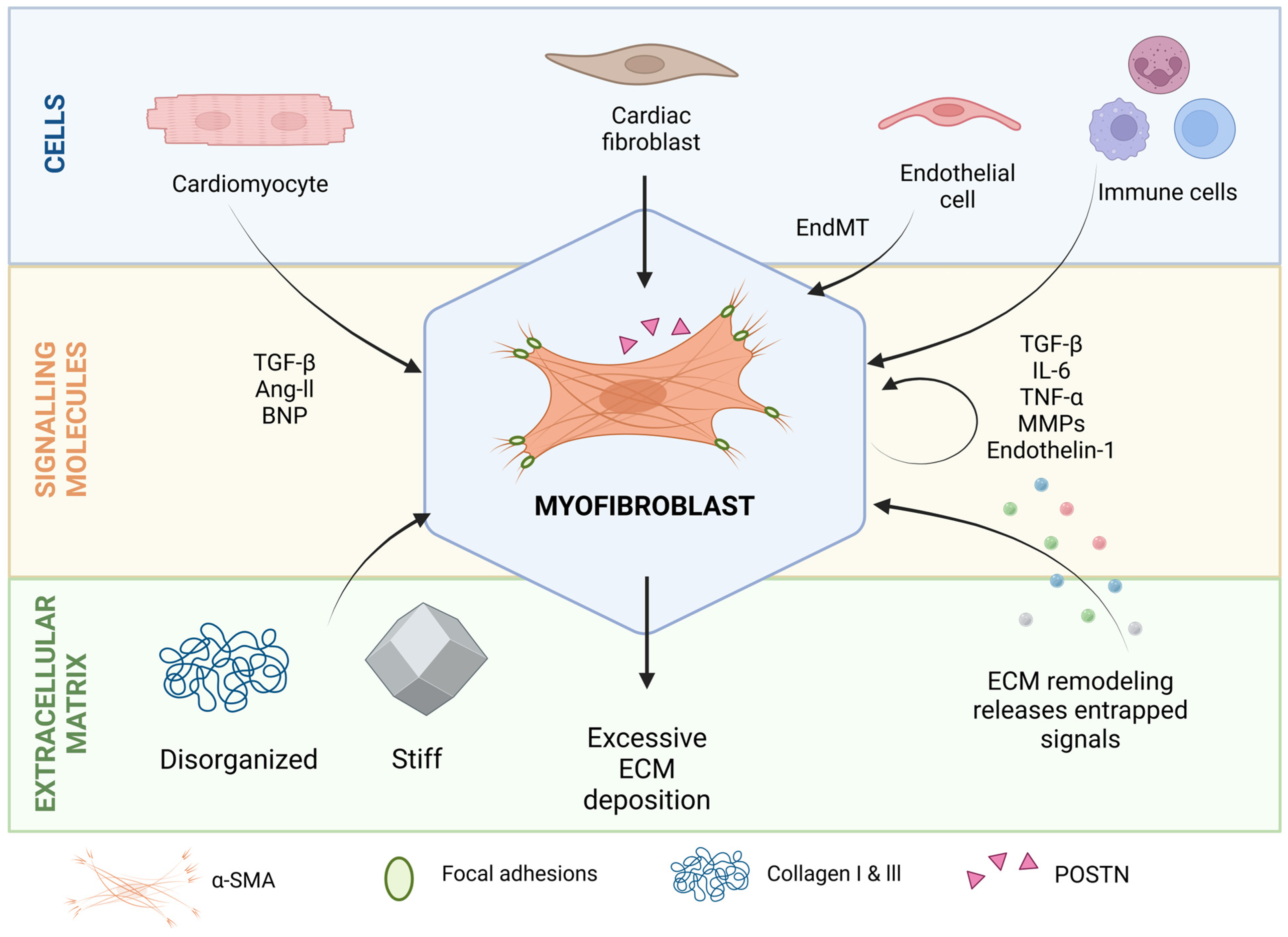
2. Molecular and Cellular Players in Cardiac Fibrosis
2.1. Myofibroblasts
2.2. Paracrine Signals
2.3. ECM as a Source of Biomechanical Cues and Signaling Molecules That Regulate Cell Profibrotic Activity
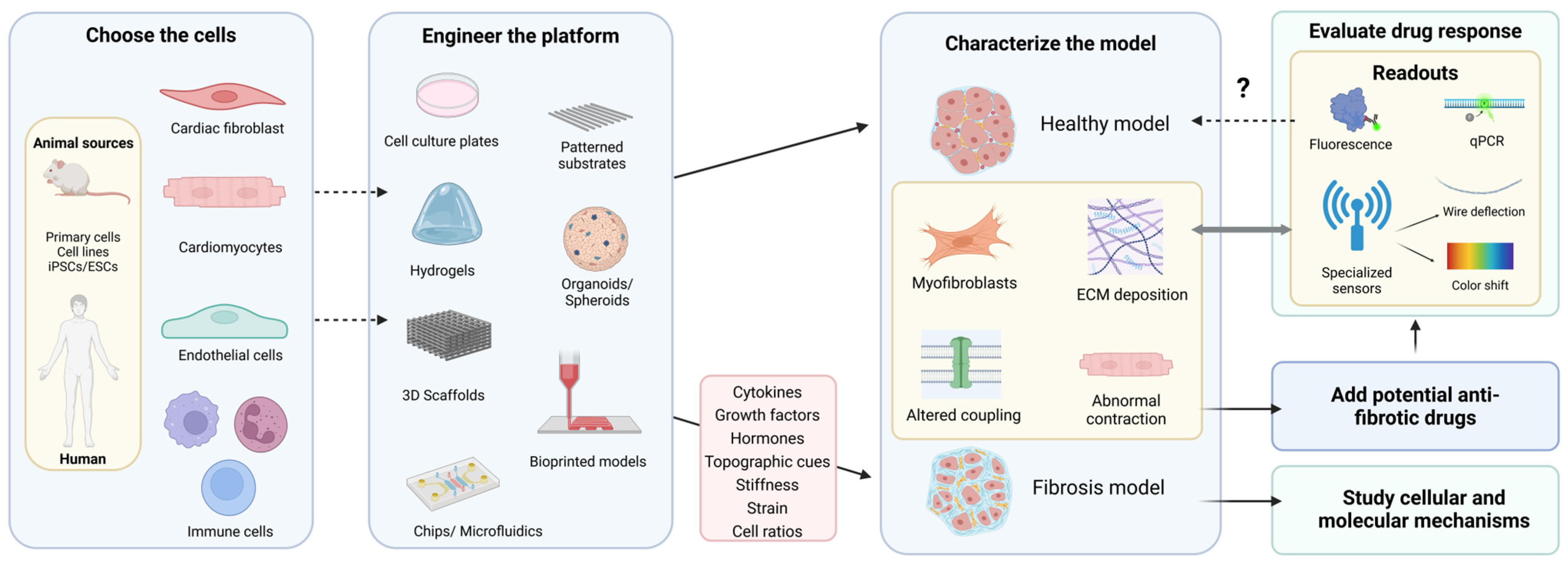
3. Main Characteristics of an Ideal In Vitro Model of Cardiac Fibrosis
- (1)
- Include cellular populations involved in cardiac fibrosis, namely CF (including myofibroblasts), CM, EC, and immune cells.
- (2)
- Accurately model the dynamic ECM remodeling with excessive collagen deposition, increased stiffness, and the release of bioactive factors from the matrix. Additionally, it should emulate the mechanical and electrical properties of fibrotic tissue.
- (3)
- Consider the variety of triggers implicated in cardiac fibrosis. This should encompass biochemical and biomechanical cues but also the hypoxia or reperfusion conditions that naturally occur during MI.
- (4)
- Have clear, measurable, and objective readouts of fibroblast activation, ECM deposition, electrical activity, and contractile behavior or an objective measure of a certain behavior (by including sensing systems).
- (5)
- Be reproducible, cost-effective, and have high-throughput capabilities.
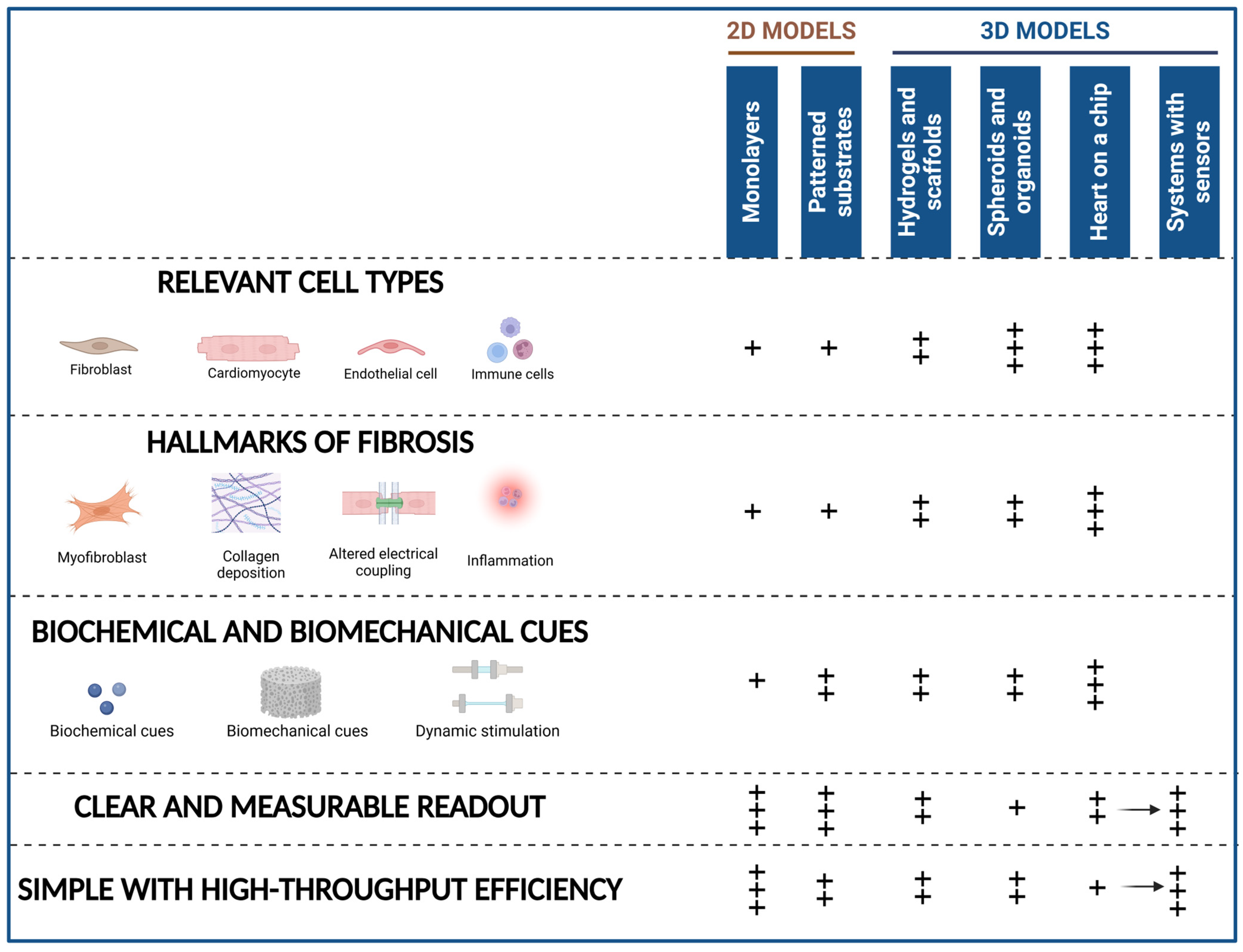
4. 2D and 3D In Vitro Models of Cardiac Fibrosis
4.1. Cellular Components

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Cells | Stimuli | Characterization | Readout | Tested Molecules | Example References | |
|---|---|---|---|---|---|---|---|
| Monolayers | CF (Human) [55,69] (Rat) [56,70] | TFG-β Ang ll [56,71] | Proliferation and Viability: Viability Cell Markers: α-SMA, POSTN, phalloidin [59] ECM Remodeling: Fibronectin, collagen type I and type III, collagen maturity (resistance to digestion, cell surface biotinylation experiments), deposition of extracellular collagen (immunofluorescence for glycosaminoglycans) [55] Inflammatory markers: Inflammatory protein expression panel, intracellular ROS [69] Secreted Molecules: TGF-β, BNP secretion Phenotypic changes throughout culture: [59] | Proliferation, viability, and migration: Cell viability, cytotoxicity, proliferation and migration [54,70,71,72] Cell Markers: α-SMA ECM Remodeling: Collagen type I, telo-collagen IαI expression and secretion [65], MMP-2 and MMP-9 activity (gelatin zymography) [72] Inflammatory markers: Inflammatory protein expression panel, intracellular ROS Activated Pathways: Wnt1, β-catenin Others: Proline incorporation [70] | Small molecules Pirfenidone [54] SB431542, 21 small molecules and 2 proteins previously reported to confer antifibrotic effects in vivo, compound ESI09 [55] Molecules based on Tranilast’s core structure (FT011) [56] Src inhibitor WH-4 023 [73] Natural compounds Ellagic acid, punicic acid [69] Curcumin [72] Quercetin dihydrate [71] Molecular similarities of bufalin and lycorine [74] Libraries of compounds Libraries of 5000 preclinical and clinical compounds [54] Library of natural compounds [75] | [54] [55] [56] [59] [71] [70] [72] [69] [75] [73] [74] | |
| CF, CM and EC (Human) | TFG-β | Proliferation and Viability: Viability Cell Markers: Vimentin, CD90, CD31, cTnT, TGF-β receptor (TGF-βR), antifibrotic factor HGF receptor (c-met) ECM Remodeling: Collagen type I, collagen type III, fibronectin, MMP-2, PCR array of ECM-related genes Secreted molecules: TFG-β expression | ECM Remodeling: Collagen type I, Collagen type III, Fibronectin, MMP-2, PCR array of ECM related genes Secreted molecules: TFG-β expression Contraction and relaxation behavior | HGF ONO-1301 Camostat mesilate Pirfenidone | [67] | ||
| Hydrogels/Scaffolds | Patterned substrates (PEG hydrogel matrix) | CF (Rat) | Stiffness | Purity of Rat CF: DDR2 expression Cell Markers: α-SMA, F-actin ECM Remodeling: Collagen, fibronectin Proliferation and Viability: Cell proliferation, cell migration | Cell Markers: α-SMA, F-actin ECM Remodeling: Collagen, fibronectin | ROCK inhibitor (Y27632) | [60] |
| Structural color hydrogels | CM and CF (Human) | CM:CF ratio (Healthy 3:1-Fibrotic 1:3) | Proliferation and Viability: Cell proliferation, cell viability Cell Markers: α-SMA ECM Remodeling: Collagen type I Mechanical Properties: Microtissue stiffness, passive tension Secreted Molecules: BNP secretion Hydrogel color shift properties | Functional/Contractile Properties (Contraction = Color shift) | Losartan Saracatinib Relaxin-2 Pirfenidone Nintedanib | [76] | |
| Hydrogels (GelMa) | CF (Human) | TFG-β | Proliferation and Viability: Cell spreading in hydrogel, viability Cell Markers: α-SMA, vimentin, fibroblast-specific protein (FSP) ECM Remodeling: Collagen IαI/IIIαI Mechanical Properties: Mechanical properties of hydrogels (elastic modulus, mass swelling ratio, and pore size) | Cell Markers: α-SMA ECM Deposition: Collagen IαI/IIIαI | Paracrine effects of cardiac progenitor cells | [26] | |
| CF and CM (Rat) [77] (Human) [78] | Stiffness TGF-β [78] | Proliferation and Viability: Cell spreading in hydrogel, viability, proliferation, metabolic activity Cell Markers: α-SMA, vimentin ECM Remodeling: Collagen type I and type III Hydrogel Characterization: Mechanical properties (elastic modulus, stiffness), mass swelling ratio, pore size, morphology (SEM) Contraction and Electrophysiological Behavior: Beating pattern (beats per minute, synchronicity), maturation phase (time for cells to become fully functional after fabrication), hydrogel contraction test [54,77] Calcium Handling/Dynamics: Cell coupling (electromechanical coupling, calcium transients) Other: Targeted proteomics | Cell Markers: α-SMA ECM Deposition: Fibronectin, collagen type I and type III, MMP-2, POSTN Contraction and Electrophysiological Behavior: Beating pattern Other: Targeted proteomics | Isoprenaline (to assess physiological beating response) [77] Pirfenidone | [54] [77] [78] | ||
| Scaffolds | CF (Human) | Random nanofibrous organization and stiffness | Proliferation and Viability: Cytocompatibility, cytotoxicity Scaffold Characterization: Nanofiber orientation, porosity, area (SEM), mechanical properties (Young modulus) Surface coating and grafting efficiency Cell Markers: Cell distribution on scaffold, α-SMA (fibroblast activation on scaffold) | Cell Markers: α-SMA ECM Deposition: Fibronectin, collagen type I, collagen type III | Tranilast [79] | [79] [80] | |
| Spheroids | Three-cell-type spheroids | CF, CM and EC (Rat) | Phenylephrine-induced cardiac hypertrophy [81] TGF-β + Doxycycline [82] | Proliferation and Viability: Cell death (Calcein/PI), mitochondrial and membrane potential, calcium concentration, apoptosis, and remodeling of the spheroid Structural/Phenotypic Markers: Markers of CMs, CFs, and ECs in the microspheres, HE staining (muscle fibers), expression of CTGF, fibronectin, TGFβ Dimensional Properties: Diameter Functional Properties: Beating frequency ECM and Histological Analysis: ECM deposition (histological analysis using PicroSirius Red staining) | Dimensional Properties: Diameter Viability: Mitochondrial and membrane potential, calcium concentration Other: RNA-seq | Guanxinning injection [81] | [82] [81] |
| Two-cell-type spheroids | CF and CM (Mice) [83] (Human) [84] | TGF-β Pre-culture fibroblasts in stiff plastic before assembling them in spheroids [84] CM:CF ratio | Cell Markers: α-actinin, vimentin ECM Remodeling: Fibronectin, laminin Mechanical Properties Dimensional Properties: Spheroid volume Secreted Molecules and Pathways: Upregulation of TGFβ in media after stimuli, activation of TGF pathway: pSMAD2, MMP-2, and MMP-9 activities | Dimensional Properties: Spheroid volume ECM Remodeling: FN, Laminin, TIMP-1 Activity: MMP-2 and MMP-9 | SB 431542 (TGFB inhibitor) [83] | [84] [83] | |
| One-cell-type spheroids | CF (Human) | High glucose media + TGF-β | Cell Markers: α-SMA ECM Remodeling: Collagen type I, MMP-2, and MMP-9 activity | - | - | [85] | |
| Bioprinted spheroids | CF and CM (Human) | CM:CF ratio (healthy 4:1—scarred 1:4) | Proliferation and Viability: Viability Cell Markers: α-actinin (sarcomere formation), cTnT, and vimentin (cell distribution and fusion of spheroids) Functional/Contractile Properties: Contraction recording, amplitude analysis Electrophysiological Properties: Connexin-43 (coupling) Calcium Handling/Dynamics: Optical mapping of intracellular Ca2⁺ (synchronization), key parameters of the cardiac cell cycle (calcium transient duration, time-to-peak, calcium flux amplitude) | Proliferation and Viability: CM and CF proliferation (EdU, Vimentin, and cTnT staining) Functional/Contractile Properties: Contraction recording/amplitude analysis Calcium Handling/Dynamics: Activation delays, calcium flux amplitude | miR302 b/c induced Hippo inhibition | [86] | |
| Organoids | CF, CM and EC (Human) | Hypoxia-induced ischemia + TGF-β [87] Cryoinjury [88] | Proliferation and Viability: Sensitivity of ischemic injury and ischemic reperfusion injury (CM apoptosis) Cell Markers: Cellular composition and distribution (FACS, Immunostaining, qPCR, and single-cell transcriptomics), integrity of sarcomeric structures ECM Remodeling: Collagen deposition, mRNA expression of fibrosis-related genes (ACTA2, POSTN, Vimentin, MMP-2), and collagen-related genes Dimensional Properties: Volume Secreted Molecules: Release of cTnT, MB, and CKM Functional/Contractile Properties: Contraction (arbitrary units) Calcium Handling/Dynamics: Calcium signaling and transients Electrophysiological Properties: Beating rate, spike amplitude, conduction velocity, field potential duration corrected by frequency (multielectrode arrays) | Functional/Contractile Properties: Contraction (arbitrary units) Calcium Handling/Dynamics: Calcium signaling and transients Cell Markers: α-SMA, VIM, CDH5, PECAM1, TNNT2, MYH7 ECM Remodeling: Connective tissue deposition Secreted Molecules: Secreted cTnT | Captopril [88] | [88] [87] | |
| dECM | Layer-by-layer 3D extrusion printed scaffold with cdECM Bioink, Laponite-XLG nanoclay, and PEG-DA [89] | CF and CM (to assess biocompatibility) (Human) | Stiffness | Rheological Characterization of Bioink Formulations: Yield stress, loss factor, tan δ, viscosity Gelation Kinetics: Rate and behavior during gelation Bioink Stability: Filament width at different timepoints Cell Viability | - | - | [89] |
| Fibrotic dECM from activated iPSC-CF [90] | CM and CF (to produce ECM) (Human) | TGF-β (to stimulate the CF producing the matrix) | Cell Markers: Purity of differentiated iPSCs: Matrix-Producing Cells (CF): α-SMA, fibroblast activation protein (FAP), vinculin; Seeded CM: α-actinin (sarcomere assembly), cTnT Secreted Molecules: TGF-β Characterization of CF-Produced dECM: Morphology: Collagen type I/type IV, fibronectin, ED-A and ED-B fibronectin isoforms, laminin Matrisome Components: Mass spectrometry Mechanical Properties: CM behavior/organization: number, proliferation, adhesion (YAP nuclear/cytoplasmic ratio), sarcomere assembly Functional Properties: Beating activity, contraction amplitude, contractility traces Calcium Handling/Dynamics: Calcium uptake, transient amplitude, and decay time | CM Behavior/Organization: Sarcomere assembly | Irreversible inhibitor of LOX, beta-aminopropionitrile (BAPN) (reduces tissue stiffness) → Used on the CF producing the dECM | [90] | |
| Porcine dECM [85] | CF (Human) | High glucose media + TGF-B + mechanical stimulation (cyclic pressure) | Proliferation and Viability: Viability Cell Markers: α-SMA ECM Remodeling: Collagen type I, MMP-2, and MMP-9 activity | - | - | [85] | |
| Models on a chip | Microtissues attached to PDMS rods/micropillars | CF and CM (Human) | CM:CF ratio (healthy 3:1-fibrotic1:3) + TGF-β [91] Laser injury [92] | Cell Viability and Proliferation: Cell apoptosis, necrosis, proliferation Cell Markers: CM alignment, α-SMA positive cells distribution, vimentin, fibronectin ECM Remodeling: Fibrillar collagen deposition, collagen type I/III ratio Mechanical Properties: Young’s modulus, passive tension Secreted Molecules: BNP secretion Electrophysiological Properties: Excitation threshold Calcium Handling/Dynamics: Calcium transient amplitudes and synchronization Functional/Contractile Properties: Beating pattern, twitch force | Secreted Molecules: BNP secretion Mechanical Properties: Stiffness, passive tension Functional/Contractile Properties: Active force ECM Remodeling: Collagen deposition Other: mRNA, miR signature | Carvedilol, Losartan [91] | [91] [92] |
| Biowire (fibrin-based hydrogel fitted with two polymer (POMaC) wires subjected to electrical conditioning) | CF and CM (Human) | CM:CF ratio (healthy 3:1-fibrotic1:3) + TGF-β1 | Cell Markers: α-SMA positive cell distribution ECM Remodeling: Collagen deposition and alignment (second harmonic generation imaging) Structural/Mechanical Properties: Sarcomeric length, Young’s modulus, passive tension Electrophysiological Properties: Excitation threshold, action potential profiles, connexin 43 Functional/Contractile Properties: Maximum capture rates, force–frequency relationships, post-rest potentiation (PRP) of force Calcium Handling/Dynamics: Calcium transient amplitudes and synchronization | Mechanical Properties: Passive tension (main screening parameter) ECM Remodeling: Collagen imaging and tissue compaction data (validation) Functional/Contractile Properties: Active force, ET, and MCR (monitoring) | Furin inhibitors: PCI(p-guanidinomethyl-phenylacetyl-Arg-Val-Arg-4-amidinobenzylamide) Fil (dec-RVKR-cmk) Fill (hexa-D-arginine amide) | [93] | |
| 3D microtissues subjected to mechanical stimulation (uniaxial strain) | CF (Human) [47,94] CM and CF (Rat) [95] | Mechanical (uniaxial strain) and biochemical stimulation (TGF-β) | Cell Viability and Proliferation: Proliferation Cellular Markers: α-SMA, cTROP Cellular Populations Distribution ECM Remodeling: Aggrecan, fibronectin, collagen type I, collagen type III, tenascin-c, MMP-2, Nppa Secreted Molecules: TGF-β Electrophysiological Properties: Excitation threshold Functional/Contractile Properties: Maximum capture rates, contraction direction, contraction probability density function Expression of Proteins Involved in Fibrotic Pathways: RhoA/ROCK pathway, hippo pathway, WNT pathway, TGFB pathway, CTGF, PAI-1, BNP, NPRA | Cellular Markers: α-SMA ECM Remodeling: Aggrecan, fibronectin, collagen type I, collagen type III Others: CF reprogramming into CM-like cells [47] | Pirfenidone [47] Tranilast [47,94] miRcombo(miR-1,miR-133,miR-208,andmiR-499) [47] | [47] [94] [95] | |
4.2. Profibrotic Stimuli
5. Exploring Cardiac Fibrosis Using 2D In Vitro Models
5.1. Cell Culture Platforms
5.2. Model Characterization and Readout Parameters
5.3. High-Throughput Screening in 2D Models
6. 3D Engineered In Vitro Models of Cardiac Fibrosis
6.1. Hydrogels and dECM Scaffolds
6.2. Spheroids and Organoids
6.3. Bioprinted 3D Models
6.4. Specialized Models on a Chip
7. Bridging the Gap Between High Throughput and Biological Relevance
8. Tested Molecules in In Vitro Systems
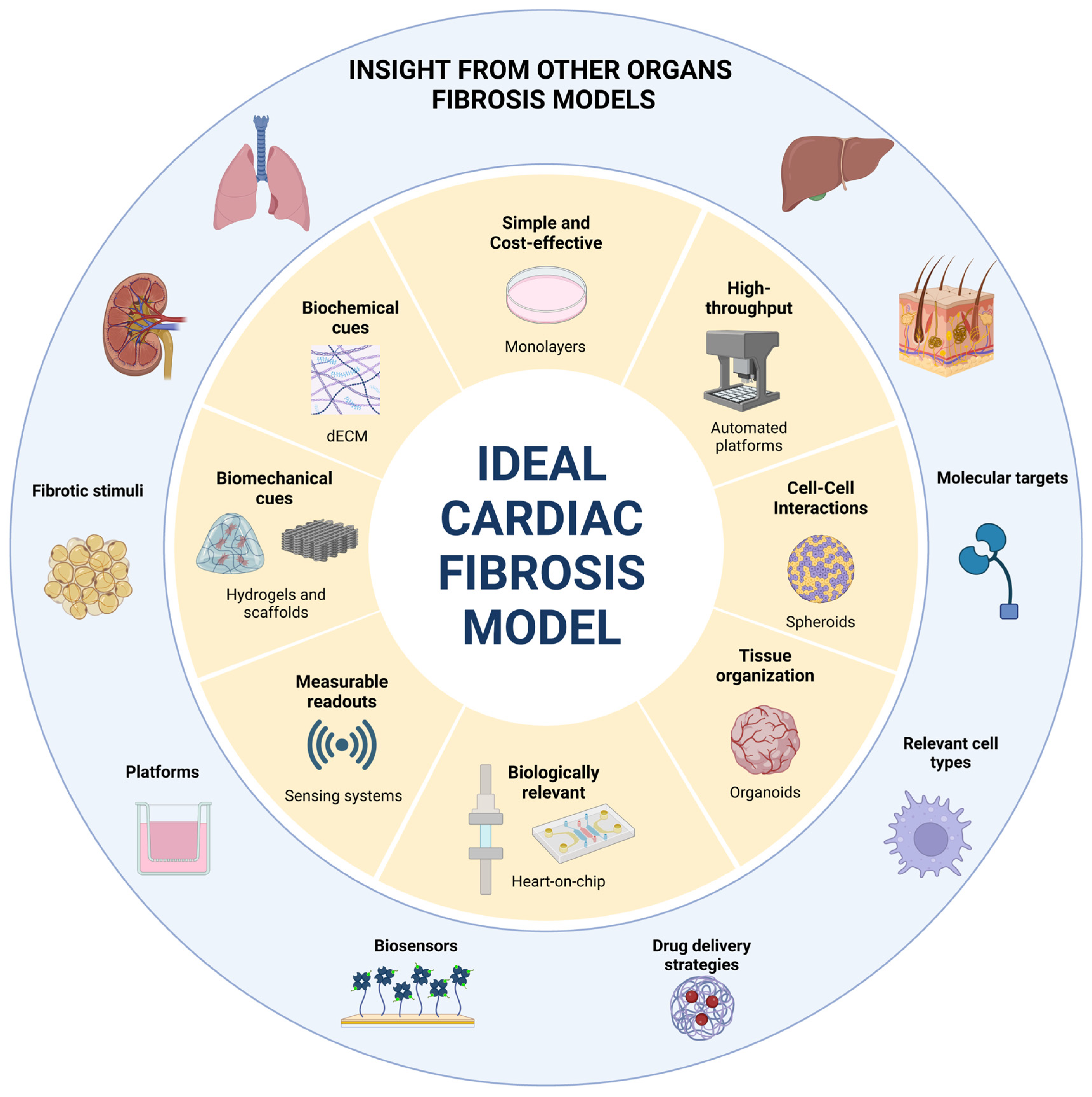
9. What Can We Learn from Other Organs?
9.1. Conserved Mechanisms and Inspirational Molecular Targets
9.2. Platforms and Culture Conditions
9.3. Sensing Systems
10. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Park, S.; Nguyen, N.B.; Pezhouman, A.; Ardehali, R. Cardiac fibrosis: Potential therapeutic targets. Transl. Res. 2019, 209, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.P.; Qu, Z.; Weiss, J.N. Cardiac fibrosis and arrhythmogenesis: The road to repair is paved with perils. J. Mol. Cell. Cardiol. 2014, 70, 83–91. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Q.; Tao, B.; Angelini, M.; Ramadoss, S.; Sun, B.; Wang, P.; Krokhaleva, Y.; Ma, F.; Gu, Y.; et al. Fibroblasts in heart scar tissue directly regulate cardiac excitability and arrhythmogenesis. Science 2023, 381, 1480–1487. [Google Scholar] [CrossRef]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013, 10, 15–26. [Google Scholar] [CrossRef]
- Sit, B.; Gutmann, D.; Iskratsch, T. dense plaques and podosomes: The cell matrix adhesions in cardiovascular mechanosensing. J. Muscle Res. Cell Motil. 2019, 40, 197–209. [Google Scholar] [CrossRef]
- Pandey, P.; Hawkes, W.; Hu, J.; Megone, W.V.; Gautrot, J.; Anilkumar, N.; Zhang, M.; Hirvonen, L.; Cox, S.; Ehler, E.; et al. Cardiomyocytes Sense Matrix Rigidity through a Combination of Muscle and Non-muscle Myosin Contractions. Dev. Cell 2018, 44, 326–336.e3. [Google Scholar] [CrossRef]
- Maruyama, K.; Imanaka-Yoshida, K. The Pathogenesis of Cardiac Fibrosis: A Review of Recent Progress. Int. J. Mol. Sci. 2022, 23, 2617. [Google Scholar] [CrossRef]
- Reed, G.W.; Rossi, J.E.; Cannon, C.P. Acute myocardial infarction. Lancet 2017, 389, 197–210. [Google Scholar]
- Kurose, H. Cardiac Fibrosis and Fibroblasts. Cells 2021, 10, 1716. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar]
- González, A.; López, B.; Ravassa, S.; José, G.S.; Latasa, I.; Butler, J.; Díez, J. Myocardial Interstitial Fibrosis in Hypertensive Heart Disease: From Mechanisms to Clinical Management. Hypertension 2024, 81, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, A.R.; Silva, A.C.; La Cruz, J.O.-D.; Martino, F.; Horváth, V.; Caluori, G.; Polanský, O.; Vinarský, V.; Azzato, G.; de Marco, G.; et al. Multiscale Analysis of Extracellular Matrix Remodeling in the Failing Heart. Circ. Res. 2021, 128, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar]
- Lancaster, L.H.; de Andrade, J.A.; Zibrak, J.D.; Padilla, M.L.; Albera, C.; Nathan, S.D.; Wijsenbeek, M.S.; Stauffer, J.L.; Kirchgaessler, K.-U.; Costabel, U. Pirfenidone safety and adverse event management in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2017, 26, 170057. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar]
- Van Nieuwenhoven, F.A.; Turner, N.A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vascul. Pharmacol. 2013, 58, 182–188. [Google Scholar]
- Czubryt, M.P. Cardiac Fibroblast to Myofibroblast Phenotype Conversion-An Unexploited Therapeutic Target. J. Cardiovasc. Dev. Dis. 2019, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tian, L.; Shen, M.; Tu, C.; Wu, H.; Gu, M.; Paik, D.T.; Wu, J.C. Generation of Quiescent Cardiac Fibroblasts From Human Induced Pluripotent Stem Cells for In Vitro Modeling of Cardiac Fibrosis. Circ. Res. 2019, 125, 552–566. [Google Scholar] [CrossRef]
- Shao, J.; Liu, J.; Zuo, S. Roles of Epigenetics in Cardiac Fibroblast Activation and Fibrosis. Cells 2022, 11, 2347. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Pan, J.; Wen, L.; Gong, B.; Li, J.; Gao, H.; Tan, W.; Liang, S.; Zhang, H.; Wang, X. MiR-590-3p regulates proliferation, migration and collagen synthesis of cardiac fibroblast by targeting ZEB1. J. Cell. Mol. Med. 2020, 24, 227–237. [Google Scholar] [CrossRef]
- Tillmanns, J.; Hoffmann, D.; Habbaba, Y.; Schmitto, J.D.; Sedding, D.; Fraccarollo, D.; Galuppo, P.; Bauersachs, J. Fibroblast activation protein alpha expression identifies activated fibroblasts after myocardial infarction. J. Mol. Cell. Cardiol. 2015, 87, 194–203. [Google Scholar] [CrossRef]
- Bracco Gartner, T.C.L.; Deddens, J.C.; Mol, E.A.; Ferrer, M.M.; van Laake, L.W.; Bouten, C.V.C.; Khademhosseini, A.; Doevendans, P.A.; Suyker, W.J.L.; Sluijter, J.P.G.; et al. Anti-fibrotic Effects of Cardiac Progenitor Cells in a 3D-Model of Human Cardiac Fibrosis. Front. Cardiovasc. Med. 2019, 6, 52. [Google Scholar] [CrossRef]
- Reichardt, I.M.; Robeson, K.Z.; Regnier, M.; Davis, J. Controlling cardiac fibrosis through fibroblast state space modulation. Cell. Signal. 2021, 79, 109888. [Google Scholar] [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar]
- Silvis, M.J.M.; Dengler, S.E.K.G.; Odille, C.A.; Mishra, M.; van der Kaaij, N.P.; Doevendans, P.A.; Sluijter, J.P.G.; de Kleijn, D.P.V.; de Jager, S.C.A.; Bosch, L.; et al. Damage-Associated Molecular Patterns in Myocardial Infarction and Heart Transplantation: The Road to Translational Success. Front. Immunol. 2020, 11, 599511. [Google Scholar] [CrossRef]
- Aujla, P.K.; Kassiri, Z. Diverse origins and activation of fibroblasts in cardiac fibrosis. Cell. Signal. 2021, 78, 109869. [Google Scholar] [CrossRef]
- Shinde, A.V.; Frangogiannis, N.G. Mechanisms of Fibroblast Activation in the Remodeling Myocardium. Curr. Pathobiol. Rep. 2017, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Transforming growth factor-beta in myocardial disease. Nat. Rev. Cardiol. 2022, 19, 435–455. [Google Scholar]
- Lee, Y.-S.; Kim, K.L.; Jang, H.-S.; Kim, J.-M.; Lee, J.-Y.; Shin, I.-S.; Lee, J.-S.; Suh, W.; Choi, J.-H.; Jeon, E.-S.; et al. Aldosterone upregulates connective tissue growth factor gene expression via p38 MAPK pathway and mineralocorticoid receptor in ventricular myocytes. J. Korean Med. Sci. 2004, 19, 805–811. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef]
- Zaidi, Y.; Aguilar, E.G.; Troncoso, M.; Ilatovskaya, D.V.; DeLeon-Pennell, K.Y. Immune regulation of cardiac fibrosis post myocardial infarction. Cell. Signal. 2021, 77, 109837. [Google Scholar] [CrossRef]
- Yousefi, F.; Shabaninejad, Z.; Vakili, S.; Derakhshan, M.; Movahedpour, A.; Dabiri, H.; Ghasemi, Y.; Mahjoubin-Tehran, M.; Nikoozadeh, A.; Savardashtaki, A.; et al. TGF-β and WNT signaling pathways in cardiac fibrosis: Non-coding RNAs come into focus. Cell Commun. Signal. 2020, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Czepiel, M.; Diviani, D.; Jaźwa-Kusior, A.; Tkacz, K.; Rolski, F.; Smolenski, R.T.; Siedlar, M.; Eriksson, U.; Kania, G.; Błyszczuk, P. Angiotensin II receptor 1 controls profibrotic Wnt/β-catenin signalling in experimental autoimmune myocarditis. Cardiovasc. Res. 2022, 118, 573–584. [Google Scholar] [CrossRef]
- Blyszczuk, P.; Müller-Edenborn, B.; Valenta, T.; Osto, E.; Stellato, M.; Behnke, S.; Glatz, K.; Basler, K.; Lüscher, T.F.; Distler, O.; et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur. Heart J. 2017, 38, 1413–1425. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef]
- Eckersley, A.; Ozols, M.; O’Cualain, R.; Keevill, E.-J.; Foster, A.; Pilkington, S.; Knight, D.; Griffiths, C.E.; Watson, R.E.; Sherratt, M.J. Proteomic fingerprints of damage in extracellular matrix assemblies. Matrix Biol. Plus 2020, 5, 100027. [Google Scholar] [CrossRef]
- Jariwala, N.; Ozols, M.; Bell, M.; Bradley, E.; Gilmore, A.; Debelle, L.; Sherratt, M.J. Matrikines as mediators of tissue remodelling. Adv. Drug Deliv. Rev. 2022, 185, 114240. [Google Scholar] [CrossRef] [PubMed]
- Creemers, E.E.; Pinto, Y.M. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc. Res. 2011, 89, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Lunde, I.G.; McCulloch, A.D.; Christensen, G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J. Clin. Med. 2017, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Landry, N.M.; Dixon, I.M.C. Fibroblast mechanosensing, SKI and Hippo signaling and the cardiac fibroblast phenotype: Looking beyond TGF-β. Cell. Signal. 2020, 76, 109802. [Google Scholar]
- Pesce, M.; Duda, G.N.; Forte, G.; Girao, H.; Raya, A.; Roca-Cusachs, P.; Sluijter, J.P.G.; Tschöpe, C.; Van Linthout, S. Cardiac fibroblasts and mechanosensation in heart development, health and disease. Nat. Rev. Cardiol. 2023, 20, 309–324. [Google Scholar] [CrossRef]
- Huang, C.; Ogawa, R. Fibroproliferative disorders and their mechanobiology. Connect. Tissue Res. 2012, 53, 187–196. [Google Scholar] [CrossRef]
- Visone, R.; Paoletti, C.; Cordiale, A.; Nicoletti, L.; Divieto, C.; Rasponi, M.; Chiono, V.; Occhetta, P. In Vitro Mechanical Stimulation to Reproduce the Pathological Hallmarks of Human Cardiac Fibrosis on a Beating Chip and Predict The Efficacy of Drugs and Advanced Therapies. Adv. Healthc. Mater. 2024, 13, e2301481. [Google Scholar] [CrossRef]
- Berry, M.F.; Engler, A.J.; Woo, Y.J.; Pirolli, T.J.; Bish, L.T.; Jayasankar, V.; Morine, K.J.; Gardner, T.J.; Discher, D.; Sweeney, H.L. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2196–H2203. [Google Scholar] [CrossRef]
- Galie, P.A.; Westfall, M.V.; Stegemann, J.P. Reduced serum content and increased matrix stiffness promote the cardiac myofibroblast transition in 3D collagen matrices. Cardiovasc. Pathol. 2011, 20, 325–333. [Google Scholar] [CrossRef]
- Zhao, X.-H.; Laschinger, C.; Arora, P.; Szászi, K.; Kapus, A.; McCulloch, C.A. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J. Cell Sci. 2007, 120 Pt 10, 1801–1809. [Google Scholar] [CrossRef]
- Conant, G.; Lai, B.F.L.; Lu, R.X.Z.; Korolj, A.; Wang, E.Y.; Radisic, M. High-Content Assessment of Cardiac Function Using Heart-on-a-Chip Devices as Drug Screening Model. Stem Cell Rev. Rep. 2017, 13, 335–346. [Google Scholar] [CrossRef]
- Picchio, V.; Floris, E.; Derevyanchuk, Y.; Cozzolino, C.; Messina, E.; Pagano, F.; Chimenti, I.; Gaetani, R. Multicellular 3D Models for the Study of Cardiac Fibrosis. Int. J. Mol. Sci. 2022, 23, 11642. [Google Scholar] [CrossRef] [PubMed]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Thai, P.N.; Shivnaraine, R.V.; Ren, L.; Wu, X.; Siepe, D.H.; Liu, Y.; Tu, C.; Shin, H.S.; Caudal, A.; et al. Multiscale drug screening for cardiac fibrosis identifies MD2 as a therapeutic target. Cell 2024, 187, 7143–7163.e22. [Google Scholar] [CrossRef]
- Palano, G.; Jansson, M.; Backmark, A.; Martinsson, S.; Sabirsh, A.; Hultenby, K.; Åkerblad, P.; Granberg, K.; Jennbacken, K.; Müllers, E.; et al. A high-content, in vitro cardiac fibrosis assay for high-throughput, phenotypic identification of compounds with anti-fibrotic activity. J. Mol. Cell. Cardiol. 2020, 142, 105–117. [Google Scholar] [CrossRef]
- Zhang, Y.; Elsik, M.; Edgley, A.J.; Cox, A.J.; Kompa, A.R.; Wang, B.; Tan, C.Y.R.; Khong, F.L.; Stapleton, D.I.; Zammit, S.; et al. A new anti-fibrotic drug attenuates cardiac remodeling and systolic dysfunction following experimental myocardial infarction. Int. J. Cardiol. 2013, 168, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.-J.; Dangerfield, A.L.; Rattan, S.G.; Bathe, K.L.; Cunnington, R.H.; Raizman, J.E.; Bedosky, K.M.; Freed, D.H.; Kardami, E.; Dixon, I.M.C. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev. Dyn. 2010, 239, 1573–1584. [Google Scholar] [CrossRef]
- Garvin, A.M.; Katwa, L.C. Primary cardiac fibroblast cell culture: Methodological considerations for physiologically relevant conditions. Am. J. Physiol. Heart Circ. Physiol. 2023, 325, H869–H881. [Google Scholar] [CrossRef]
- Zhou, Y.; Richards, A.M.; Wang, P. Characterization and Standardization of Cultured Cardiac Fibroblasts for Ex Vivo Models of Heart Fibrosis and Heart Ischemia. Tissue Eng. Part C Methods 2017, 23, 422–433. [Google Scholar] [CrossRef]
- Zhao, H.; Li, X.; Zhao, S.; Zeng, Y.; Zhao, L.; Ding, H.; Sun, W.; Du, Y. Microengineered in vitro model of cardiac fibrosis through modulating myofibroblast mechanotransduction. Biofabrication 2014, 6, 045009. [Google Scholar] [CrossRef]
- Goldsmith, E.C.; Hoffman, A.; Morales, M.O.; Potts, J.D.; Price, R.L.; McFadden, A.; Rice, M.; Borg, T.K. Organization of fibroblasts in the heart. Dev. Dyn. 2004, 230, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Gao, M.; Liao, S.; Zhou, Z.; Luo, G.; Zhou, Y. Role and mechanism of CD90(+) fibroblasts in inflammatory diseases and malignant tumors. Mol. Med. 2023, 29, 20. [Google Scholar] [CrossRef] [PubMed]
- Ivey, M.J.; Kuwabara, J.T.; Riggsbee, K.L.; Tallquist, M.D. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H330–H344. [Google Scholar] [CrossRef]
- Soussi, S.; Savchenko, L.; Rovina, D.; Iacovoni, J.S.; Gottinger, A.; Vialettes, M.; Pioner, J.-M.; Farini, A.; Mallia, S.; Rabino, M.; et al. IPSC derived cardiac fibroblasts of DMD patients show compromised actin microfilaments, metabolic shift and pro-fibrotic phenotype. Biol. Direct 2023, 18, 41. [Google Scholar]
- Palano, G.; Foinquinos, A.; Müllers, E. In vitro Assays and Imaging Methods for Drug Discovery for Cardiac Fibrosis. Front. Physiol. 2021, 12, 697270. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Liu, Z.; Alimohamadi, S.; Yin, C.; Liu, J.; Qian, L. Comparative Gene Expression Analyses Reveal Distinct Molecular Signatures between Differentially Reprogrammed Cardiomyocytes. Cell Rep. 2017, 20, 3014–3024. [Google Scholar] [CrossRef]
- Iseoka, H.; Miyagawa, S.; Sakai, Y. Cardiac fibrosis models using human induced pluripotent stem cell-derived cardiac tissues allow anti-fibrotic drug screening in vitro. Stem Cell Res. 2021, 54, 102420. [Google Scholar] [CrossRef]
- Pellman, J.; Zhang, J.; Sheikh, F. Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: Mechanisms and model systems. J. Mol. Cell. Cardiol. 2016, 94, 22–31. [Google Scholar] [CrossRef]
- Mannino, F.; Imbesi, C.; Bitto, A.; Minutoli, L.; Squadrito, F.; D’angelo, T.; Booz, C.; Pallio, G.; Irrera, N. Anti-oxidant and anti-inflammatory effects of ellagic and punicic acid in an in vitro model of cardiac fibrosis. Biomed. Pharmacother. 2023, 162, 114666. [Google Scholar] [CrossRef]
- Zhang, Y.; Edgley, A.J.; Cox, A.J.; Powell, A.K.; Wang, B.; Kompa, A.R.; Stapleton, D.I.; Zammit, S.C.; Williams, S.J.; Krum, H.; et al. FT011, a new anti-fibrotic drug, attenuates fibrosis and chronic heart failure in experimental diabetic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 549–562. [Google Scholar] [CrossRef]
- Wang, L.; Tan, A.; An, X.; Xia, Y.; Xie, Y. Quercetin Dihydrate inhibition of cardiac fibrosis induced by angiotensin II in vivo and in vitro. Biomed. Pharmacother. 2020, 127, 110205. [Google Scholar] [CrossRef]
- Ji, X.; Xiao, J.; Sheng, X.; Zhang, X.; Guo, M. Curcumin protects against myocardial infarction-induced cardiac fibrosis via SIRT1 activation in vivo and in vitro. Drug Des. Dev. Ther. 2016, 10, 1267–1277. [Google Scholar] [CrossRef]
- Nelson, A.R.; Christiansen, S.L.; Naegle, K.M.; Saucerman, J.J. Logic-based mechanistic machine learning on high-content images reveals how drugs differentially regulate cardiac fibroblasts. Proc. Natl. Acad. Sci. USA 2024, 121, e2303513121. [Google Scholar] [CrossRef] [PubMed]
- Kreutzer, F.P.; Meinecke, A.; Mitzka, S.; Hunkler, H.J.; Hobuß, L.; Abbas, N.; Geffers, R.; Weusthoff, J.; Xiao, K.; Jonigk, D.D.; et al. Development and characterization of anti-fibrotic natural compound similars with improved effectivity. Basic Res. Cardiol. 2022, 117, 9. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, K.; Jung, M.; Foinquinos, A.; José, G.S.; Beaumont, J.; Bock, K.; Grote-Levi, L.; Xiao, K.; Bär, C.; Pfanne, A.; et al. Natural Compound Library Screening Identifies New Molecules for the Treatment of Cardiac Fibrosis and Diastolic Dysfunction. Circulation 2020, 141, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Xu, D.; Zhao, Y.; Sun, L. A Biomimetic Optical Cardiac Fibrosis-on-a-Chip for High-Throughput Anti-Fibrotic Drug Screening. Research 2024, 7, 0471. [Google Scholar] [CrossRef]
- Sadeghi, A.H.; Shin, S.R.; Deddens, J.C.; Fratta, G.; Mandla, S.; Yazdi, I.K.; Prakash, G.; Antona, S.; Demarchi, D.; Buijsrogge, M.P.; et al. Engineered 3D Cardiac Fibrotic Tissue to Study Fibrotic Remodeling. Adv. Healthc. Mater. 2017, 6, 1601434. [Google Scholar] [CrossRef]
- Bracco Gartner, T.C.L.; Crnko, S.; Leiteris, L.; van Adrichem, I.; van Laake, L.W.; Bouten, C.V.C.; Goumans, M.J.; Suyker, W.J.L.; Sluijter, J.P.G.; Hjortnaes, J. Pirfenidone Has Anti-fibrotic Effects in a Tissue-Engineered Model of Human Cardiac Fibrosis. Front. Cardiovasc. Med. 2022, 9, 854314. [Google Scholar] [CrossRef]
- Ruocco, G.; Zoso, A.; Mortati, L.; Carmagnola, I.; Chiono, V. Biomimetic Electrospun Scaffold-Based In Vitro Model Resembling the Hallmarks of Human Myocardial Fibrotic Tissue. ACS Biomater. Sci. Eng. 2023, 9, 4368–4380. [Google Scholar] [CrossRef]
- Spedicati, M.; Ruocco, G.; Zoso, A.; Mortati, L.; Lapini, A.; Delledonne, A.; Divieto, C.; Romano, V.; Castaldo, C.; Di Meglio, F.; et al. Biomimetic design of bioartificial scaffolds for the in vitro modelling of human cardiac fibrosis. Front. Bioeng. Biotechnol. 2022, 10, 983872. [Google Scholar] [CrossRef]
- Fan, S.; Xiao, G.; Ni, J.; Zhao, Y.; Du, H.; Liang, Y.; Lv, M.; He, S.; Fan, G.; Zhu, Y. Guanxinning injection ameliorates cardiac remodeling in HF mouse and 3D heart spheroid models via p38/FOS/MMP1-mediated inhibition of myocardial hypertrophy and fibrosis. Biomed. Pharmacother. 2023, 162, 114642. [Google Scholar] [CrossRef]
- Figtree, G.A.; Bubb, K.J.; Tang, O.; Kizana, E.; Gentile, C. Vascularized Cardiac Spheroids as Novel 3D in vitro Models to Study Cardiac Fibrosis. Cells Tissues Organs 2017, 204, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Ferrão, P.M.; Nisimura, L.M.; Moreira, O.C.; Land, M.G.; Pereira, M.C.; de Mendonça-Lima, L.; Araujo-Jorge, T.C.; Waghabi, M.C.; Garzoni, L.R. Inhibition of TGF-beta pathway reverts extracellular matrix remodeling in T. cruzi-infected cardiac spheroids. Exp. Cell Res. 2018, 362, 260–267. [Google Scholar] [CrossRef]
- Zanetti, M.; Braidotti, N.; Khumar, M.; Montelongo, E.; Lombardi, R.; Sbaizero, O.; Mestroni, L.; Taylor, M.R.; Baj, G.; Lazzarino, M.; et al. Investigations of cardiac fibrosis rheology by in vitro cardiac tissue modeling with 3D cellular spheroids. J. Mech. Behav. Biomed. Mater. 2024, 155, 106571. [Google Scholar] [CrossRef]
- Marsh, S.; Raudat, M.; Lefeber, B.; Herndon, L.B.; Herbert, H.; Mccallum, L.; Simionescu, A. Dynamic Bioreactor Model to Mimic Early Cardiac Fibrosis in Diabetes. J. Mech. Med. Biol. 2021, 21, 2150047. [Google Scholar] [CrossRef]
- Daly, A.C.; Davidson, M.D.; Burdick, J.A. 3D bioprinting of high cell-density heterogeneous tissue models through spheroid fusion within self-healing hydrogels. Nat. Commun. 2021, 12, 753. [Google Scholar] [CrossRef]
- Song, M.; Bin Choi, D.; Im, J.S.; Na Song, Y.; Kim, J.H.; Lee, H.; An, J.; Kim, A.; Choi, H.; Kim, J.-C.; et al. Modeling acute myocardial infarction and cardiac fibrosis using human induced pluripotent stem cell-derived multi-cellular heart organoids. Cell Death Dis. 2024, 15, 308. [Google Scholar] [CrossRef]
- Yang, J.; Lei, W.; Xiao, Y.; Tan, S.; Yang, J.; Lin, Y.; Yang, Z.; Zhao, D.; Zhang, C.; Shen, Z.; et al. Generation of human vascularized and chambered cardiac organoids for cardiac disease modelling and drug evaluation. Cell Prolif. 2024, 57, e13631. [Google Scholar]
- Shin, Y.J.; Shafranek, R.T.; Tsui, J.H.; Walcott, J.; Nelson, A.; Kim, D.-H. 3D bioprinting of mechanically tuned bioinks derived from cardiac decellularized extracellular matrix. Acta Biomater. 2021, 119, 75–88. [Google Scholar] [CrossRef]
- Niro, F.; Fernandes, S.; Cassani, M.; Apostolico, M.; La Cruz, J.O.-D.; Pereira-Sousa, D.; Pagliari, S.; Vinarsky, V.; Zdráhal, Z.; Potesil, D.; et al. Fibrotic extracellular matrix impacts cardiomyocyte phenotype and function in an iPSC-derived isogenic model of cardiac fibrosis. Transl. Res. 2024, 273, 58–77. [Google Scholar] [CrossRef]
- Mastikhina, O.; Moon, B.-U.; Williams, K.; Hatkar, R.; Gustafson, D.; Mourad, O.; Sun, X.; Koo, M.; Lam, A.Y.; Sun, Y.; et al. Human cardiac fibrosis-on-a-chip model recapitulates disease hallmarks and can serve as a platform for drug testing. Biomaterials 2020, 233, 119741. [Google Scholar] [CrossRef]
- Das, S.L.; Sutherland, B.P.; Lejeune, E.; Eyckmans, J.; Chen, C.S. Mechanical response of cardiac microtissues to acute localized injury. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H738–H748. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.Y.; Rafatian, N.; Zhao, Y.; Lee, A.; Lai, B.F.L.; Lu, R.X.; Jekic, D.; Huyer, L.D.; Knee-Walden, E.J.; Bhattacharya, S.; et al. Biowire Model of Interstitial and Focal Cardiac Fibrosis. ACS Cent. Sci. 2019, 5, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Lee, J.; Yazdi, I.K.; Miri, A.K.; Lin, Y.; Seo, J.; Zhang, Y.S.; Khademhosseini, A.; Shin, S.R. Cardiac Fibrotic Remodeling on a Chip with Dynamic Mechanical Stimulation. Adv. Healthc. Mater. 2019, 8, e1801146. [Google Scholar] [CrossRef]
- Mainardi, A.; Carminati, F.; Ugolini, G.S.; Occhetta, P.; Isu, G.; Diaz, D.R.; Reid, G.; Visone, R.; Rasponi, M.; Marsano, A. A dynamic microscale mid-throughput fibrosis model to investigate the effects of different ratios of cardiomyocytes and fibroblasts. Lab Chip 2021, 21, 4177–4195. [Google Scholar] [CrossRef]
- Kerkelä, R.; Ulvila, J.; Magga, J. Natriuretic Peptides in the Regulation of Cardiovascular Physiology and Metabolic Events. J. Am. Heart Assoc. 2015, 4, e002423. [Google Scholar] [CrossRef]
- Tsai, Y.-T.; Lin, F.-Y.; Lin, C.-S.; Loh, S.-H.; Li, C.-Y.; Lin, C.-Y.; Lin, Y.-W.; Tsai, C.-S. B-type natriuretic peptide enhances fibrotic effects via matrix metalloproteinase-2 expression in the mouse atrium in vivo and in human atrial myofibroblasts in vitro. Transl. Res. 2019, 208, 30–46. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, M.; Wu, J.C. Generation of Quiescent Cardiac Fibroblasts Derived from Human Induced Pluripotent Stem Cells. Methods Mol. Biol. 2022, 2454, 109–115. [Google Scholar]
- Herum, K.M.; Choppe, J.; Kumar, A.; Engler, A.J.; McCulloch, A.D. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell 2017, 28, 1871–1882. [Google Scholar] [CrossRef]
- Chen, C.Z.; Peng, Y.; Wang, Z.; Fish, P.; Kaar, J.; Koepsel, R.; Russell, A.; Lareu, R.; Raghunath, M. The Scar-in-a-Jar: Studying potential antifibrotic compounds from the epigenetic to extracellular level in a single well. Br. J. Pharmacol. 2009, 158, 1196–1209. [Google Scholar] [CrossRef]
- Sharma, P.; Ando, D.; Daub, A.; Kaye, J.A.; Finkbeiner, S. High-throughput screening in primary neurons. Methods Enzymol. 2012, 506, 331–360. [Google Scholar] [PubMed]
- Iversen, P.W.; Eastwood, B.J.; Sittampalam, G.S.; Cox, K.L. A comparison of assay performance measures in screening assays: Signal window, Z′ factor, and assay variability ratio. J. Biomol. Screen. 2006, 11, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s biochemical and cell-based approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef]
- Luo, J.; Zhu, Z. Sensitive and High-Throughput Time-Resolved Luminescence Detection of Tetracycline in Milk for Eliminating Background Fluorescence on a Miniaturized Apparatus. Anal. Chem. 2024, 96, 11115–11120. [Google Scholar] [CrossRef]
- Imamura, R.M.; Kumagai, K.; Nakano, H.; Okabe, T.; Nagano, T.; Kojima, H. Inexpensive High-Throughput Screening of Kinase Inhibitors Using One-Step Enzyme-Coupled Fluorescence Assay for ADP Detection. SLAS Discov. 2019, 24, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, G.; Misteli, T. High-Throughput Imaging for the Discovery of Cellular Mechanisms of Disease. Trends Genet. 2017, 33, 604–615. [Google Scholar] [CrossRef]
- Hillsley, A.; Santos, J.E.; Rosales, A.M. A deep learning approach to identify and segment alpha-smooth muscle actin stress fiber positive cells. Sci. Rep. 2021, 11, 21855. [Google Scholar] [CrossRef]
- Barbulescu, G.I.; Bojin, F.M.; Ordodi, V.L.; Goje, I.D.; Barbulescu, A.S.; Paunescu, V. Decellularized Extracellular Matrix Scaffolds for Cardiovascular Tissue Engineering: Current Techniques and Challenges. Int. J. Mol. Sci. 2022, 23, 13040. [Google Scholar] [CrossRef]
- Filippo Buono, M.; von Boehmer, L.; Strang, J.; Hoerstrup, S.P.; Emmert, M.Y.; Nugraha, B. Human Cardiac Organoids for Modeling Genetic Cardiomyopathy. Cells 2020, 9, 1733. [Google Scholar] [CrossRef]
- Basara, G.; Celebi, L.E.; Ronan, G.; Santos, V.D.; Zorlutuna, P. 3D bioprinted aged human post-infarct myocardium tissue model. Health Sci. Rep. 2024, 7, e1945. [Google Scholar] [CrossRef] [PubMed]
- Basara, G.; Ozcebe, S.G.; Ellis, B.W.; Zorlutuna, P. Tunable Human Myocardium Derived Decellularized Extracellular Matrix for 3D Bioprinting and Cardiac Tissue Engineering. Gels 2021, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, Y.; Ogata, T.; Mochizuki, K.; Tamura, S.; Morishita, Y.; Takamatsu, T.; Matoba, S.; Tanaka, H. Myofibroblasts impair myocardial impulse propagation by heterocellular connexin43 gap-junctional coupling through micropores. Front. Physiol. 2024, 15, 1352911. [Google Scholar] [CrossRef]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Massé, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef]
- Tiburcy, M.; Hudson, J.E.; Balfanz, P.; Schlick, S.; Meyer, T.; Liao, M.-L.C.; Levent, E.; Raad, F.; Zeidler, S.; Wingender, E.; et al. Defined Engineered Human Myocardium With Advanced Maturation for Applications in Heart Failure Modeling and Repair. Circulation 2017, 135, 1832–1847. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Rafatian, N.; Feric, N.T.; Cox, B.J.; Aschar-Sobbi, R.; Wang, E.Y.; Aggarwal, P.; Zhang, B.; Conant, G.; Ronaldson-Bouchard, K.; et al. A Platform for Generation of Chamber-Specific Cardiac Tissues and Disease Modeling. Cell 2019, 176, 913–927.e18. [Google Scholar] [CrossRef]
- Ricketts, S.N.; Qian, L. The heart of cardiac reprogramming: The cardiac fibroblasts. J. Mol. Cell. Cardiol. 2022, 172, 90–99. [Google Scholar] [CrossRef]
- Wang, Y.; Jeon, H. 3D cell cultures toward quantitative high-throughput drug screening. Trends Pharmacol. Sci. 2022, 43, 569–581. [Google Scholar] [CrossRef]
- Lind, J.U.; Busbee, T.A.; Valentine, A.D.; Pasqualini, F.S.; Yuan, H.; Yadid, M.; Park, S.-J.; Kotikian, A.; Nesmith, A.P.; Campbell, P.H.; et al. Instrumented cardiac microphysiological devices via multimaterial three-dimensional printing. Nat. Mater. 2017, 16, 303–308. [Google Scholar] [CrossRef]
- Shang, Y.; Gan, J.; Xu, D.; Zhao, Y.; Sun, L. A Biomimetic Cardiac Fibrosis-on-a-Chip as a Visible Disease Model for Evaluating Mesenchymal Stem Cell-Derived Exosome Therapy. ACS Nano 2024, 18, 829–838. [Google Scholar] [CrossRef]
- Myllarniemi, M.; Kaarteenaho, R. Pharmacological treatment of idiopathic pulmonary fibrosis—Preclinical and clinical studies of pirfenidone, nintedanib, and N-acetylcysteine. Eur. Clin. Respir. J. 2015, 2, 26385. [Google Scholar] [CrossRef] [PubMed]
- Kreidieh, M.; Hamadi, R.; Alsheikh, M.; Al Moussawi, H.; Deeb, L. Statin Use in Patients With Chronic Liver Disease and Cirrhosis: Current Evidence and Future Directions. Gastroenterol. Res. 2022, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Bettenworth, D.; Rieder, F. Medical therapy of stricturing Crohn’s disease: What the gut can learn from other organs—A systematic review. Fibrogenesis Tissue Repair 2014, 7, 5. [Google Scholar] [CrossRef]
- Steiner, C.A.; Higgins, P.D.R. Anti-Fibrotic Therapies from Other Organs: What the Gut Can Learn from the Liver, Skin, Lung and Heart. In Fibrostenotic Inflammatory Bowel Disease; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Erasmus, M.; Samodien, E.; Lecour, S.; Cour, M.; Lorenzo, O.; Dludla, P.; Pheiffer, C.; Johnson, R. Linking LOXL2 to Cardiac Interstitial Fibrosis. Int. J. Mol. Sci. 2020, 21, 5913. [Google Scholar] [CrossRef]
- Axelsson Raja, A.; Wakimoto, H.; DeLaughter, D.M.; Reichart, D.; Gorham, J.; Conner, D.A.; Lun, M.; Probst, C.K.; Sakai, N.; Knipe, R.S.; et al. Ablation of lysophosphatidic acid receptor 1 attenuates hypertrophic cardiomyopathy in a mouse model. Proc. Natl. Acad. Sci. USA 2022, 119, e2204174119. [Google Scholar] [CrossRef]
- Janssen, W.; Schymura, Y.; Novoyatleva, T.; Kojonazarov, B.; Boehm, M.; Wietelmann, A.; Luitel, H.; Murmann, K.; Krompiec, D.R.; Tretyn, A.; et al. 5-HT2B receptor antagonists inhibit fibrosis and protect from RV heart failure. BioMed Res. Int. 2015, 2015, 438403. [Google Scholar] [CrossRef]
- Ma, Z.-G.; Yuan, Y.-P.; Zhang, X.; Xu, S.-C.; Wang, S.-S.; Tang, Q.-Z. Piperine Attenuates Pathological Cardiac Fibrosis Via PPAR-gamma/AKT Pathways. EBioMedicine 2017, 18, 179–187. [Google Scholar] [CrossRef]
- Anan, A.; Baskin-Bey, E.S.; Bronk, S.F.; Werneburg, N.W.; Shah, V.H.; Gores, G.J. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology 2006, 43, 335–344. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef]
- He, S.; Yao, L.; Li, J. Role of MCP-1/CCR2 axis in renal fibrosis: Mechanisms and therapeutic targeting. Medicine 2023, 102, e35613. [Google Scholar]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.C.; Xu, J.; Cheng, C.; Xiang, X.-J.; Hong, B.-Y.; Zhang, M.; Gong, C.; Ma, L.K. Macrophages promote the transition from myocardial ischemia reperfusion injury to cardiac fibrosis in mice through GMCSF/CCL2/CCR2 and phenotype switching. Acta Pharmacol. Sin. 2024, 45, 959–974. [Google Scholar] [PubMed]
- Song, X.; Jiang, C.; Yu, M.; Lu, C.; He, X. CCR2/CCR5 antagonist cenicriviroc reduces colonic inflammation and fibrosis in experimental colitis. J. Gastroenterol. Hepatol. 2024, 39, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Thiam, F.; Phogat, S.; Abokor, F.A.; Osei, E.T. In vitro co-culture studies and the crucial role of fibroblast-immune cell crosstalk in IPF pathogenesis. Respir. Res. 2023, 24, 298. [Google Scholar] [CrossRef]
- Osei, E.T.; Booth, S.; Hackett, T.-L. What Have In Vitro Co-Culture Models Taught Us about the Contribution of Epithelial-Mesenchymal Interactions to Airway Inflammation and Remodeling in Asthma? Cells 2020, 9, 1694. [Google Scholar] [CrossRef]
- Ali, M.F.; Egan, A.M.; Shaughnessy, G.F.; Anderson, D.K.; Kottom, T.J.; Dasari, H.; Van Keulen, V.P.; Aubry, M.-C.; Yi, E.S.; Limper, A.H.; et al. Antifibrotics Modify B-Cell-induced Fibroblast Migration and Activation in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 722–733. [Google Scholar] [CrossRef]
- Smithmyer, M.E.; Sawicki, L.A.; Kloxin, A.M. Hydrogel scaffolds as in vitro models to study fibroblast activation in wound healing and disease. Biomater. Sci. 2014, 2, 634–650. [Google Scholar]
- Thiam, F.; Al Yazeedi, S.; Feng, K.; Phogat, S.; Demirsoy, E.; Brussow, J.; Abokor, F.A.; Osei, E.T. Understanding fibroblast-immune cell interactions via co-culture models and their role in asthma pathogenesis. Front. Immunol. 2023, 14, 1128023. [Google Scholar] [CrossRef]
- Wygrecka, M.; Dahal, B.; Kosanovic, D.; Petersen, F.; Taborski, B.; Preissner, K.; Schermuly, R.; Markart, P. Mast cells and fibroblasts work in concert to aggravate pulmonary fibrosis: Role of transmembrane SCF and the PAR-2/PKC-alpha/Raf-1/p44/42 signaling pathway. Am. J. Pathol. 2013, 182, 2094–2108. [Google Scholar]
- Norona, L.M.; Nguyen, D.G.; Gerber, D.A.; Presnell, S.C.; Mosedale, M.; Watkins, P.B. Bioprinted liver provides early insight into the role of Kupffer cells in TGF-beta1 and methotrexate-induced fibrogenesis. PLoS ONE 2019, 14, e0208958. [Google Scholar] [CrossRef]
- Jain, N.; Shashi Bhushan, B.L.; Natarajan, M.; Mehta, R.; Saini, D.K.; Chatterjee, K. Advanced 3D In Vitro Lung Fibrosis Models: Contemporary Status, Clinical Uptake, and Prospective Outlooks. ACS Biomater. Sci. Eng. 2024, 10, 1235–1261. [Google Scholar] [CrossRef] [PubMed]
- Barron, S.L.; Wyatt, O.; O’connor, A.; Mansfield, D.; Cohen, E.S.; Witkos, T.M.; Strickson, S.; Owens, R.M. Modelling bronchial epithelial-fibroblast cross-talk in idiopathic pulmonary fibrosis (IPF) using a human-derived in vitro air liquid interface (ALI) culture. Sci. Rep. 2024, 14, 240. [Google Scholar] [CrossRef]
- Mimouni, M.; Lajoix, A.-D.; Desmetz, C. Experimental Models to Study Endothelial to Mesenchymal Transition in Myocardial Fibrosis and Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 25, 382. [Google Scholar] [CrossRef]
- Pekor, C.; Gerlach, J.C.; Nettleship, I.; Schmelzer, E. Induction of Hepatic and Endothelial Differentiation by Perfusion in a Three-Dimensional Cell Culture Model of Human Fetal Liver. Tissue Eng. Part C Methods 2015, 21, 705–715. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.; Choi, Y.; Cho, D.-W. Application of Gelatin Bioinks and Cell-Printing Technology to Enhance Cell Delivery Capability for 3D Liver Fibrosis-on-a-Chip Development. ACS Biomater. Sci. Eng. 2020, 6, 2469–2477. [Google Scholar] [CrossRef] [PubMed]
- Noor, N.; Shapira, A.; Edri, R.; Gal, I.; Wertheim, L.; Dvir, T. 3D Printing of Personalized Thick and Perfusable Cardiac Patches and Hearts. Adv. Sci. 2019, 6, 1900344. [Google Scholar] [CrossRef]
- McQuitty, C.E.; Williams, R.; Chokshi, S.; Urbani, L. Immunomodulatory Role of the Extracellular Matrix Within the Liver Disease Microenvironment. Front. Immunol. 2020, 11, 574276. [Google Scholar] [CrossRef]
- Silva, A.C.; Pereira, C.; Fonseca, A.C.R.G.; Pinto-Do-Ó, P.; Nascimento, D.S. Bearing My Heart: The Role of Extracellular Matrix on Cardiac Development, Homeostasis, and Injury Response. Front. Cell Dev. Biol. 2020, 8, 621644. [Google Scholar] [CrossRef]
- O’Reilly, P.J.; Ding, Q.; Akthar, S.; Cai, G.; Genschmer, K.R.; Patel, D.F.; Jackson, P.L.; Viera, L.; Roda, M.; Locy, M.L.; et al. Angiotensin-converting enzyme defines matrikine-regulated inflammation and fibrosis. JCI Insight 2017, 2, e91923. [Google Scholar] [CrossRef]
- Reese-Petersen, A.L.; Genovese, F.; Zhao, L.; Banks, G.; Gordon, D.A.; Karsdal, M.A. may be a driver of fibroblast activation in fibro-inflammatory diseases. Front. Mol. Biosci. 2023, 10, 1228232. [Google Scholar] [CrossRef]
- Alsafadi, H.N.; Staab-Weijnitz, C.A.; Lehmann, M.; Lindner, M.; Peschel, B.; Königshoff, M.; Wagner, D.E. An ex vivo model to induce early fibrosis-like changes in human precision-cut lung slices. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L896–L902. [Google Scholar] [CrossRef]
- Stribos, E.G.; Hillebrands, J.-L.; Olinga, P.; Mutsaers, H.A. Renal fibrosis in precision-cut kidney slices. Eur. J. Pharmacol. 2016, 790, 57–61. [Google Scholar] [CrossRef]
- Kang, C.; Qiao, Y.; Li, G.; Baechle, K.; Camelliti, P.; Rentschler, S.; Efimov, I.R. Human Organotypic Cultured Cardiac Slices: New Platform For High Throughput Preclinical Human Trials. Sci. Rep. 2016, 6, 28798. [Google Scholar] [CrossRef]
- Wang, K.; Lee, P.; Mirams, G.R.; Sarathchandra, P.; Borg, T.K.; Gavaghan, D.J.; Kohl, P.; Bollensdorff, C. Cardiac tissue slices: Preparation, handling, and successful optical mapping. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1112–H1125. [Google Scholar] [CrossRef] [PubMed]
- Schinner, C.; Xu, L.; Franz, H.; Zimmermann, A.; Wanuske, M.-T.; Rathod, M.; Hanns, P.; Geier, F.; Pelczar, P.; Liang, Y.; et al. Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy by Involving an Integrin-alphaVbeta6/TGF-beta Signaling Cascade. Circulation 2022, 146, 1610–1626. [Google Scholar] [PubMed]
- Perbellini, F.; Watson, S.A.; Scigliano, M.; Alayoubi, S.; Tkach, S.; Bardi, I.; Quaife, N.; Kane, C.; Dufton, N.P.; Simon, A.; et al. Investigation of cardiac fibroblasts using myocardial slices. Cardiovasc. Res. 2018, 114, 77–89. [Google Scholar] [CrossRef]
- Nunez-Toldra, R.; Kirwin, T.; Ferraro, E.; Pitoulis, F.G.; Nicastro, L.; Bardi, I.; Kit-Anan, W.; Gorelik, J.; Simon, A.R.; Terracciano, C.M. Mechanosensitive molecular mechanisms of myocardial fibrosis in living myocardial slices. ESC Heart Fail. 2022, 9, 1400–1412. [Google Scholar] [CrossRef]
- Watson, S.A.; Duff, J.; Bardi, I.; Zabielska, M.; Atanur, S.S.; Jabbour, R.J.; Simon, A.; Tomas, A.; Smolenski, R.T.; Harding, S.E.; et al. Biomimetic electromechanical stimulation to maintain adult myocardial slices in vitro. Nat. Commun. 2019, 10, 2168. [Google Scholar] [CrossRef]
- Schneider-Warme, F.; Johnston, C.M.; Kohl, P. Organotypic myocardial slices as model system to study heterocellular interactions. Cardiovasc. Res. 2018, 114, 3–6. [Google Scholar] [CrossRef]
- Dewyse, L.; De Smet, V.; Verhulst, S.; Eysackers, N.; Kunda, R.; Messaoudi, N.; Reynaert, H.; van Grunsven, L.A. Improved Precision-Cut Liver Slice Cultures for Testing Drug-Induced Liver Fibrosis. Front. Med. 2022, 9, 862185. [Google Scholar] [CrossRef]
- Carvalho, A.M.; Bansal, R.; Barrias, C.C.; Sarmento, B. The Material World of 3D-Bioprinted and Microfluidic-Chip Models of Human Liver Fibrosis. Adv. Mater. 2024, 36, e2307673. [Google Scholar] [CrossRef] [PubMed]
- Son, K.J.; Gheibi, P.; Stybayeva, G.; Rahimian, A.; Revzin, A. Detecting cell-secreted growth factors in microfluidic devices using bead-based biosensors. Microsyst. Nanoeng. 2017, 3, 17025. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Patel, D.; Kwa, T.; Haque, A.; Matharu, Z.; Stybayeva, G.; Gao, Y.; Diehl, A.M.; Revzin, A. Liver injury-on-a-chip: Microfluidic co-cultures with integrated biosensors for monitoring liver cell signaling during injury. Lab Chip 2015, 15, 4467–4478. [Google Scholar] [CrossRef]
- Zhuang, T.; Chen, M.-H.; Wu, R.-X.; Wang, J.; Hu, X.-D.; Meng, T.; Wu, A.-H.; Yang, Y.-F.; Lei, Y.; Hu, D.-H.; et al. ALKBH5-mediated m6A modification of IL-11 drives macrophage-to-myofibroblast transition and pathological cardiac fibrosis in mice. Nat. Commun. 2024, 15, 1995. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, A.K.; Shah, G. Exploring in vivo and in vitro models for heart failure with biomarker insights: A review. Egypt. Heart J. 2024, 76, 141. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardona-Timoner, M.; Gomes, R.N.; Nascimento, D.S. Dressed in Collagen: 2D and 3D Cardiac Fibrosis Models. Int. J. Mol. Sci. 2025, 26, 3038. https://doi.org/10.3390/ijms26073038
Cardona-Timoner M, Gomes RN, Nascimento DS. Dressed in Collagen: 2D and 3D Cardiac Fibrosis Models. International Journal of Molecular Sciences. 2025; 26(7):3038. https://doi.org/10.3390/ijms26073038
Chicago/Turabian StyleCardona-Timoner, Maria, Rita N. Gomes, and Diana S. Nascimento. 2025. "Dressed in Collagen: 2D and 3D Cardiac Fibrosis Models" International Journal of Molecular Sciences 26, no. 7: 3038. https://doi.org/10.3390/ijms26073038
APA StyleCardona-Timoner, M., Gomes, R. N., & Nascimento, D. S. (2025). Dressed in Collagen: 2D and 3D Cardiac Fibrosis Models. International Journal of Molecular Sciences, 26(7), 3038. https://doi.org/10.3390/ijms26073038