
Shared Genetics in Celiac Disease and Inflammatory Bowel Disease Specify a Greater Role for Intestinal Epithelial Cells


Abstract
1. Introduction
2. Intestinal Barrier Disfunction in CeD and IBD
3. Genes in Shared CeD and IBD Loci Contribute to Intestinal Epithelial Barrier Function
3.1. IECs as Non-Conventional Antigen-Presenting Cells
3.2. Regulation of Immune Response in IECs
3.3. IECs Express T-Cell-Regulating Genes
3.4. Interferon Signaling Genes in IECs
3.5. Genetic Impact on Intestinal Epithelial Cell Differentiation
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CeD | Celiac disease |
| IBD | Inflammatory bowel disease |
| CD | Chron’s disease |
| UC | Ulcerative colitis |
| IEC | Intestinal epithelial cell |
| IEL | Intraepithelial lymphocyte |
Appendix A
Glossary
- Apicobasal polarity: Cell polarity of epithelial cells in the intestine. The apical side faces the lumen. The basolateral side faces the lamina propria.
- cis-expression Quantitative Trait Locus (cis-eQTL): A genomic locus that affects the expression of a gene located nearby in the genome.
- Crypt hyperplasia: Elongation of the intestinal crypt.
- Dysbiosis: Disruption in the body’s microbiota composition and/or function.
- Enhancer: A DNA region to which transcription factors can bind to regulate the genes with which they interact.
- Epitope: Part of an antigen molecule that is recognized by the immune system.
- Genome-Wide Association Study (GWAS): Observational study that looks at the whole genome of individuals to identify genetic variation associated with a trait.
- Gliadin: Protein component of gluten and main trigger of CeD. Deaminated gliadin is recognized by HLA-QD2/8 and presented to gluten-specific CD4+ T cells.
- Gluten: A group of proteins naturally found in cereal grains such as wheat, barley, and rye
- Haploinsufficiency: When one copy of a gene is inactivated or deleted and the remaining functional copy is not enough to maintain normal function.
- HLA: The group of genes that encode cell-surface proteins of the major histocompatibility complexes (MHC) that are responsible for the presentation of antigens.
- Ileitis: Inflammation of the ileum, the last part of the small intestine.
- JAK-STAT: A highly conserved signal transduction pathway involved in cell division, cell death, inflammation response, and carcinogenesis. This pathway responds to stimuli by cytokines and growth factors.
- Locus: (Plural: Loci), A genomic locus is a specific region of the genome that is defined based on an area of interest, such as a transcribed RNA, a single exon, or a region associated with differences in expression. It is typically chosen to encompass the feature of interest while minimizing the inclusion of unrelated genomic regions.
- M-like cell: Microfold cells (M cells) are epithelial cells specialized in antigen uptake that are found in the Peyer’s patches of the small intestine and the mucosa-associated lymphoid tissue along the gastrointestinal tract.
- Monogenic: Describes a genetic trait that is influenced only by one gene.
- NETosis: A type of cell death mediated by neutrophils that release a web-like structure made of DNA and bactericidal proteins.
- NLRP3 inflammasome: Component of the innate immune system that mediates caspase activation and release of proinflammatory cytokines in response to microbial infection and cellular damage.
- Over-representation analysis: A statistical method to determine whether a predefined gene set is present more than would be expected by chance in a subset of genes.
- Polygenic: Describes a genetic trait that is influenced by two or more genes.
- Promoter: A DNA region to which regulatory proteins bind to initiate RNA transcription of a gene.
- Pyroptosis: A type of programmed cell death that leads to cell lysis and release of inflammatory signal. Pyroptosis is less controlled than apoptosis.
- Single nucleotide polymorphism (SNP): A variation in a single nucleotide at a specific position in the genome.
- Stricturing: An abnormal narrowing of a bodily passage.
Appendix B
Immunopathology of Celiac Disease and Inflammatory Bowel Disease
- Celiac Disease (CeD): An autoimmune disorder that affects the small intestine. In CeD patients, gluten-derived peptides initiate a cascade of immune reactions. HLA-DQ2 or -DQ8 encoded MHC class II receptors on the surface of antigen-presenting cells present the peptides to gluten-specific CD4+ T cells, which then produce elevated levels of proinflammatory cytokines including IFN-γ and IL-21 [102]. Subsequently, plasma cells are generated, producing anti-gliadin and anti-tissue transglutaminase antibodies (Figure A1). Gliadin may also interact directly with intestinal cells and impair tight junctions, increasing gut permeability [103]. Another hallmark of CeD is the accumulation of activated CD8+ intraepithelial lymphocytes (IELs), which kill intestinal epithelial cells [104]. All these immune activities eventually cause villus atrophy and crypt hyperplasia, leading to intestinal and extra-intestinal symptoms.
- Inflammatory Bowel Disease (IBD): A chronic inflammatory condition comprising two clinical features: Crohn’s disease (CD) and ulcerative colitis (UC). Although the exact trigger for IBD is unknown, genetics, gut microbiota, other environmental factors, and immunological abnormalities all likely contribute to the disease. IBD is marked by episodes of abdominal pain, diarrhea, bloody stools, weight loss, and the infiltration of neutrophils and macrophages. Immune cells produce cytokines, proteolytic enzymes, and free radicals, leading to inflammation and ulceration [105]. Despite common features between CD and UC, there are several distinct pathophysiological attributes. CD can occur anywhere in the intestine, in a discontinuous pattern, whereas UC is restricted to the large intestine in a continuous pattern [105]. In contrast to CD, which is primarily Th1-/Th17-driven with an elevated expression of major cytokines like IL-12, IL-23, IFN-γ, and IL-17, UC is more associated with a Th2/Th9 response and their cytokines, including IL-13, IL-5, and IL-9 [106,107] (Figure A1). However, these Th cell types are not exclusive to UC or CD and may be present in both. Another important IBD hallmark is that the composition of the gut microbiota is altered, influencing gut homeostasis [108].
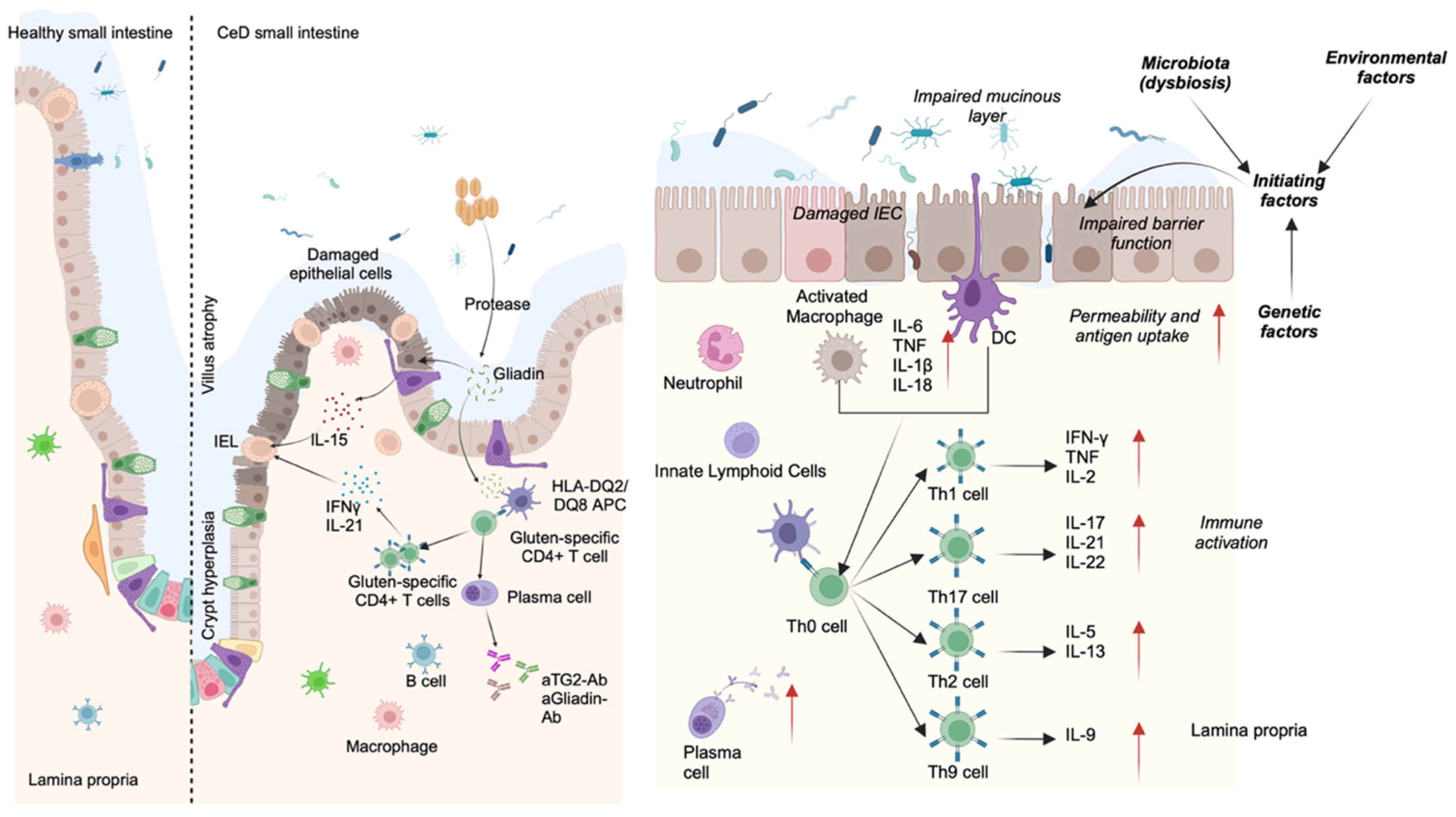
Appendix C
Intestinal Epithelial Cell Barrier
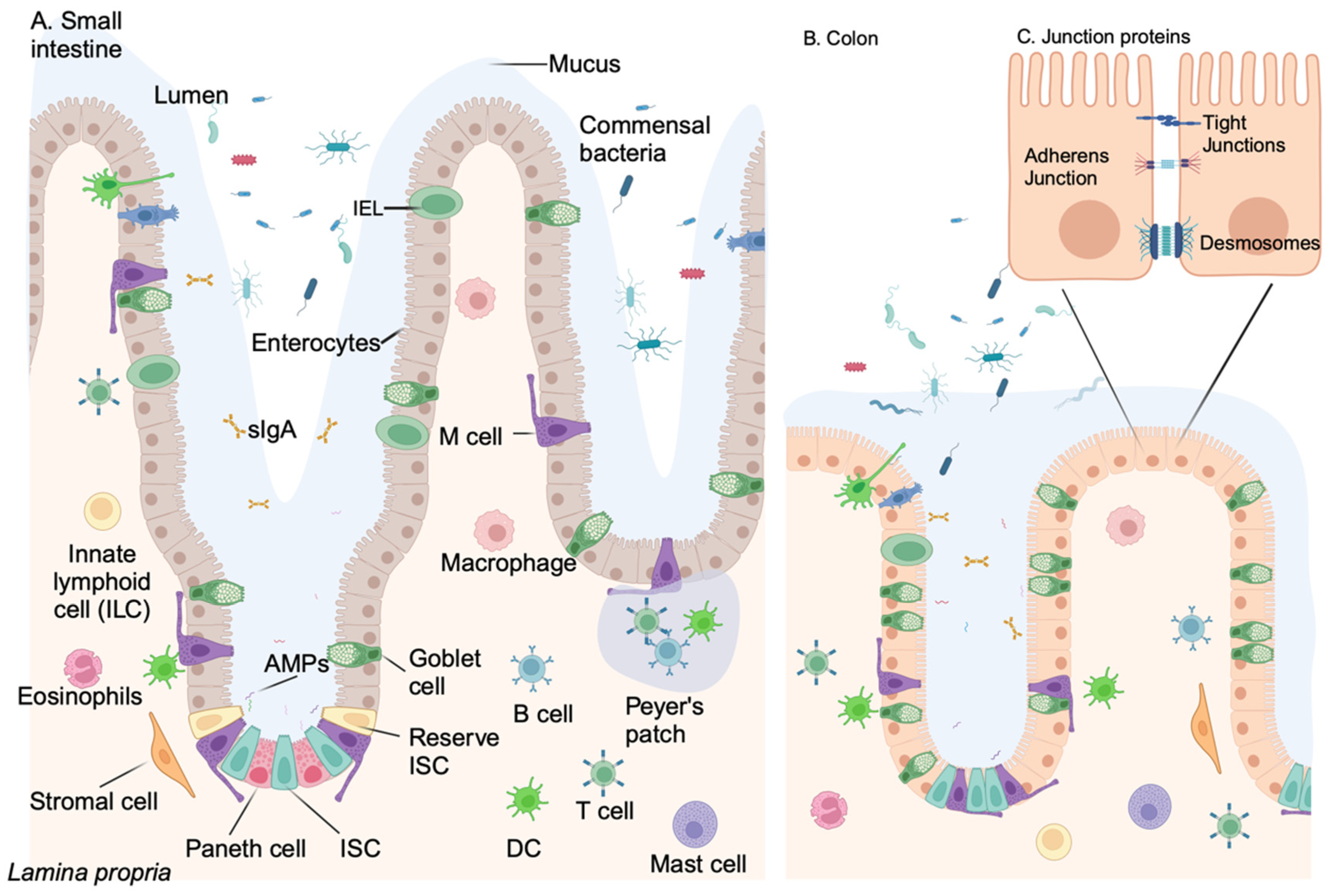
Appendix D
References
- Windsor, J.W.; Kaplan, G.G. Evolving Epidemiology of IBD. Curr. Gastroenterol. Rep. 2019, 21, 40. [Google Scholar] [CrossRef] [PubMed]
- Makharia, G.K.; Chauhan, A.; Singh, P.; Ahuja, V. Review article: Epidemiology of coeliac disease. Aliment. Pharmacol. Ther. 2022, 56 (Suppl. 1), S3–S17. [Google Scholar] [CrossRef]
- Jonkers, I.H.; Wijmenga, C. Context-specific effects of genetic variants associated with autoimmune disease. Hum. Mol. Genet. 2017, 26, R185–R192. [Google Scholar] [CrossRef]
- Stolfi, C.; Maresca, C.; Monteleone, G.; Laudisi, F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines 2022, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. All disease begins in the (leaky) gut: Role of zonulin-mediated gut permeability in the pathogenesis of some chronic inflammatory diseases. F1000Research 2020, 9, F1000-Faculty. [Google Scholar] [CrossRef]
- Fasano, A.; Shea-Donohue, T. Mechanisms of disease: The role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 416–422. [Google Scholar] [CrossRef]
- Jardine, S.; Dhingani, N.; Muise, A.M. TTC7A: Steward of Intestinal Health. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 555–570. [Google Scholar] [CrossRef]
- Hoefkens, E.; Nys, K.; John, J.M.; Van Steen, K.; Arijs, I.; Van der Goten, J.; Van Assche, G.; Agostinis, P.; Rutgeerts, P.; Vermeire, S.; et al. Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress. Autophagy 2013, 9, 2046–2055. [Google Scholar] [CrossRef]
- Mirkov, M.U.; Verstockt, B.; Cleynen, I. Genetics of inflammatory bowel disease: Beyond NOD2. Lancet Gastroenterol. Hepatol. 2017, 2, 224–234. [Google Scholar] [CrossRef]
- Rahmani, S.; Galipeau, H.J.; Clarizio, A.V.; Wang, X.; Hann, A.; Rueda, G.H.; Kirtikar, U.N.; Constante, M.; Wulczynski, M.; Su, H.M.; et al. Gluten dependent activation of CD4(+) T cells by MHC class II-expressing epithelium. Gastroenterology 2024, 167, 1113–1128. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Schwab, M.; Schaffeler, E.; Schlee, M.; Herrlinger, K.R.; Stallmach, A.; Noack, F.; Fritz, P.; et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004, 53, 1658–1664. [Google Scholar] [CrossRef]
- Salem, M.; Ammitzboell, M.; Nys, K.; Seidelin, J.B.; Nielsen, O.H. ATG16L1: A multifunctional susceptibility factor in Crohn disease. Autophagy 2015, 11, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Nayar, S.; Morrison, J.K.; Giri, M.; Gettler, K.; Chuang, L.S.; Walker, L.A.; Ko, H.M.; Kenigsberg, E.; Kugathasan, S.; Merad, M.; et al. A myeloid-stromal niche and gp130 rescue in NOD2-driven Crohn’s disease. Nature 2021, 593, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Manzanillo, P.; Mouchess, M.; Ota, N.; Dai, B.; Ichikawa, R.; Wuster, A.; Haley, B.; Alvarado, G.; Kwon, Y.; Caothien, R.; et al. Inflammatory Bowel Disease Susceptibility Gene C1ORF106 Regulates Intestinal Epithelial Permeability. Immunohorizons 2018, 2, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, V.; Nakata, T.; Desch, A.N.; Levesque, C.; Boroughs, A.; Guzman, G.; Cao, Z.; Creasey, E.; Yao, J.; Boucher, G.; et al. C1orf106 is a colitis risk gene that regulates stability of epithelial adherens junctions. Science 2018, 359, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, M.C.; Monsuur, A.J.; van Bodegraven, A.A.; Weersma, R.K.; Bevova, M.R.; Linskens, R.K.; Howdle, P.; Holmes, G.; Mulder, C.J.; Dijkstra, G.; et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut 2008, 57, 463–467. [Google Scholar] [CrossRef]
- van Sommeren, S.; Visschedijk, M.C.; Festen, E.A.; de Jong, D.J.; Ponsioen, C.Y.; Wijmenga, C.; Weersma, R.K. HNF4alpha and CDH1 are associated with ulcerative colitis in a Dutch cohort. Inflamm. Bowel Dis. 2011, 17, 1714–1718. [Google Scholar] [CrossRef]
- McCole, D.F. IBD candidate genes and intestinal barrier regulation. Inflamm. Bowel Dis. 2014, 20, 1829–1849. [Google Scholar] [CrossRef]
- Schumann, M.; Gunzel, D.; Buergel, N.; Richter, J.F.; Troeger, H.; May, C.; Fromm, A.; Sorgenfrei, D.; Daum, S.; Bojarski, C.; et al. Cell polarity-determining proteins Par-3 and PP-1 are involved in epithelial tight junction defects in coeliac disease. Gut 2012, 61, 220–228. [Google Scholar] [CrossRef]
- Jauregi-Miguel, A.; Santin, I.; Garcia-Etxebarria, K.; Olazagoitia-Garmendia, A.; Romero-Garmendia, I.; Sebastian-delaCruz, M.; Irastorza, I.; Spanish Consortium for the Genetics of Celiac, D.; Castellanos-Rubio, A.; Bilbao, J.R. MAGI2 Gene Region and Celiac Disease. Front. Nutr. 2019, 6, 187. [Google Scholar] [CrossRef]
- Muise, A.M.; Walters, T.D.; Glowacka, W.K.; Griffiths, A.M.; Ngan, B.Y.; Lan, H.; Xu, W.; Silverberg, M.S.; Rotin, D. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn’s disease. Gut 2009, 58, 1121–1127. [Google Scholar] [CrossRef]
- Dubois, P.C.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.; Adany, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- Hunt, K.A.; Zhernakova, A.; Turner, G.; Heap, G.A.; Franke, L.; Bruinenberg, M.; Romanos, J.; Dinesen, L.C.; Ryan, A.W.; Panesar, D.; et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008, 40, 395–402. [Google Scholar] [CrossRef]
- Elmentaite, R.; Kumasaka, N.; Roberts, K.; Fleming, A.; Dann, E.; King, H.W.; Kleshchevnikov, V.; Dabrowska, M.; Pritchard, S.; Bolt, L.; et al. Cells of the human intestinal tract mapped across space and time. Nature 2021, 597, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, C.; Pott, J.; Maloy, K.J. Why do intestinal epithelial cells express MHC class II? Immunology 2021, 162, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Fais, S.; Maiuri, L.; Pallone, F.; De Vincenzi, M.; De Ritis, G.; Troncone, R.; Auricchio, S. Gliadin induced changes in the expression of MHC-class II antigens by human small intestinal epithelium. Organ culture studies with coeliac disease mucosa. Gut 1992, 33, 472–475. [Google Scholar] [CrossRef]
- Kornberg, A.; Botella, T.; Moon, C.S.; Rao, S.; Gelbs, J.; Cheng, L.; Miller, J.; Bacarella, A.M.; Garcia-Vilas, J.A.; Vargas, J.; et al. Gluten induces rapid reprogramming of natural memory alphabeta and gammadelta intraepithelial T cells to induce cytotoxicity in celiac disease. Sci. Immunol. 2023, 8, eadf4312. [Google Scholar] [CrossRef]
- Dotan, I.; Allez, M.; Nakazawa, A.; Brimnes, J.; Schulder-Katz, M.; Mayer, L. Intestinal epithelial cells from inflammatory bowel disease patients preferentially stimulate CD4+ T cells to proliferate and secrete interferon-gamma. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1630–G1640. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef]
- McDonald, G.B.; Jewell, D.P. Class II antigen (HLA-DR) expression by intestinal epithelial cells in inflammatory diseases of colon. J. Clin. Pathol. 1987, 40, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Hirata, I.; Austin, L.L.; Blackwell, W.H.; Weber, J.R.; Dobbins, W.O., 3rd. Immunoelectron microscopic localization of HLA-DR antigen in control small intestine and colon and in inflammatory bowel disease. Dig. Dis. Sci. 1986, 31, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, C.E.; Janney, A.; Ilott, N.; Bertocchi, A.; Pott, S.; Gu, Y.; Pohin, M.; Friedrich, M.; Mann, E.H.; Pearson, C.; et al. MHC class II antigen presentation by intestinal epithelial cells fine-tunes bacteria-reactive CD4 T-cell responses. Mucosal Immunol. 2024, 17, 416–430. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef]
- Didriksen, B.J.; Eshleman, E.M.; Alenghat, T. Epithelial regulation of microbiota-immune cell dynamics. Mucosal Immunol. 2024, 17, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Hehlgans, T.; Pfeffer, K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: Players, rules and the games. Immunology 2005, 115, 1–20. [Google Scholar] [CrossRef]
- Sandberg, W.J.; Halvorsen, B.; Yndestad, A.; Smith, C.; Otterdal, K.; Brosstad, F.R.; Froland, S.S.; Olofsson, P.S.; Damas, J.K.; Gullestad, L.; et al. Inflammatory interaction between LIGHT and proteinase-activated receptor-2 in endothelial cells: Potential role in atherogenesis. Circ. Res. 2009, 104, 60–68. [Google Scholar] [CrossRef]
- Shui, J.W.; Larange, A.; Kim, G.; Vela, J.L.; Zahner, S.; Cheroutre, H.; Kronenberg, M. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature 2012, 488, 222–225. [Google Scholar] [CrossRef]
- Steinberg, M.W.; Turovskaya, O.; Shaikh, R.B.; Kim, G.; McCole, D.F.; Pfeffer, K.; Murphy, K.M.; Ware, C.F.; Kronenberg, M. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J. Exp. Med. 2008, 205, 1463–1476. [Google Scholar] [CrossRef]
- Ramírez-Sánchez, A.D.; Zühlke, S.; Aguirre-Gamboa, R.; Vochteloo, M.; Franke, L.; Lundin, K.E.A.; Withoff, S.; Jonkers, I.H. Gene expression and eQTL analysis reflect the heterogeneity in the inflammatory status of the duodenal epithelial lining in coeliac disease. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kong, L.; Pokatayev, V.; Lefkovith, A.; Carter, G.T.; Creasey, E.A.; Krishna, C.; Subramanian, S.; Kochar, B.; Ashenberg, O.; Lau, H.; et al. The landscape of immune dysregulation in Crohn’s disease revealed through single-cell transcriptomic profiling in the ileum and colon. Immunity 2023, 56, 444–458.e5. [Google Scholar] [CrossRef] [PubMed]
- Kabakchiev, B.; Silverberg, M.S. Expression quantitative trait loci analysis identifies associations between genotype and gene expression in human intestine. Gastroenterology 2013, 144, 1488–1496.e3. [Google Scholar] [CrossRef]
- Cielo, D.; Galatola, M.; Fernandez-Jimenez, N.; De Leo, L.; Garcia-Etxebarria, K.; Loganes, C.; Tommasini, A.; Not, T.; Auricchio, R.; Greco, L.; et al. Combined Analysis of Methylation and Gene Expression Profiles in Separate Compartments of Small Bowel Mucosa Identified Celiac Disease Patients’ Signatures. Sci. Rep. 2019, 9, 10020. [Google Scholar] [CrossRef]
- Loberman-Nachum, N.; Sosnovski, K.; Di Segni, A.; Efroni, G.; Braun, T.; BenShoshan, M.; Anafi, L.; Avivi, C.; Barshack, I.; Shouval, D.S.; et al. Defining the Celiac Disease Transcriptome using Clinical Pathology Specimens Reveals Biologic Pathways and Supports Diagnosis. Sci. Rep. 2019, 9, 16163. [Google Scholar] [CrossRef]
- Dotsenko, V.; Oittinen, M.; Taavela, J.; Popp, A.; Peraaho, M.; Staff, S.; Sarin, J.; Leon, F.; Isola, J.; Maki, M.; et al. Genome-Wide Transcriptomic Analysis of Intestinal Mucosa in Celiac Disease Patients on a Gluten-Free Diet and Postgluten Challenge. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 13–32. [Google Scholar] [CrossRef]
- Huang, B.; Chen, Z.; Geng, L.; Wang, J.; Liang, H.; Cao, Y.; Chen, H.; Huang, W.; Su, M.; Wang, H.; et al. Mucosal Profiling of Pediatric-Onset Colitis and IBD Reveals Common Pathogenics and Therapeutic Pathways. Cell 2019, 179, 1160–1176.e24. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Uniken Venema, W.T.; Westra, H.J.; Vich Vila, A.; Barbieri, R.; Voskuil, M.D.; Blokzijl, T.; Jansen, B.H.; Li, Y.; Daly, M.J.; et al. Inflammation status modulates the effect of host genetic variation on intestinal gene expression in inflammatory bowel disease. Nat. Commun. 2021, 12, 1122. [Google Scholar] [CrossRef]
- GTEx Portal. Colon Transverse—GTEx Analysis Release V8. Available online: https://gtexportal.org/home/tissue/Colon_Transverse (accessed on 13 August 2024).
- GTEx Portal. Small Intestine—Terminal Ileum—GTEx Analysis Release V8. Available online: https://gtexportal.org/home/tissue/Small_Intestine_Terminal_Ileum (accessed on 13 August 2024).
- Bradford, E.M.; Ryu, S.H.; Singh, A.P.; Lee, G.; Goretsky, T.; Sinh, P.; Williams, D.B.; Cloud, A.L.; Gounaris, E.; Patel, V.; et al. Epithelial TNF Receptor Signaling Promotes Mucosal Repair in Inflammatory Bowel Disease. J. Immunol. 2017, 199, 1886–1897. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Aljamaei, H.M.; Stadnyk, A.W. The Production and Function of Endogenous Interleukin-10 in Intestinal Epithelial Cells and Gut Homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1343–1352. [Google Scholar] [CrossRef]
- Wei, H.X.; Wang, B.; Li, B. IL-10 and IL-22 in Mucosal Immunity: Driving Protection and Pathology. Front. Immunol. 2020, 11, 1315. [Google Scholar] [CrossRef]
- Zhu, H.; Lei, X.; Liu, Q.; Wang, Y. Interleukin-10-1082A/G polymorphism and inflammatory bowel disease susceptibility: A meta-analysis based on 17,585 subjects. Cytokine 2013, 61, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yuan, W.; Park, S. Association between IL-10 rs3024505 and susceptibility to inflammatory bowel disease: A systematic review and meta-analysis. Cytokine 2022, 149, 155721. [Google Scholar] [CrossRef]
- Lin, Z.; Chen, Q.; Ruan, H.B. To die or not to die: Gasdermins in intestinal health and disease. Semin. Immunol. 2024, 71, 101865. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, S.; Broz, P. Regulation of Lytic and Non-Lytic Functions of Gasdermin Pores. J. Mol. Biol. 2022, 434, 167246. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef] [PubMed]
- Canale, V.; Spalinger, M.R.; Alvarez, R.; Sayoc-Becerra, A.; Sanati, G.; Manz, S.; Chatterjee, P.; Santos, A.N.; Lei, H.; Jahng, S.; et al. PTPN2 Is a Critical Regulator of Ileal Paneth Cell Viability and Function in Mice. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 39–62. [Google Scholar] [CrossRef]
- Festen, E.A.; Goyette, P.; Green, T.; Boucher, G.; Beauchamp, C.; Trynka, G.; Dubois, P.C.; Lagace, C.; Stokkers, P.C.; Hommes, D.W.; et al. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn’s disease and celiac disease. PLoS Genet. 2011, 7, e1001283. [Google Scholar] [CrossRef]
- Spalinger, M.R.; Kasper, S.; Chassard, C.; Raselli, T.; Frey-Wagner, I.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Mair, F.; et al. PTPN2 controls differentiation of CD4(+) T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol. 2015, 8, 918–929. [Google Scholar] [CrossRef]
- Spalinger, M.R.; Crawford, M.; Bobardt, S.D.; Li, J.; Sayoc-Becerra, A.; Santos, A.N.; Shawki, A.; Chatterjee, P.; Nair, M.G.; McCole, D.F. Loss of protein tyrosine phosphatase non-receptor type 2 reduces IL-4-driven alternative macrophage activation. Mucosal Immunol. 2022, 15, 74–83. [Google Scholar] [CrossRef]
- Bao, X.; Qin, Y.; Lu, L.; Zheng, M. Transcriptional Regulation of Early T-Lymphocyte Development in Thymus. Front. Immunol. 2022, 13, 884569. [Google Scholar] [CrossRef]
- Li, X.; Thyssen, G.; Beliakoff, J.; Sun, Z. The novel PIAS-like protein hZimp10 enhances Smad transcriptional activity. J. Biol. Chem. 2006, 281, 23748–23756. [Google Scholar] [CrossRef]
- Medrano, L.M.; Pascual, V.; Bodas, A.; Lopez-Palacios, N.; Salazar, I.; Espino-Paisan, L.; Gonzalez-Perez, B.; Urcelay, E.; Mendoza, J.L.; Nunez, C. Expression patterns common and unique to ulcerative colitis and celiac disease. Ann. Hum. Genet. 2019, 83, 86–94. [Google Scholar] [CrossRef]
- Lan, X.; Lan, X.; Chang, Y.; Zhang, X.; Liu, J.; Vikash, V.; Wang, W.; Huang, M.; Wang, X.; Zhou, F.; et al. Identification of Two Additional Susceptibility Loci for Inflammatory Bowel Disease in a Chinese Population. Cell. Physiol. Biochem. 2017, 41, 2077–2090. [Google Scholar] [CrossRef]
- De Vries, L.C.S.; Ghiboub, M.; van Hamersveld, P.H.P.; Welting, O.; Verseijden, C.; Bell, M.J.; Rioja, I.; Prinjha, R.K.; Koelink, P.J.; Strobl, B.; et al. Tyrosine Kinase 2 Signalling Drives Pathogenic T cells in Colitis. J. Crohn’s Colitis 2021, 15, 617–630. [Google Scholar] [CrossRef]
- Hainzl, E.; Stockinger, S.; Rauch, I.; Heider, S.; Berry, D.; Lassnig, C.; Schwab, C.; Rosebrock, F.; Milinovich, G.; Schlederer, M.; et al. Intestinal Epithelial Cell Tyrosine Kinase 2 Transduces IL-22 Signals To Protect from Acute Colitis. J. Immunol. 2015, 195, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Stankey, C.T.; Bourges, C.; Haag, L.M.; Turner-Stokes, T.; Piedade, A.P.; Palmer-Jones, C.; Papa, I.; Silva Dos Santos, M.; Zhang, Q.; Cameron, A.J.; et al. A disease-associated gene desert directs macrophage inflammation through ETS2. Nature 2024, 630, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Butler, L.; Perkins, A.; Kershaw, N.J.; Babon, J.J. The Role of LNK (SH2B3) in the Regulation of JAK-STAT Signalling in Haematopoiesis. Pharmaceuticals 2021, 15, 24. [Google Scholar] [CrossRef]
- Takizawa, H.; Kubo-Akashi, C.; Nobuhisa, I.; Kwon, S.M.; Iseki, M.; Taga, T.; Takatsu, K.; Takaki, S. Enhanced engraftment of hematopoietic stem/progenitor cells by the transient inhibition of an adaptor protein, Lnk. Blood 2006, 107, 2968–2975. [Google Scholar] [CrossRef]
- Devalliere, J.; Chatelais, M.; Fitau, J.; Gerard, N.; Hulin, P.; Velazquez, L.; Turner, C.E.; Charreau, B. LNK (SH2B3) is a key regulator of integrin signaling in endothelial cells and targets alpha-parvin to control cell adhesion and migration. FASEB J. 2012, 26, 2592–2606. [Google Scholar] [CrossRef]
- Langer, V.; Vivi, E.; Regensburger, D.; Winkler, T.H.; Waldner, M.J.; Rath, T.; Schmid, B.; Skottke, L.; Lee, S.; Jeon, N.L.; et al. IFN-gamma drives inflammatory bowel disease pathogenesis through VE-cadherin-directed vascular barrier disruption. J. Clin. Investig. 2019, 129, 4691–4707. [Google Scholar] [CrossRef]
- Malik, A.; Sharma, D.; Aguirre-Gamboa, R.; McGrath, S.; Zabala, S.; Weber, C.; Jabri, B. Epithelial IFNgamma signalling and compartmentalized antigen presentation orchestrate gut immunity. Nature 2023, 623, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhang, Y.B.; Gui, J.F.; Lemon, S.M.; Yamane, D. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PLoS Pathog. 2021, 17, e1009220. [Google Scholar] [CrossRef]
- Oshima, S.; Nakamura, T.; Namiki, S.; Okada, E.; Tsuchiya, K.; Okamoto, R.; Yamazaki, M.; Yokota, T.; Aida, M.; Yamaguchi, Y.; et al. Interferon regulatory factor 1 (IRF-1) and IRF-2 distinctively up-regulate gene expression and production of interleukin-7 in human intestinal epithelial cells. Mol. Cell. Biol. 2004, 24, 6298–6310. [Google Scholar] [CrossRef]
- Kim, T.S.; Rha, M.S.; Shin, E.C. IFN-gamma Induces IL-15 Trans-Presentation by Epithelial Cells via IRF1. J. Immunol. 2022, 208, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Diegelmann, J.; Brand, S. Identification of IL-27 as a novel regulator of major histocompatibility complex class I and class II expression, antigen presentation, and processing in intestinal epithelial cells. Front. Immunol. 2023, 14, 1226809. [Google Scholar] [CrossRef]
- Tan, G.; Huang, C.; Chen, J.; Chen, B.; Shi, Y.; Zhi, F. An IRF1-dependent Pathway of TNFalpha-induced Shedding in Intestinal Epithelial Cells. J. Crohn’s Colitis 2022, 16, 133–142. [Google Scholar] [CrossRef]
- Abadie, V.; Kim, S.M.; Lejeune, T.; Palanski, B.A.; Ernest, J.D.; Tastet, O.; Voisine, J.; Discepolo, V.; Marietta, E.V.; Hawash, M.B.F.; et al. IL-15, gluten and HLA-DQ8 drive tissue destruction in coeliac disease. Nature 2020, 578, 600–604. [Google Scholar] [CrossRef]
- Santos, A.J.M.; van Unen, V.; Lin, Z.; Chirieleison, S.M.; Ha, N.; Batish, A.; Chan, J.E.; Cedano, J.; Zhang, E.T.; Mu, Q.; et al. A human autoimmune organoid model reveals IL-7 function in coeliac disease. Nature 2024, 632, 401–410. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ito, M.; Mise-Omata, S.; Ando, M. SOCS: Negative regulators of cytokine signaling for immune tolerance. Int. Immunol. 2021, 33, 711–716. [Google Scholar] [CrossRef]
- Davey, G.M.; Heath, W.R.; Starr, R. SOCS1: A potent and multifaceted regulator of cytokines and cell-mediated inflammation. Tissue Antigens 2006, 67, 1–9. [Google Scholar] [CrossRef]
- Rodari, M.M.; Cazals-Hatem, D.; Uzzan, M.; Martin Silva, N.; Khiat, A.; Ta, M.C.; Lhermitte, L.; Touzart, A.; Hanein, S.; Rouillon, C.; et al. Insights into the expanding intestinal phenotypic spectrum of SOCS1 haploinsufficiency and therapeutic options. J. Clin. Immunol. 2023, 43, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Shan, X.; Qian, J.; Ji, Q.; Wang, L.; Wang, X.; Li, M.; Ding, H.; Liu, Q.; Chen, L.; et al. The suppressor of cytokine signaling SOCS1 promotes apoptosis of intestinal epithelial cells via p53 signaling in Crohn’s disease. Exp. Mol. Pathol. 2016, 101, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Richmond, C.A.; Rickner, H.; Shah, M.S.; Ediger, T.; Deary, L.; Zhou, F.; Tovaglieri, A.; Carlone, D.L.; Breault, D.T. JAK/STAT-1 Signaling Is Required for Reserve Intestinal Stem Cell Activation during Intestinal Regeneration Following Acute Inflammation. Stem Cell Rep. 2018, 10, 17–26. [Google Scholar] [CrossRef]
- Stolzer, I.; Schickedanz, L.; Chiriac, M.T.; Lopez-Posadas, R.; Grassl, G.A.; Mattner, J.; Wirtz, S.; Winner, B.; Neurath, M.F.; Gunther, C. STAT1 coordinates intestinal epithelial cell death during gastrointestinal infection upstream of Caspase-8. Mucosal Immunol. 2022, 15, 130–142. [Google Scholar] [CrossRef]
- Vooijs, M.; Liu, Z.; Kopan, R. Notch: Architect, landscaper, and guardian of the intestine. Gastroenterology 2011, 141, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Yang, L.; Ou, Y.N.; Wu, B.S.; Liu, W.S.; Deng, Y.T.; He, X.Y.; Chen, Y.L.; Kang, J.; Fei, C.J.; Zhu, Y.; et al. Large-scale whole-exome sequencing analyses identified protein-coding variants associated with immune-mediated diseases in 350,770 adults. Nat. Commun. 2024, 15, 5924. [Google Scholar] [CrossRef]
- Lee, C.; Hong, S.N.; Kim, E.R.; Chang, D.K.; Kim, Y.H. Depletion of Intestinal Stem Cell Niche Factors Contributes to the Alteration of Epithelial Differentiation in SAMP1/YitFcsJ Mice with Crohn Disease-Like Ileitis. Inflamm. Bowel Dis. 2021, 27, 667–676. [Google Scholar] [CrossRef]
- Ueo, T.; Imayoshi, I.; Kobayashi, T.; Ohtsuka, T.; Seno, H.; Nakase, H.; Chiba, T.; Kageyama, R. The role of Hes genes in intestinal development, homeostasis and tumor formation. Development 2012, 139, 1071–1082. [Google Scholar] [CrossRef]
- Strott, C.A.; Higashi, Y. Cholesterol sulfate in human physiology: What’s it all about? J. Lipid Res. 2003, 44, 1268–1278. [Google Scholar] [CrossRef]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martin, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220.e4. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Ma, R.; Ju, Y.; Song, X.; Niu, B.; Hong, W.; Wang, R.; Yang, Q.; Zhao, Z.; Zhang, Y.; et al. Cholesterol sulfate alleviates ulcerative colitis by promoting cholesterol biosynthesis in colonic epithelial cells. Nat. Commun. 2022, 13, 4428. [Google Scholar] [CrossRef]
- Minikel, E.V.; Painter, J.L.; Dong, C.C.; Nelson, M.R. Refining the impact of genetic evidence on clinical success. Nature 2024, 629, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Collij, V.; Festen, E.A.; Alberts, R.; Weersma, R.K. Drug Repositioning in Inflammatory Bowel Disease Based on Genetic Information. Inflamm. Bowel Dis. 2016, 22, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 765474. [Google Scholar] [CrossRef]
- Okamoto, R.; Watanabe, M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J. Gastroenterol. 2016, 51, 11–21. [Google Scholar] [CrossRef]
- Discepolo, V.; Kelly, C.P.; Koning, F.; Schuppan, D. How Future Pharmacologic Therapies for Celiac Disease Will Complement the Gluten-Free Diet. Gastroenterology 2024, 167, 90–103. [Google Scholar] [CrossRef]
- Sollid, L.M.; Koning, F. Lack of relationship of AT1001 to zonulin and prehaptoglobin-2: Clinical implications. Gut 2021, 70, 2211–2212. [Google Scholar] [CrossRef]
- Moerkens, R.; Mooiweer, J.; Ramirez-Sanchez, A.D.; Oelen, R.; Franke, L.; Wijmenga, C.; Barrett, R.J.; Jonkers, I.H.; Withoff, S. An iPSC-derived small intestine-on-chip with self-organizing epithelial, mesenchymal, and neural cells. Cell Rep. 2024, 43, 114247. [Google Scholar] [CrossRef]
- Bjorck, S.; Lindehammer, S.R.; Fex, M.; Agardh, D. Serum cytokine pattern in young children with screening detected coeliac disease. Clin. Exp. Immunol. 2015, 179, 230–235. [Google Scholar] [CrossRef]
- Visser, J.; Rozing, J.; Sapone, A.; Lammers, K.; Fasano, A. Tight junctions, intestinal permeability, and autoimmunity: Celiac disease and type 1 diabetes paradigms. Ann. N. Y. Acad. Sci. 2009, 1165, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Malamut, G.; Cording, S.; Cerf-Bensussan, N. Recent advances in celiac disease and refractory celiac disease. F1000Research 2019, 8, F1000-Faculty. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef]
- de Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, M.F.; Liang, Y.J.; Xu, J.; Xu, H.M.; Nie, Y.Q.; Wang, L.S.; Yao, J.; Li, D.F. Immunology of Inflammatory Bowel Disease: Molecular Mechanisms and Therapeutics. J. Inflamm. Res. 2022, 15, 1825–1844. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Untersmayr, E.; Brandt, A.; Koidl, L.; Bergheim, I. The Intestinal Barrier Dysfunction as Driving Factor of Inflammaging. Nutrients 2022, 14, 949. [Google Scholar] [CrossRef] [PubMed]
- Mayassi, T.; Jabri, B. Human intraepithelial lymphocytes. Mucosal Immunol. 2018, 11, 1281–1289. [Google Scholar] [CrossRef]
- Cardoso-Silva, D.; Delbue, D.; Itzlinger, A.; Moerkens, R.; Withoff, S.; Branchi, F.; Schumann, M. Intestinal Barrier Function in Gluten-Related Disorders. Nutrients 2019, 11, 2325. [Google Scholar] [CrossRef]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]

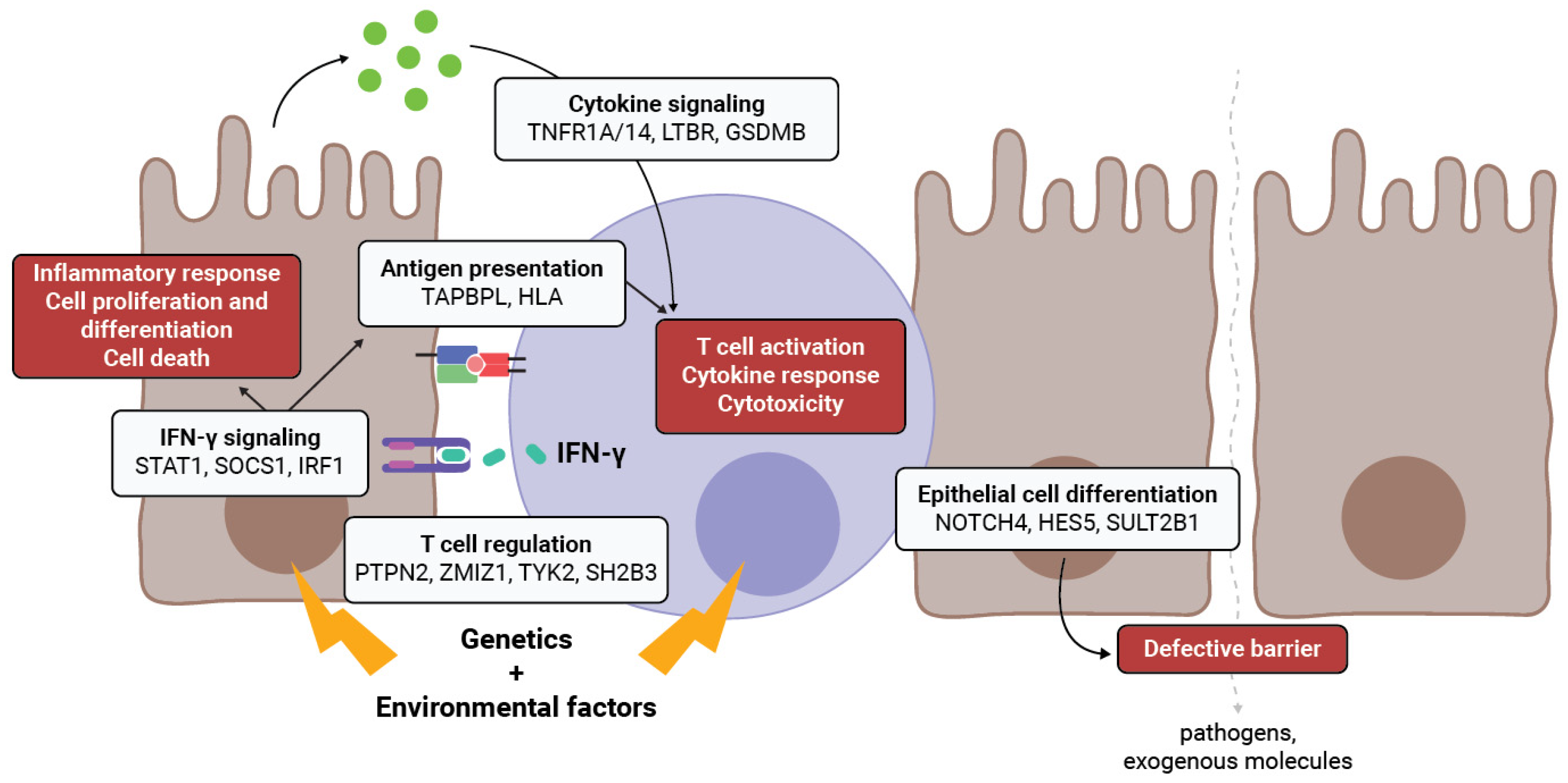

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Function 1 | Differential Expression Status in Epithelial Cells | SNPs with cis-eQTL Effect | ||
|---|---|---|---|---|---|
| CeD 2 | IBD 3 | CeD | IBD | ||
| TAPBPL | Links MHC I molecules to TAP transporter | ||||
| HLA-DQA2 | Antigen-presentation via MHC class II | ||||
| HLA-DRB5 | Antigen-presentation via MHC class II | Down [26] | Up [25,27] | rs424232 [28] | rs3135005, rs693797, rs9267911 [28] |
| HLA-DRB1 | Antigen-presentation via MHC class II | Up [27] | |||
| HLA-DQA1 | Antigen-presentation via MHC class II | Down [26] | Up (7) | rs3135005, rs9268403, rs693797 [28] | |
| TNFRSF14 | TNF receptor superfamily | Down [27] | |||
| IL10 | Immunoregulatory cytokine | Up [26] | |||
| ZMIZ1 | Transcriptional coactivator | Up [27] | rs1250567 [26] | ||
| ETS1 | Transcription factor | ||||
| SH2B3 | Negative regulator of cytokine signaling | Down [26], Up [29] | |||
| EFNB2 | Ephrin receptor | Up [27] | |||
| SOCS1 | Negative regulator of interferon signaling | Up [27] | |||
| PTPN2 | Protein tyrosine phosphatase (signaling) | Down [27] | |||
| TYK2 | Tyrosine kinase (signaling) | ||||
| IRF1 | Transcription factor of interferon genes | Up [26,30,31] | Up [27] | ||
| NOTCH4 | Membrane receptor of Notch signaling | ||||
| CDC37 | Molecular chaperone | Down [27,32] | |||
| STAT1 | Signal transducer in response to interferon | Up [26,30,31] | Up [25,27,32] | ||
| HES5 | Transcription repressor | ||||
| TNFRSF1A | TNF receptor superfamily | Up [32] | rs2364484 [26] | ||
| DENND1B | Regulates T-cell receptor internalization | Up [27] | |||
| INAVA/C1ORF106 | Cytokine production, adherens junctions | ||||
| MAPKAPK2 | Stress-activated serine/threonine-protein kinase | ||||
| SPHK2 | Catalyzes the phosphorylation of sphingosine | ||||
| AGPAT1 | Converts lysophosphatidic acid into phosphatidic acid | Up [27] | |||
| GPSM3 | Regulator of GTPase activity | Up [32] | |||
| REL | Proto-oncogene involved in lymphopoiesis | Down [27] | |||
| GSDMB | Pore-forming protein | Up [27] | rs2305480, rs2872507, rs10852936, rs2305479, rs11557467, rs12950743, rs8067378 [28] | rs8069176, rs2305480, rs2872507, rs10852936, rs2305479, rs10445308, rs11557467, rs12950743, rs8067378, rs4795405, rs3902025, rs11078927 [28,33,34] | |
| SEH1L | Component of nuclear pore complex | ||||
| CCR5 | Beta chemokine receptor family | ||||
| SLC22A5 | Cation transporter | Down [26] | Down [27] | ||
| RNF5 | Membrane-bound ubiquitin ligase | Down [27] | |||
| KEAP1 | Sensor of oxidative stress | ||||
| SULT2B1 | Sulfation of cholesterol | Down [26] | Up (colon), Down (ileum) [27] | ||
| LTBR | TNF receptor superfamily | Down [27] | rs10849448 [35], rs2364480, rs9669611, rs12354 [28], rs2364484 [26,28] | ||
| LURAP1L | Regulation of NFkB signaling | ||||
| GPR35 | G-protein coupled receptor | Down [26] | Down [27] | ||
| CCRL2 | Chemokine receptor | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, N.V.; Anwar, S.; Withoff, S.; Jonkers, I.H. Shared Genetics in Celiac Disease and Inflammatory Bowel Disease Specify a Greater Role for Intestinal Epithelial Cells. Int. J. Mol. Sci. 2025, 26, 2982. https://doi.org/10.3390/ijms26072982
Ribeiro NV, Anwar S, Withoff S, Jonkers IH. Shared Genetics in Celiac Disease and Inflammatory Bowel Disease Specify a Greater Role for Intestinal Epithelial Cells. International Journal of Molecular Sciences. 2025; 26(7):2982. https://doi.org/10.3390/ijms26072982
Chicago/Turabian StyleRibeiro, Nathan Vinícius, Sajid Anwar, Sebo Withoff, and Iris H. Jonkers. 2025. "Shared Genetics in Celiac Disease and Inflammatory Bowel Disease Specify a Greater Role for Intestinal Epithelial Cells" International Journal of Molecular Sciences 26, no. 7: 2982. https://doi.org/10.3390/ijms26072982
APA StyleRibeiro, N. V., Anwar, S., Withoff, S., & Jonkers, I. H. (2025). Shared Genetics in Celiac Disease and Inflammatory Bowel Disease Specify a Greater Role for Intestinal Epithelial Cells. International Journal of Molecular Sciences, 26(7), 2982. https://doi.org/10.3390/ijms26072982