
Genome Editing in Mouse Embryo Using the CRISPR/Cas12i3 System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
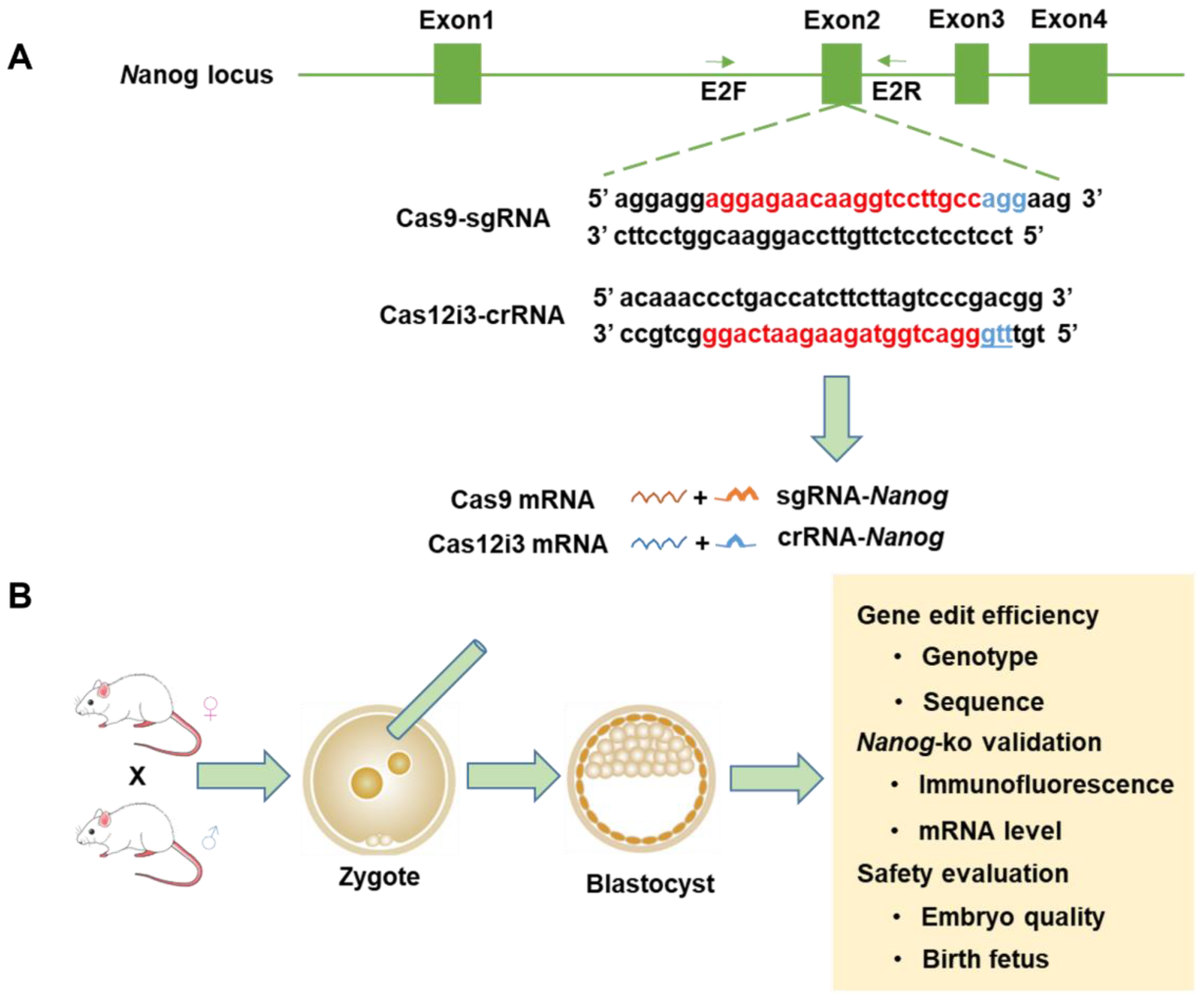
2.1. Design and Preparation of Mouse Nanog Knockout Using the Cas12i3 System
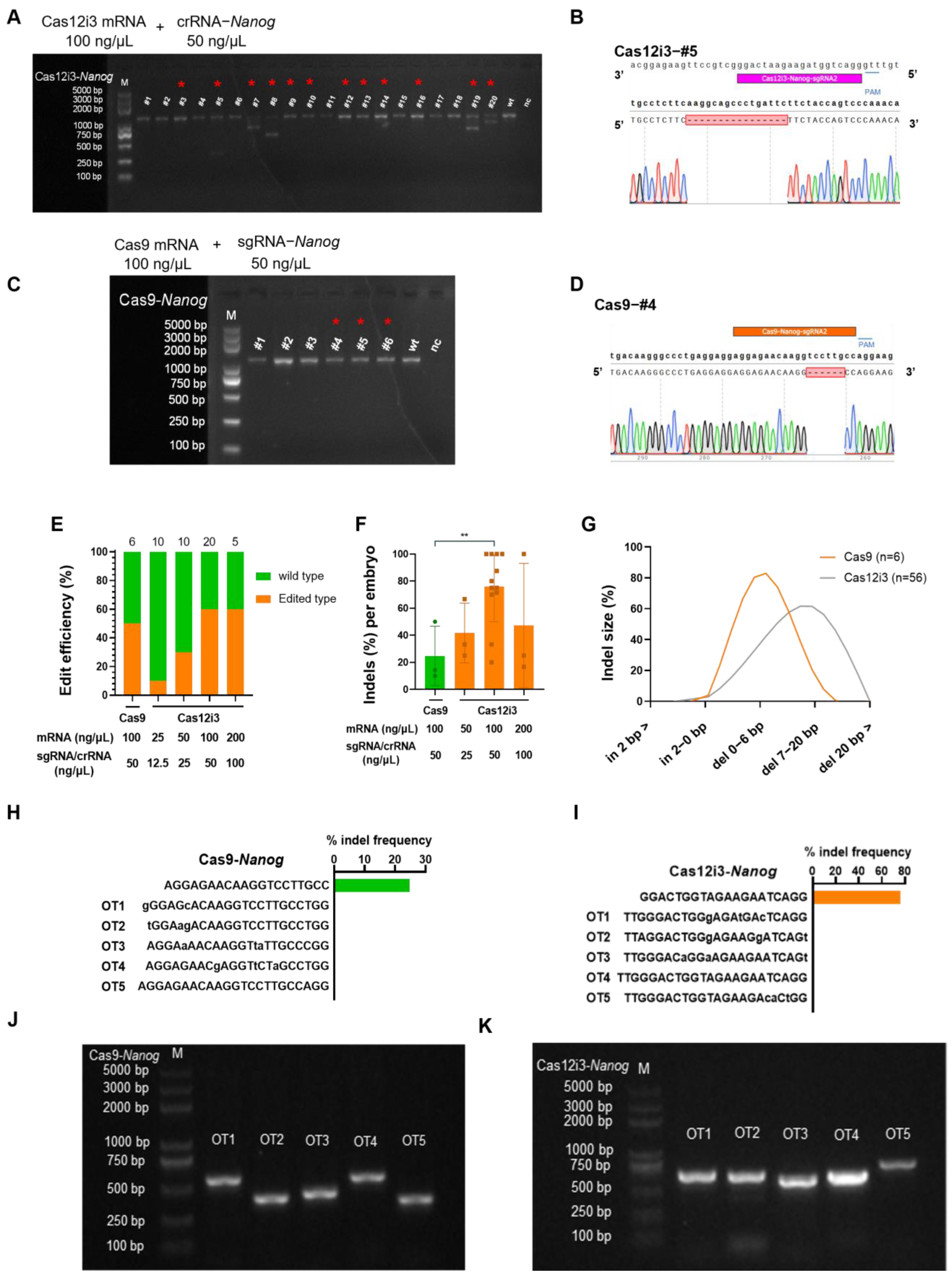
2.2. Optimization and Efficacy of the CRISPR/Cas12i3 System in Mouse Embryo
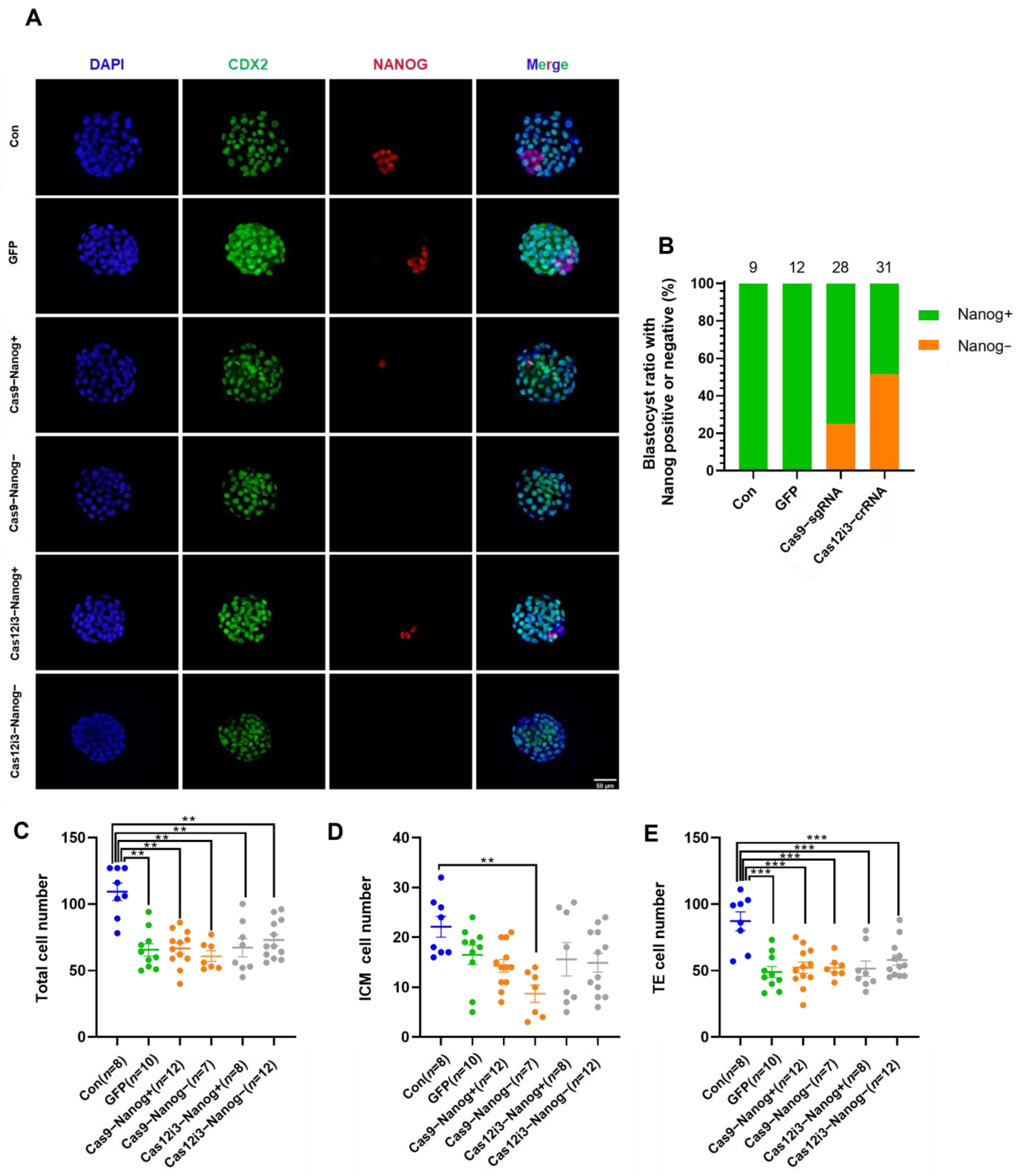
2.3. Construction of the Mouse Preimplantation Embryo Model of Nanog Knockout Using the CRISPR/Cas12i3 System
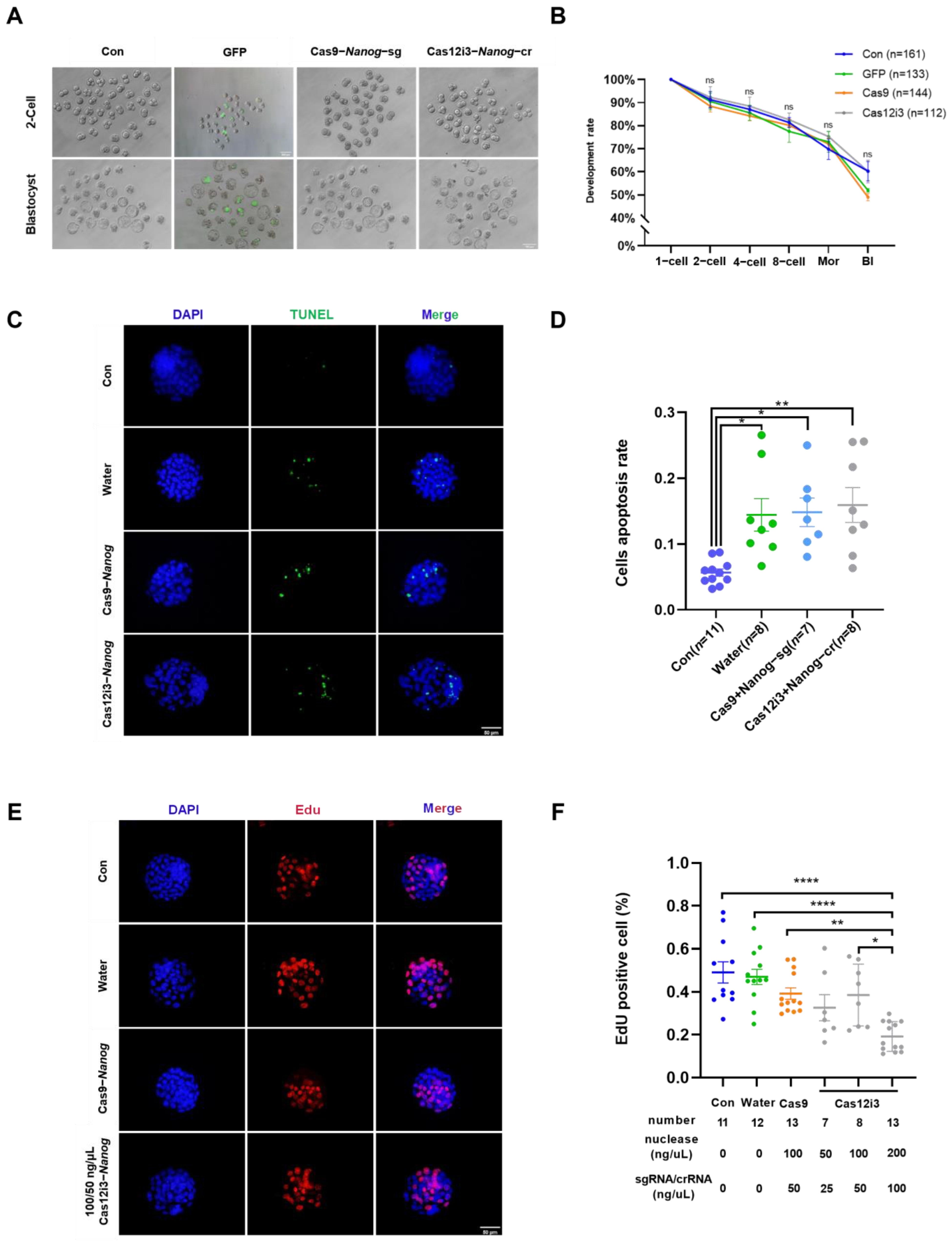
2.4. Effects of Nanog Knockdown on Preimplantation Embryo Development in Mice by the CRISPR/Cas12i3 System
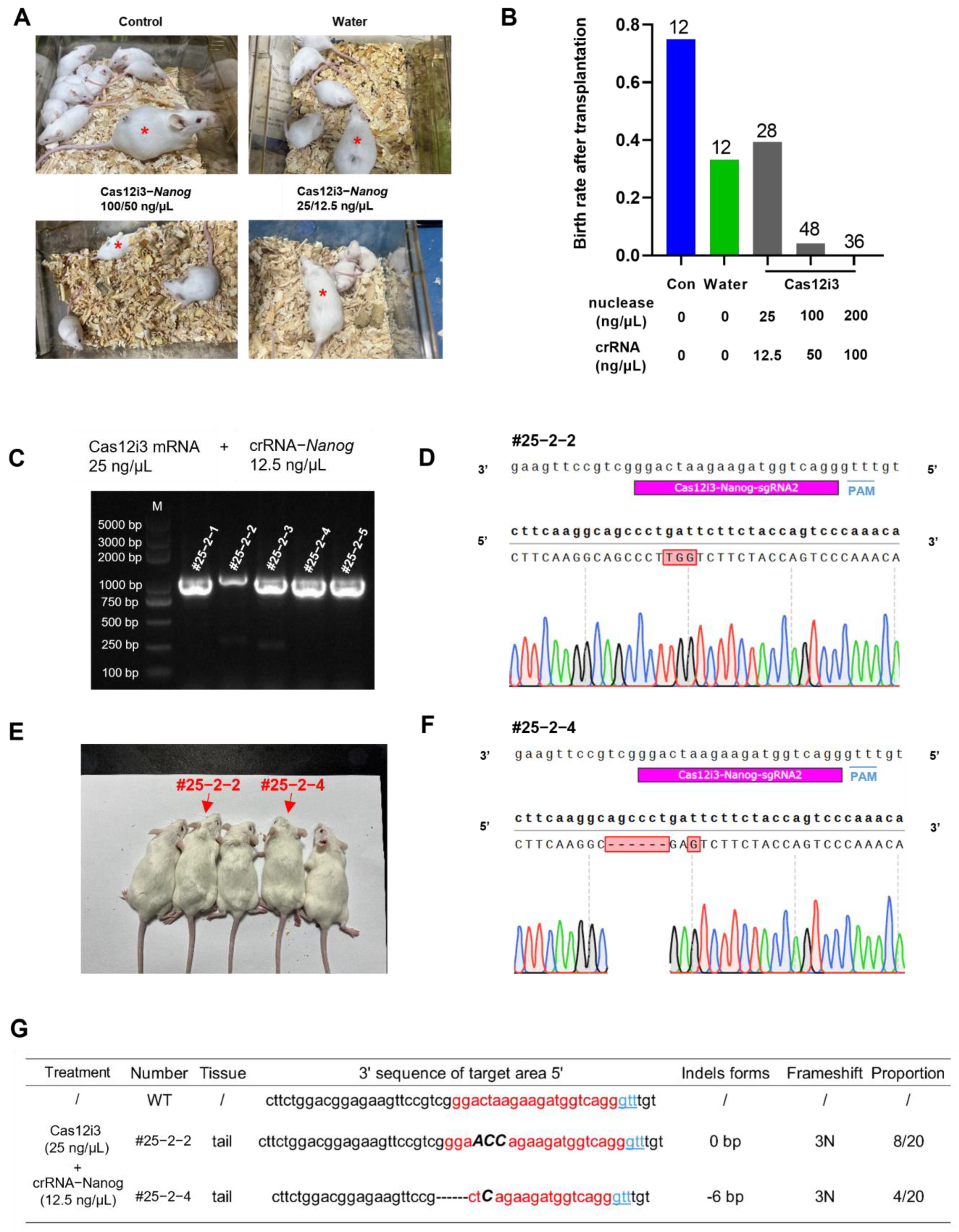
2.5. Generation of Nanog-Targeted Gene-Edited Mice Using the CRISPR/Cas12i3 System
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. The sgRNA/crRNA Design for Nanog
4.3. Construction of sgRNA/crRNA Expression Vectors
4.4. In Vitro Transcription of crRNA/sgRNA and Cas12i3 mRNA
4.5. Mouse Embryo Collection and In Vitro Culture (IVC)
4.6. Embryo Cytoplasmic Microinjection
4.7. Single Embryo DNA Extraction
4.8. Mouse Tail DNA Extraction
4.9. Mouse Embryo Transfer
4.10. Detection of Gene Editing Efficiency and Types
4.11. Off-Target Effect Prediction and Detection
4.12. Embryo Immunofluorescence Staining
4.13. TUNEL Staining
4.14. EdU Staining
4.15. Data Statistics and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| ZFN | Zinc finger nuclease |
| TALENs | Transcription activator-like effector nucleases |
| ICM | Inner cell mass |
| TE | Trophectoderm |
| ESC | Embryonic stem cell |
References
- Barrangou, R.; Marraffini, L.A. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol. Cell 2014, 54, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, L.P.; Chandrasegaran, S. Functional domains in Fok I restriction endonuclease. Proc. Natl. Acad. Sci. USA 1992, 89, 4275–4279. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Bibikova, M.; Whitby, F.G.; Reddy, A.R.; Chandrasegaran, S.; Carroll, D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000, 28, 3361–3369. [Google Scholar]
- Pattanayak, V.; Ramirez, C.L.; Joung, J.K.; Liu, D.R. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods 2011, 8, 765–770. [Google Scholar] [CrossRef]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Bogdanove, A.J.; Voytas, D.F. TAL effectors: Customizable proteins for DNA targeting. Science 2011, 333, 1843–1846. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar]
- Chylinski, K.; Le Rhun, A.; Charpentier, E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013, 10, 726–737. [Google Scholar] [PubMed]
- Garneau, J.E.; Dupuis, M.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [PubMed]
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 212. [Google Scholar] [CrossRef]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar]
- Yan, W.X.; Hunnewell, P.; Alfonse, L.E.; Carte, J.M.; Keston-Smith, E.; Sothiselvam, S.; Garrity, A.J.; Chong, S.; Makarova, K.S.; Koonin, E.V.; et al. Functionally diverse type V CRISPR-Cas systems. Science 2019, 363, 88–91. [Google Scholar]
- Huang, X.; Sun, W.; Cheng, Z.; Chen, M.; Li, X.; Wang, J.; Sheng, G.; Gong, W.; Wang, Y. Structural basis for two metal-ion catalysis of DNA cleavage by Cas12i2. Nat. Commun. 2020, 11, 5241. [Google Scholar]
- Zhang, R.; Chai, N.; Liu, T.; Zheng, Z.; Lin, Q.; Xie, X.; Wen, J.; Yang, Z.; Liu, Y.G.; Zhu, Q. The type V effectors for CRISPR/Cas-mediated genome engineering in plants. Biotechnol. Adv. 2024, 74, 108382. [Google Scholar]
- Paul, B.; Montoya, G. CRISPR-Cas12a: Functional overview and applications. Biomed. J. 2020, 43, 8–17. [Google Scholar]
- Zhang, B.; Luo, D.; Li, Y.; Perčulija, V.; Chen, J.; Lin, J.; Ye, Y.; Ouyang, S. Mechanistic insights into the R-loop formation and cleavage in CRISPR-Cas12i1. Nat. Commun. 2021, 12, 3476. [Google Scholar]
- McGaw, C.; Garrity, A.J.; Munoz, G.Z.; Haswell, J.R.; Sengupta, S.; Keston-Smith, E.; Hunnewell, P.; Ornstein, A.; Bose, M.; Wessells, Q.; et al. Engineered Cas12i2 is a versatile high-efficiency platform for therapeutic genome editing. Nat. Commun. 2022, 13, 2833. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liu, S.; Bai, Y.; Xu, Z.; Zhao, H.; Zhao, H.; Lai, J.; Liu, Y.; Song, W. Application of CRISPR/Cas12i.3 for targeted mutagenesis in broomcorn millet (Panicum miliaceum L.). J. Integr. Plant Biol. 2024, 66, 1544–1547. [Google Scholar] [CrossRef]
- Wang, W.; Yan, L.; Li, J.; Zhang, C.; He, Y.; Li, S.; Xia, L. Engineering a robust Cas12i3 variant-mediated wheat genome editing system. Plant Biotechnol. J. 2025, 23, 860–873. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Wang, X.; Luo, S.; Yang, N.; Chen, Y.; Li, Z.; Zhou, Q.; Li, W. Synergistic engineering of CRISPR-Cas nucleases enables robust mammalian genome editing. Innovation 2022, 3, 100264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, X.; Xue, M.; Hu, J.; Wang, Z.; Wei, Y.; Wang, H.; Zhou, J.; Zhang, W.; Xu, M.; et al. An engineered xCas12i with high activity, high specificity, and broad PAM range. Protein Cell 2023, 14, 538–543. [Google Scholar] [CrossRef]
- Han, X.; Chen, Y.; Liu, R.; Zhu, J.-K.; Duan, Z.J.T.I.L. Engineering hfCas12Max for improved gene editing efficiency. Innov. Life 2024, 2, 100067. [Google Scholar] [CrossRef]
- Ren, J.; Hai, T.; Chen, Y.; Sun, K.; Han, Z.; Wang, J.; Li, C.; Wang, Q.; Wang, L.; Zhu, H.; et al. Improve meat production and virus resistance by simultaneously editing multiple genes in livestock using Cas12i(Max). Sci. China Life Sci. 2024, 67, 555–564. [Google Scholar] [CrossRef]
- Lv, P.; Su, F.; Chen, F.; Yan, C.; Xia, D.; Sun, H.; Li, S.; Duan, Z.; Ma, C.; Zhang, H.; et al. Genome editing in rice using CRISPR/Cas12i3. Plant Biotechnol. J. 2024, 22, 379–385. [Google Scholar] [CrossRef]
- Xie, H.; Song, M.; Cao, X.; Niu, Q.; Zhu, J.; Li, S.; Wang, X.; Niu, X.; Zhu, J.K. Breeding exceptionally fragrant soybeans for soy milk with strong aroma. J. Integr. Plant Biol. 2024, 66, 642–644. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [PubMed]
- Pan, G.; Thomson, J.A. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007, 17, 42–49. [Google Scholar] [PubMed]
- Komatsu, K.; Fujimori, T. Multiple phases in regulation of Nanog expression during pre-implantation development. Dev. Growth Differ. 2015, 57, 648–656. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; Kemler, R. Nanog is required for primitive endoderm formation through a non-cell autonomous mechanism. Dev. Biol. 2010, 344, 129–137. [Google Scholar] [PubMed]
- Li, D.; Qiu, Z.; Shao, Y.; Chen, Y.; Guan, Y.; Liu, M.; Li, Y.; Gao, N.; Wang, L.; Lu, X.; et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 681–683. [Google Scholar]
- Xu, J.; Zhang, L.; Ye, Z.; Chang, B.; Tu, Z.; Du, X.; Wen, X.; Teng, Y. A 3D “sandwich” co-culture system with vascular niche supports mouse embryo development from E3.5 to E7.5 in vitro. Stem Cell Res. Ther. 2023, 14, 349. [Google Scholar]
- Bedzhov, I.; Graham, S.J.; Leung, C.Y.; Zernicka-Goetz, M. Developmental plasticity, cell fate specification and morphogenesis in the early mouse embryo. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130538. [Google Scholar] [CrossRef]
- Kojima, Y.; Tam, O.H.; Tam, P.P. Timing of developmental events in the early mouse embryo. Semin. Cell Dev. Biol. 2014, 34, 65–75. [Google Scholar] [CrossRef]
- Rivera-Pérez, J.A.; Hadjantonakis, A.K. The Dynamics of Morphogenesis in the Early Mouse Embryo. Cold Spring Harb. Perspect. Biol. 2014, 7, a015867. [Google Scholar]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar]
- Zhang, H.; Li, Z.; Xiao, R.; Chang, L. Mechanisms for target recognition and cleavage by the Cas12i RNA-guided endonuclease. Nat. Struct. Mol. Biol. 2020, 27, 1069–1076. [Google Scholar] [PubMed]
- Duan, Z.; Liang, Y.; Sun, J.; Zheng, H.; Lin, T.; Luo, P.; Wang, M.; Liu, R.; Chen, Y.; Guo, S.; et al. An engineered Cas12i nuclease that is an efficient genome editing tool in animals and plants. Innovation 2024, 5, 100564. [Google Scholar] [PubMed]
- Zuo, E.; Cai, Y.J.; Li, K.; Wei, Y.; Wang, B.A.; Sun, Y.; Liu, Z.; Liu, J.; Hu, X.; Wei, W.; et al. One-step generation of complete gene knockout mice and monkeys by CRISPR/Cas9-mediated gene editing with multiple sgRNAs. Cell Res. 2017, 27, 933–945. [Google Scholar] [PubMed]
- Tian, X.; Wang, F.; Zhang, L.; Ji, P.; Wang, J.; Lv, D.; Li, G.; Chai, M.; Lian, Z.; Liu, G. Melatonin Promotes the In Vitro Development of Microinjected Pronuclear Mouse Embryos via Its Anti-Oxidative and Anti-Apoptotic Effects. Int. J. Mol. Sci. 2017, 18, 988. [Google Scholar] [CrossRef]
- Takahashi, G.; Gurumurthy, C.B.; Wada, K.; Miura, H.; Sato, M.; Ohtsuka, M. GONAD: Genome-editing via Oviductal Nucleic Acids Delivery system: A novel microinjection independent genome engineering method in mice. Sci. Rep. 2015, 5, 11406. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Liu, J.; Yue, Y.; Wang, L.; Liu, Z.; Xi, G.; An, L.; Tian, J.; Wang, Y. Genome Editing in Mouse Embryo Using the CRISPR/Cas12i3 System. Int. J. Mol. Sci. 2025, 26, 3036. https://doi.org/10.3390/ijms26073036
He J, Liu J, Yue Y, Wang L, Liu Z, Xi G, An L, Tian J, Wang Y. Genome Editing in Mouse Embryo Using the CRISPR/Cas12i3 System. International Journal of Molecular Sciences. 2025; 26(7):3036. https://doi.org/10.3390/ijms26073036
Chicago/Turabian StyleHe, Jiale, Juan Liu, Yuan Yue, Lin Wang, Zhize Liu, Guangyin Xi, Lei An, Jianhui Tian, and Yinjuan Wang. 2025. "Genome Editing in Mouse Embryo Using the CRISPR/Cas12i3 System" International Journal of Molecular Sciences 26, no. 7: 3036. https://doi.org/10.3390/ijms26073036
APA StyleHe, J., Liu, J., Yue, Y., Wang, L., Liu, Z., Xi, G., An, L., Tian, J., & Wang, Y. (2025). Genome Editing in Mouse Embryo Using the CRISPR/Cas12i3 System. International Journal of Molecular Sciences, 26(7), 3036. https://doi.org/10.3390/ijms26073036