Functional Analysis of Mannosyltransferase-Related Genes UvALGs in Ustilaginoidea virens

Abstract
1. Introduction
- (1)
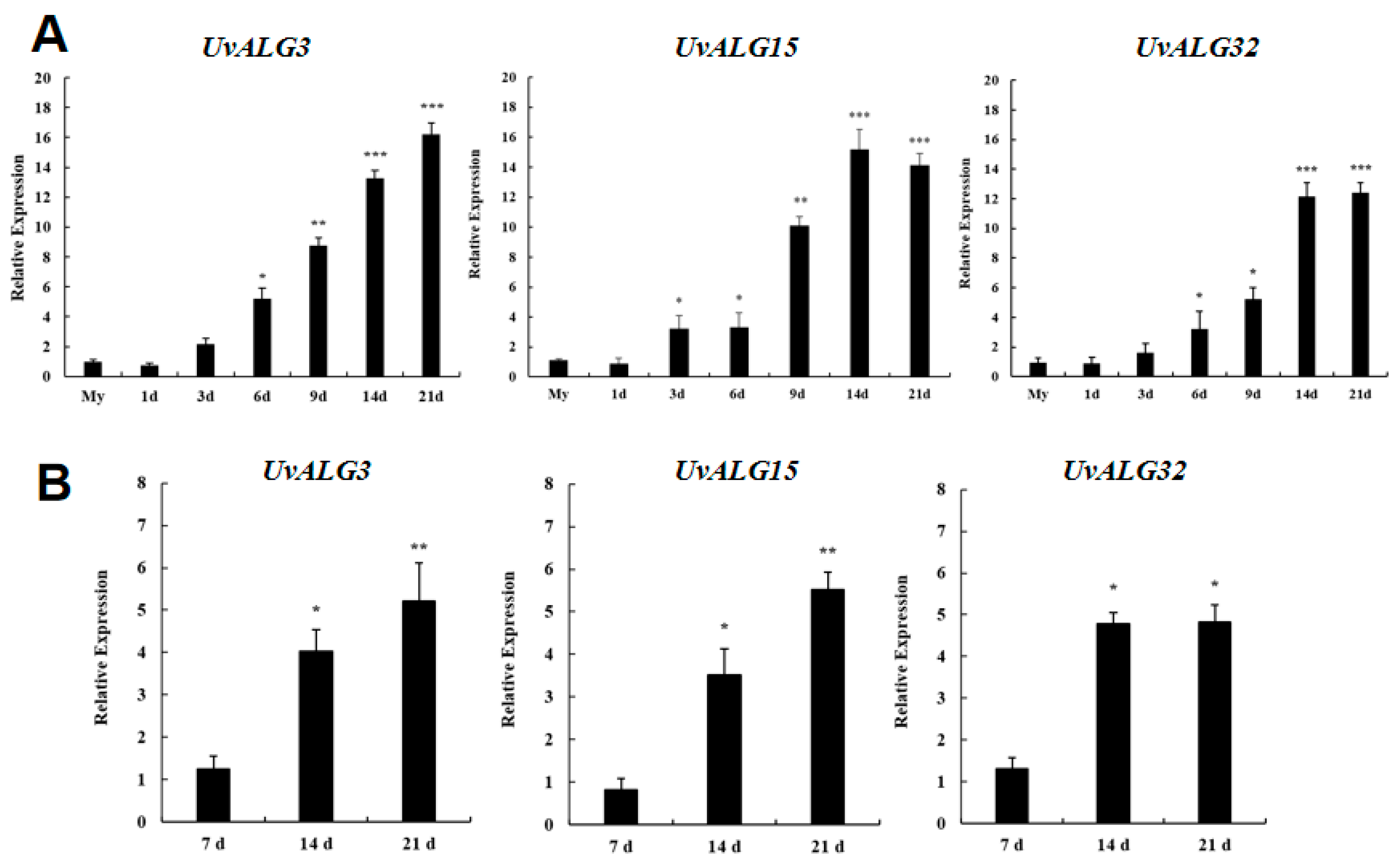
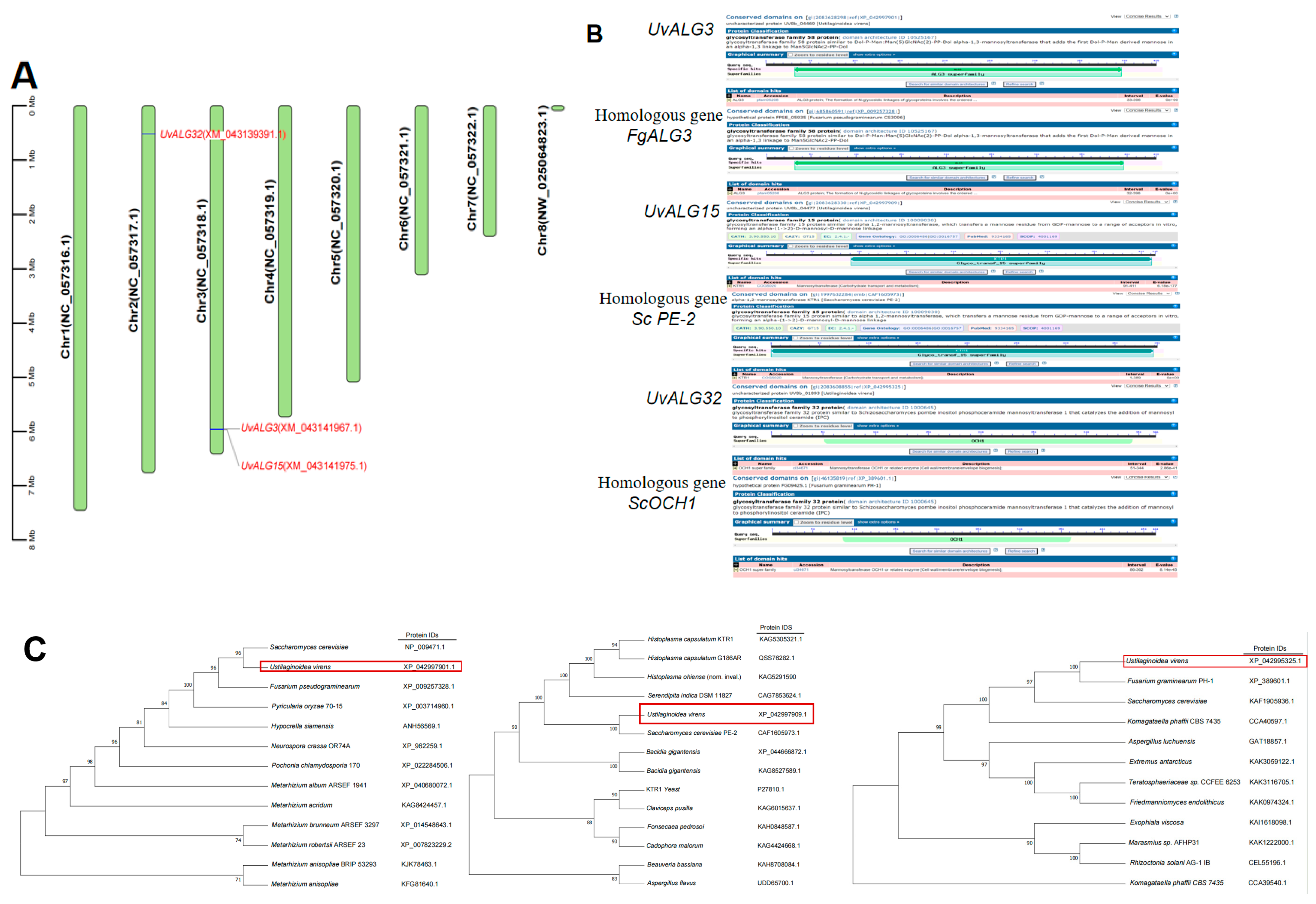
- Thirty-eight genes encoding mannosyltransferases in Ustilaginoidea virens were screened by gene annotation. The expression levels of these genes at different stages of infection of Ustilaginoidea virens and different days of growth of Ustilaginoidea virens on medium were analyzed. The significantly upregulated genes Uv8b_04477, Uv8b_01893, and Uv8b_04469 encoding mannosyltransferase were screened and analyzed by bioinformatics. The protein domain and physicochemical properties of these genes, the location of coding genes on chromosomes, the prediction of protein function, and the construction of a phylogenetic tree were predicted and analyzed. It was found that these three proteins contained different domains, which were ALG3, GT15, and OCH1, respectively. The proteins are consistent with their corresponding homologous gene domains. The amino acid lengths of the three proteins are about 400 aa, which is relatively stable. This included two hydrophilic proteins and one water-transporting protein. All chromosome sequences of Ustilaginoidea virens were downloaded from NCBI. There are seven chromosomes in Ustilaginoidea virens. UvALG32 is located on chromosome 2, and UvALG3 and UvALG15 are located on chromosome 3. Homologous alignment and phylogenetic analysis of UvALG3, UvALG15, and UvALG32 with mannosyltransferases from Fusarium graminearum and Saccharomyces cerevisiae were performed. We found that UvALG3 had the highest homology with S. cerevisiae ALG3 (96%), UvALG15 had the highest homology with S. cerevisiae KTR1 (100%), and UvALG32 had the highest homology with Fusarium graminearum OCH1 (100%).
- (2)
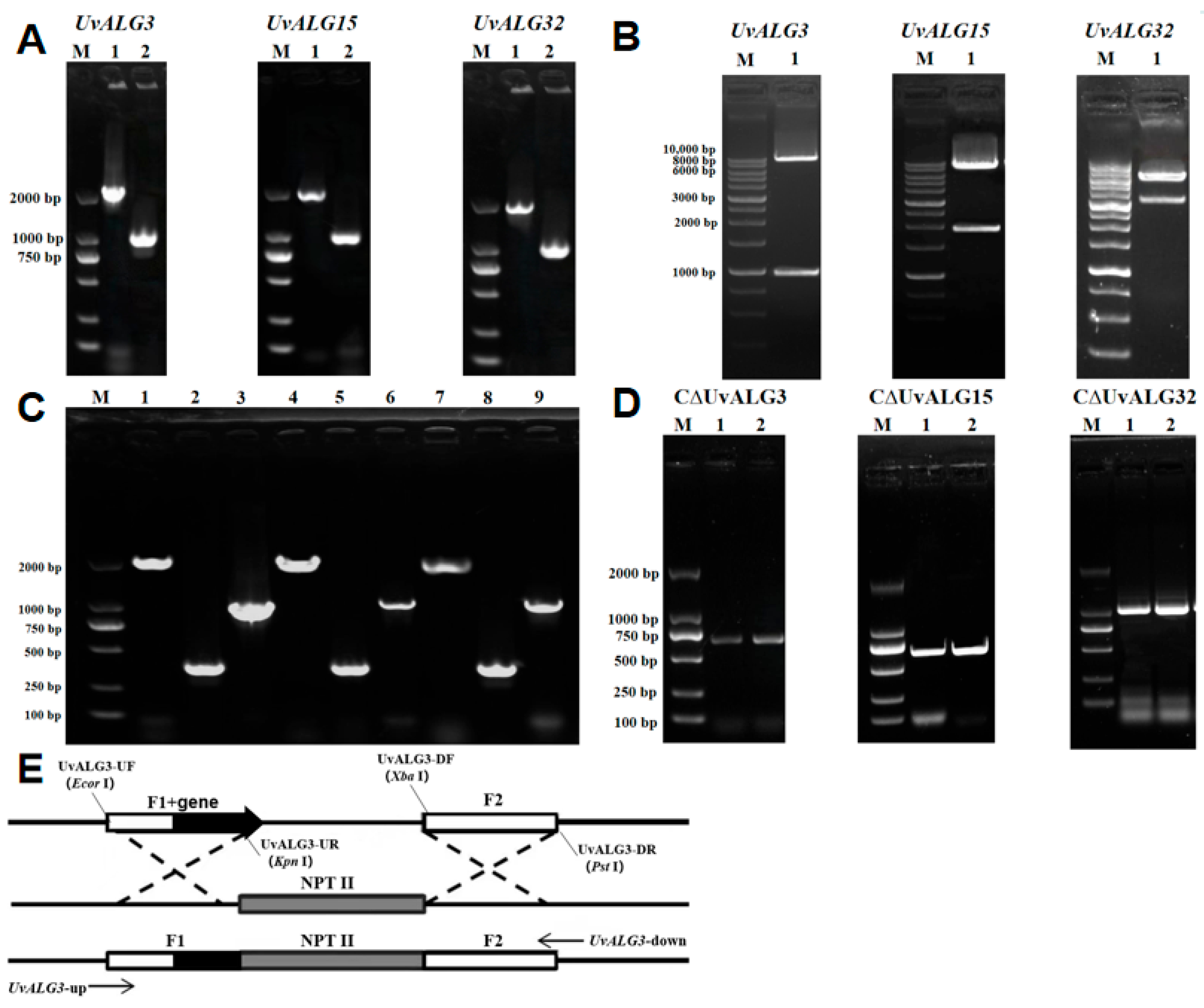
- In order to further study the function of the mannosyltransferase-encoding genes UvALG3, UvALG15, and UvALG32, based on the vector PXEH, the gene knockout vectors PXEHA3, PXEHA15, and PXEHA32 were constructed by Agrobacterium-mediated genetic transformation. The knockout transformants were obtained by Agrobacterium-mediated transformation and verified by PCR experiments. Finally, the knockout mutants ΔUvALG3, ΔUvALG15, and ΔUvALG32 were selected for subsequent experiments. Based on the vector PXNPTII, UvALG3, UvALG15, and UvALG32 complement vectors CPXEHA3, CPXEHA15, and CPXEHA32 were constructed. Using the same techniques and methods as gene knockout, the gene complement mutants CΔUvALG3, CΔUvALG15, and CΔUvALG32 were finally screened.
- (3)
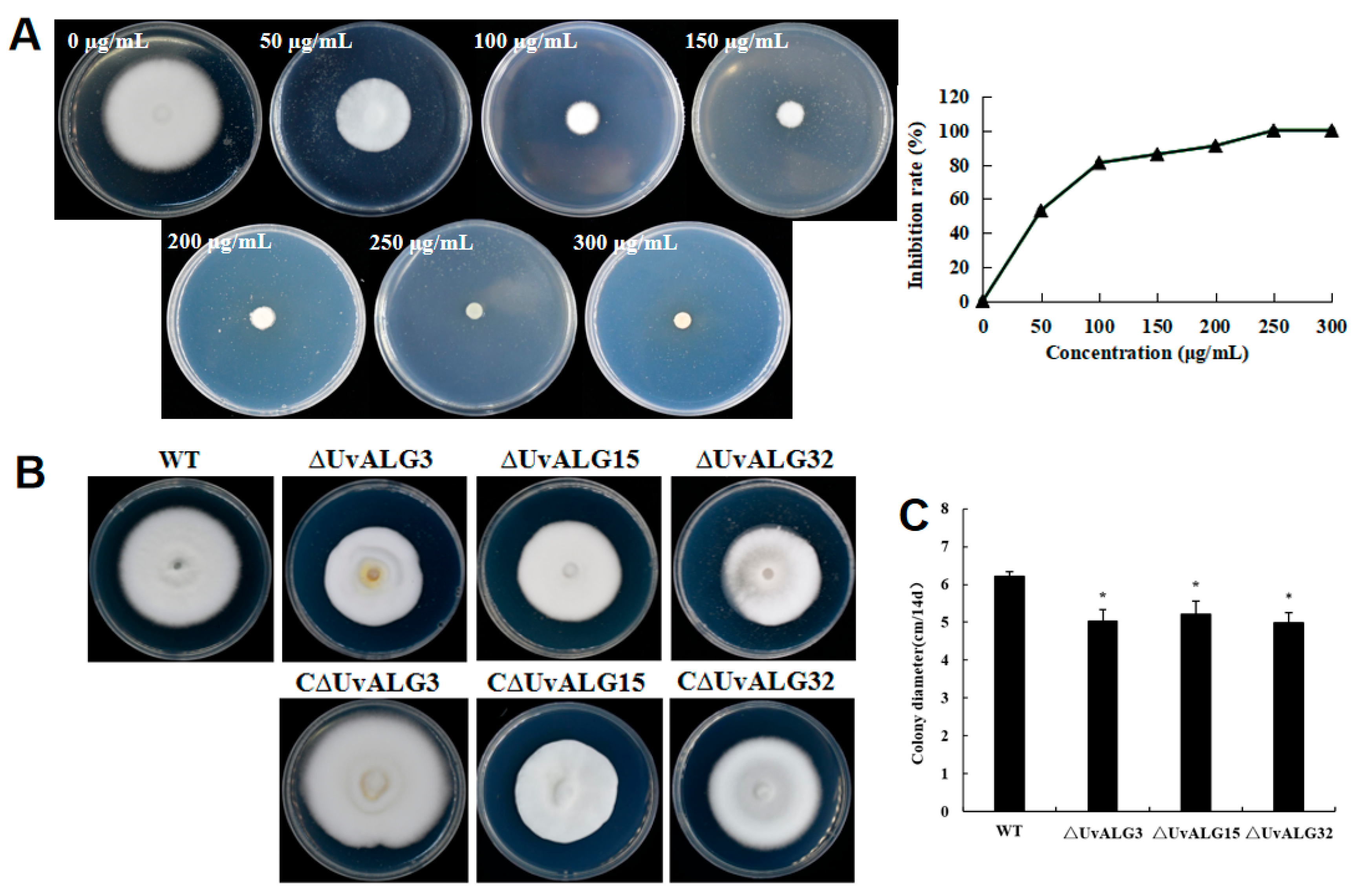
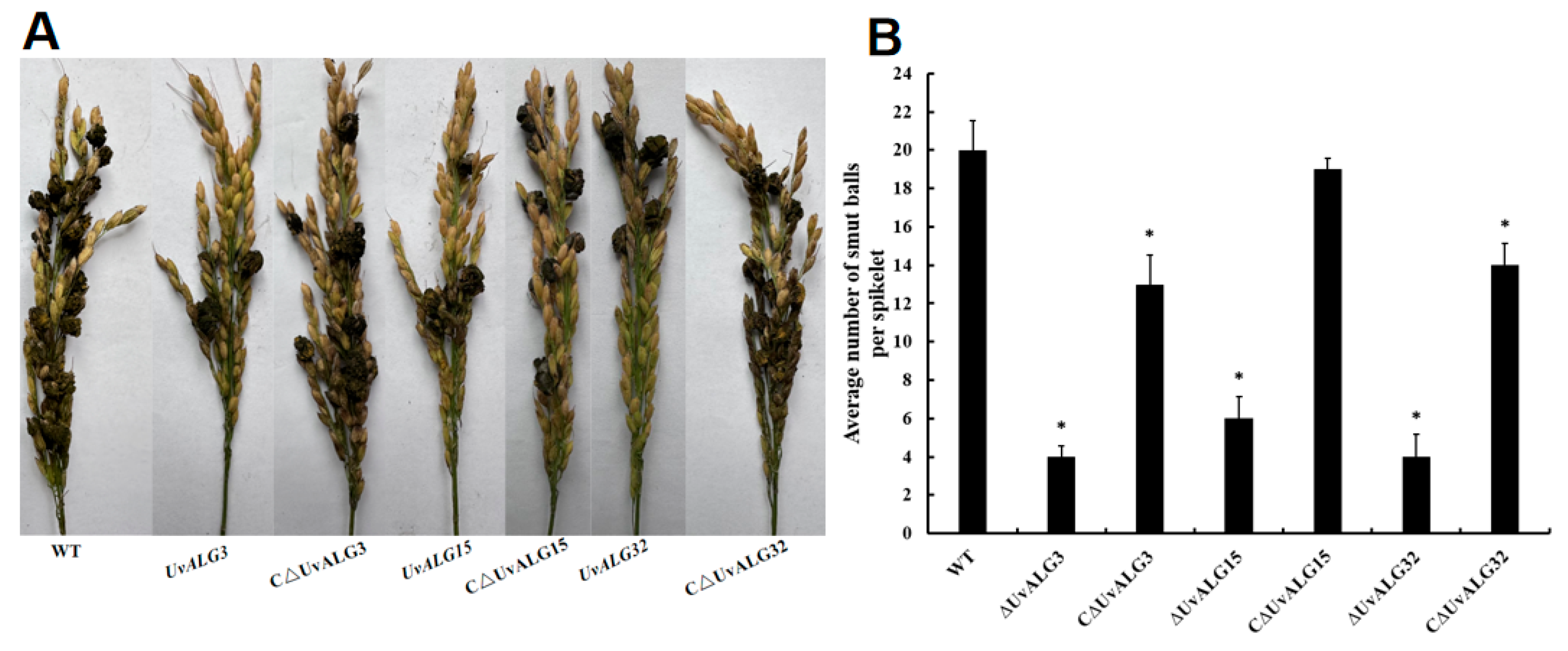
- The biological characteristics such as the growth rate, sporulation, and pathogenicity of knockout and complementation mutants were determined. It was found that the growth rate of U. virens was significantly reduced after the deletion of UvALG3, UvALG15, and UvALG32, indicating that the mannosyltransferase-encoding gene UvALGs plays a certain regulatory role in the vegetative growth of U. virens. There was no significant change in sporulation except for UvALG3, and the pathogenicity was significantly weakened, indicating that these three genes regulate the growth, sporulation, and pathogenicity of U. virens and are the pathogenic genes of the pathogen. Combining the protein domains encoded by these three genes, it was found that their protein domains were similar and their genetic relationships were similar. It is speculated that this unique domain is related to the pathogenicity of pathogens. Subsequently, the structural domains of these three proteins can be studied to further explore the relationship between the structure and function of mannosyltransferase.
2. Results
2.1. Acquisition of Mannosyltransferase Gene of U. virens and Expression Quantity Analysis
2.2. UvALGs’ Annotation and Bioinformatics Analysis
2.3. Isolation and Identification of UvALG Mutants and Complements
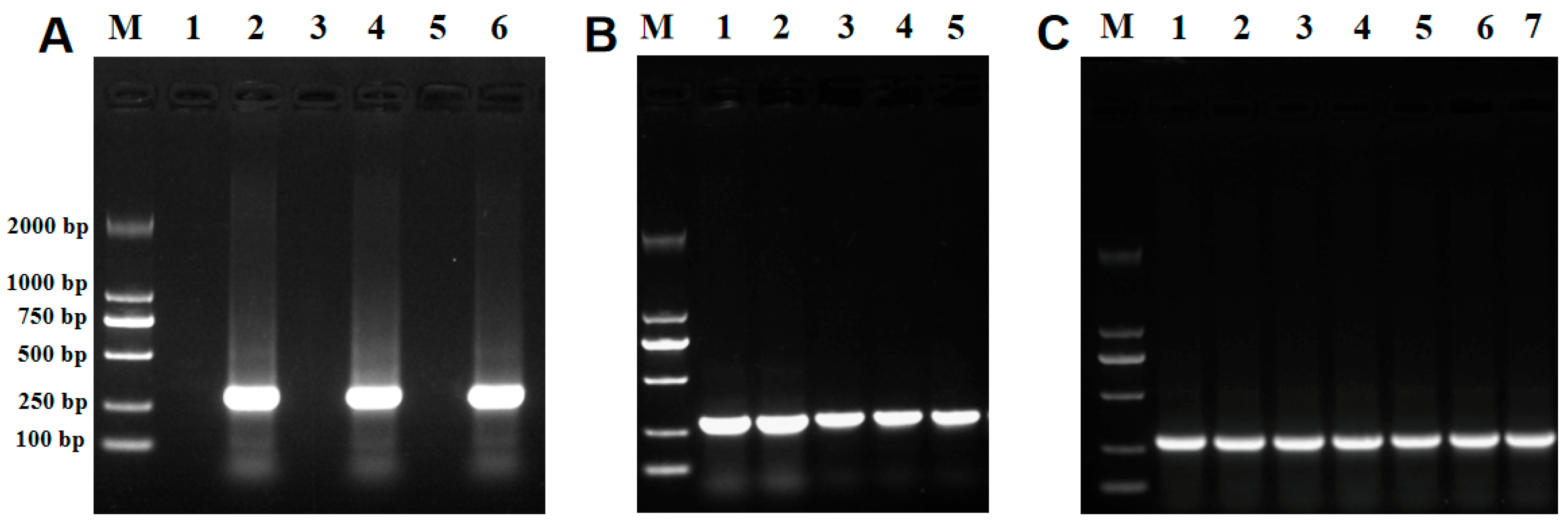
2.3.1. UvALG Mutation and Complementary Transformants Were Verified by RT-PCR
2.3.2. Pathogenicity Determination of Knockout and Complementation Mutant Strains
2.3.3. Sensitivity of the Knockout and Complement Mutant Strains to Different Pressure Stresses Was Determined
3. Discussion
4. Materials and Methods
4.1. Strains and Culture Conditions
4.2. Analysis of UvALGs’ Gene Expression
4.3. Gene Disruption and Complementation of the UvALGs
4.3.1. Acquisition of Flanking Sequences F1 and F2
4.3.2. Linearized Carrier
4.3.3. Gel Recovery and Purification
4.3.4. Carrier Recombination Reaction System and Escherichia Coli Transformation
4.3.5. Plasmid Extraction
4.3.6. Verification of Vector Restriction Enzyme Digestion of the Upstream Fragment
4.3.7. Knockout of the Connection of the Downstream Fragment of the Vector
4.3.8. Validation of Knockout Vectors
4.3.9. Agrobacterium Competent Preparation and Agrobacterium Transformation
4.3.10. Agrobacterium Verification
4.3.11. Screening of the Minimum Tolerance of Ustilaginoidea virens to Hygromycin
4.3.12. Preparation of Conidia of Ustilaginoidea virens
4.3.13. Agrobacterium Mediated Genetic Transformation of Ustilaginoidea virens
4.3.14. RT-PCR Verification of Knockout Mutants and Complemented Mutants
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andargie, M.; Li, J. Expression of the Arabidopsis SWEET genes during rice false smut infection in the transgenic Arabidopsis thaliana containing increased levels of ATP and sucrose. Curr. Plant Biol. 2019, 28, 509–520. [Google Scholar] [CrossRef]
- Andargie, M.; Li, J.X. Arabidopsis thaliana: A model host plant to study plant-pathogen interaction using rice false smut isolates of Ustilaginoidea virens. Front. Plant Sci. 2016, 7, 192. [Google Scholar] [CrossRef]
- Andargie, M.; Li, L.Y.; Feng, A.Q.; Zhu, X.Y.; Li, J.X. Development of a GFP-expressing Ustilaginoidea virens strain to study fungal invasion and colonization in rice spikelets. S. Afr. J. Bot. 2015, 97, 16–24. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, J.Y.; Fang, A.F.; Wang, J.Y.; Li, D.Y.; Li, Y.J.; Wang, S.Z.; Cui, F.H.; Yu, J.J.; Liu, Y.F.; et al. The essential effector SCRE1 in Ustilaginoidea virens suppresses rice immunity via a small peptide region. Mol. Plant Pathol. 2020, 21, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Ladhalakshmi, D.; Srinivas Prasad, M.; Prakasam, V.; Krishnaveni, D.; Sailaja, B.; Ram, S.; Vindeswari, P.; Jagjeet Singh, L.; Jyoti, J.; Surendran, M.; et al. Geographic distribution of false smut disease of rice in India and efficacy of selected fungicides for its management. Int. J. Pest Manag. 2019, 65, 177–185. [Google Scholar] [CrossRef]
- Hiremath, S.S.; Bhatia, D.; Jain, J.; Hunjan, M.S.; Kaur, R.; Zaidi, N.W.; Singh, U.S.; Zhou, B.; Lore, J.S. Identification of potential donors and QTLs for resistance to false smut in a subset of rice diversity panel. Eur. J. Plant Pathol. 2020, 159, 461–470. [Google Scholar] [CrossRef]
- Brooks, S.A.; Anders, M.M.; Yeater, K.M. Effect of furrow irrigation on the severity of false smut in susceptible rice varieties. Plant Dis. 2010, 94, 570–574. [Google Scholar] [CrossRef]
- Fan, L.L.; Yong, M.L.; Li, D.Y.; Liu, Y.J.; Lai, C.H.; Chen, H.M.; Cheng, F.M.; Hu, D.W. Effect of temperature on the development of sclerotia in Villosiclava virens. J. Integr. Agric. 2016, 15, 2550–2555. [Google Scholar] [CrossRef]
- Yong, M.L.; Deng, Q.D.; Fan, L.L.; Miao, J.K. The role of Ustilaginoidea virens sclerotia in increasing incidence of rice false smut disease in the subtropical zone in China. Eur. J. Plant Pathol. 2018, 150, 669–677. [Google Scholar] [CrossRef]
- Singh, A.K.; Pophaly, D.J. An unusual rice false smut epidemic reported in Raigarh District, Chhattisgarh, India. Int. Rice Res. 2010, 35, 1–3. [Google Scholar]
- Wang, X.H.; Wang, J.; Lai, D.W.; Wang, W.X.; Dai, J.G.; Zhou, L.G.; Liu, Y. Ustiloxin G, a new cyclopeptide mycotoxin from rice false smut balls. Toxins 2017, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.X.; Fan, J.; Fang, A.F.; Li, Y.J.; Tariqjaveed, M.; Li, D.Y.; Hu, D.W.; Wang, W.M. Ustilaginoidea virens: Insights into an emerging rice pathogen. Annu. Rev. Phytopathol. 2020, 58, 363–385. [Google Scholar] [CrossRef]
- Koiso, Y.; Li, Y.; Iwasaki, S.; Hanaka, K.; Kobayashi, T.; Sonoda, R.; Yaegashi, H.; Sato, Z. Ustiloxins, antimitotic cyclic peptides from false smut balls on rice panicles caused by Ustilaginoidea virens. J. Antibiot. 1994, 47, 765–773. [Google Scholar] [CrossRef]
- Fan, J.; Yang, J.; Wang, Y.Q.; Li, G.B.; Li, Y.; Huang, F.; Wang, W.M. Current understanding on Villosiclava virens, a unique flower-infecting fungus causing rice false smut disease. Mol. Plant Pathol. 2016, 17, 1321–1330. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef]
- Snider, M.D.; Sultzman, L.A.; Robbins, P.W. Transmembrane location of oligosaccharide-lipid synthesis in microsomal vesicles. Cell 1980, 21, 385–392. [Google Scholar] [CrossRef]
- Helenius, J.; Aebi, M. Transmembrane movement of dolichol linked carbohydrates during N-glycoprotein biosynthesis in the endoplasmic reticulum. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2002; Volume 13, pp. 171–178. [Google Scholar] [CrossRef]
- Huffaker, T.C.; Robbins, P.W. Yeast mutants deficient in protein glycosylation. Proc. Natl. Acad. Sci. USA 1983, 80, 7466–7470. [Google Scholar] [CrossRef]
- Kelleher, D.J.; Gilmore, R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 2006, 16, 47R–62R. [Google Scholar] [CrossRef]
- Dean, N. Asparagine-linked glycosylation in the yeast Golgi. Biochim. Biophys. Acta 1999, 1426, 309–322. [Google Scholar] [CrossRef]
- Roth, J. Protein N-glycosylation along the secretory pathway: Relationship to organelle topography and function, protein quality control and cell interactions. Chem. Rev. 2002, 102, 285–303. [Google Scholar] [CrossRef] [PubMed]
- Lehle, L.; Strahl, S.; Tanner, W. Protein glycosylation, conserved from yeast to man: A model organism helps elucidate congenital human diseases. Angew. Chem. Int. Ed. 2006, 45, 6802–6818. [Google Scholar] [CrossRef]
- Burda, P.; Aebi, M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta 1999, 1426, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Poulain, D.; Jouault, T. Candida albicans cell wall glycans, host receptors and responses: Elements for a decisive crosstalk. Curr. Opin. Microbiol. 2004, 7, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Mora-Montes, H.M.; Bates, S.; Netea, M.G.; Castillo, L.; Brand, A.; Buurman, E.T.; Díaz-Jiménez, D.F.; Jan Kullberg, B.; Brown, A.J.; Odds, F.C.; et al. A multifunctional mannosyltransferase family in Candida albicans determines cell wall mannan structure and host-fungus interactions. J. Biol. Chem. 2010, 285, 12087–12095. [Google Scholar] [CrossRef]
- Hiller, E.; Zavrel, M.; Hauser, N.; Sohn, K.; Burger-Kentischer, A.; Lemuth, K.; Rupp, S. Adaptation, adhesion and invasion during interaction of Candida albicans with the host—Focus on the function of cell wall proteins. Int. J. Med. Microbiol. 2011, 301, 384–389. [Google Scholar] [CrossRef]
- Schirawski, J.; Böhnert, H.U.; Steinberg, G.; Snetselaar, K.; Adamikowa, L.; Kahmann, R. Endoplasmic reticulum glucosidase II is required for pathogenicity of Ustilago maydis. Plant Cell 2005, 17, 3532–3543. [Google Scholar] [CrossRef]
- Motteram, J.; Lovegrove, A.; Pirie, E.; Marsh, J.; Devonshire, J.; van de Meene, A.; Hammond-Kosack, K.; Rudd, J.J. Aberrant protein N-glycosylation impacts upon infection-related growth transitions of the haploid plant-pathogenic fungus Mycosphaerella graminicola. Mol. Microbiol. 2011, 81, 415–433. [Google Scholar]
- Fernández-Álvarez, A.; Elías-Villalobos, A.; Jiménez-Martín, A.; Marín-Menguiano, M.; Ibeas, J.I. Endoplasmic reticulum glucosidases and protein quality control factors cooperate to establish biotrophy in Ustilago maydis. Plant Cell 2013, 25, 4676–4690. [Google Scholar] [CrossRef]
- Hemetsberger, C.; Herrberger, C.; Zechmann, B.; Hillmer, M.; Doehlemann, G. The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathogens 2012, 8, e1002684. [Google Scholar] [CrossRef]
- Horton, R.M. PCR-mediated recombination and mutagenesis. Mol. Biotechnol. 1995, 3, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.C.; Ivatt, R.J. Synthesis and processing of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1981, 50, 555–583. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.J.; Ferrari, M.A.; Roach, D.H.; Money, N.P. Penetration of hard substrates by a fungus employing enormousturgor pressures. Proc. Natl. Acad. Sci. USA 1991, 88, 11281–11284. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Suttapitugsakul, S.; Wu, R. Effective Method for Accurate and Sensitive Quantitation of Rapid Changes of Newly Synthesized Proteins. Anal. Chem. 2020, 92, 10048–10057. [Google Scholar] [CrossRef]
- Yang, R.; He, X.; Niu, G.; Meng, F.; Lu, Q.; Liu, Z.; Yu, X. A Single Fluorescent pH Probe for Simultaneous Two-Color Visualization of Nuclei and Mitochondria and Monitoring Cell Apoptosis. ACS Sens. 2021, 6, 1552–1559. [Google Scholar] [CrossRef]
- Yin, K.; Tong, M.; Sun, F.; Wu, R. Quantitative Structural Proteomics Unveils the Conformational Changes of Proteins under the Endoplasmic Reticulum Stress. Anal. Chem. 2022, 94, 13250–13260. [Google Scholar] [CrossRef]
- Wang, D.; Ma, M.; Huang, J.; Gu, T.-J.; Cui, Y.; Li, M.; Wang, Z.; Zetterberg, H.; Li, L. Boost-DiLeu: Enhanced Isobaric N,N-Dimethyl Leucine Tagging Strategy for a Comprehensive Quantitative Glycoproteomic Analysis. Anal. Chem. 2022, 94, 11773–11782. [Google Scholar] [CrossRef]
- Williams, G.T.; Kedge, J.L.; Fossey, J.S. Molecular Boronic Acid-Based Saccharide Sensors. ACS Sens. 2021, 6, 1508–1528. [Google Scholar] [CrossRef]
- Long, A.; Perraud, O.; Albalat, M.; Robert, V.; Dutasta, J.-P.; Martinez, A. Helical Chirality Induces a Substrate-Selectivity Switch in Carbohydrates Recognitions. J. Org. Chem. 2018, 83, 6301–6306. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Encodes | Annotation | Domain |
|---|---|---|
| Uv8b_07818 | Alpha 1,2 mannosyltransferase | Glyco_transf_15 |
| Uv8b_06540 | Dolichyl-phosphate-mannose-protein | PMT_2 |
| Uv8b_05581 | alpha-1,3/1,6-Mannosyltransferase ALG2 and similar proteins | GT4_ALG2-like |
| Uv8b_04477 | alpha-1,2-Mannosyltransferase | Glyco_transf_15 |
| Uv8b_03749 | Dolichyl-phosphate-mannose-protein mannosyltransferase | PMT_2 |
| Uv8b_03634 | Dolichyl-phosphate-mannose-protein | PMT_2 |
| Uv8b_03269 | Mannosyltransferase OCH1 or related enzyme | OCH1 |
| Uv8b_03119 | alpha 1,2-Mannosyltransferase | Glyco_transf_15 |
| Uv8b_02986 | alpha-1,3-Mannosyltransferase CMT1 | CAP59_mtransfer |
| Uv8b_02466 | Dolichyl-phosphate-mannose-protein | PMT_2 |
| Uv8b_01893 | Mannosyltransferase OCH1 or related enzyme | OCH1 |
| Uv8b_01686 | Dolichyl-phosphate-mannose-protein | PMT_2 |
| Uv8b_01474 | Mannosyltransferase OCH1 or related enzyme | OCH1 |
| Uv8b_01342 | alpha-1,2-Mannosyltransferase ALG11 | GT4_ALG11-like |
| Uv8b_00803 | Dolichol-phosphate-mannosyltransferase subunit 3 (DPM3) | DPM3 |
| Uv8b_00648 | Dolichyl-phosphate-mannose-protein | PMT_2 |
| Uv8b_00068 | Dolichyl-phosphate-beta-D-mannosyltransferase | PLN02726 |
| Uv8b_07818 | alpha-1,2-Mannosyltransferase | Glyco_transf_15 |
| Uv8b_06540 | Dolichyl-phosphate-mannose-protein | PMT_2 |
| Uv8b_05581 | alpha-1,3/1,6-Mannosyltransferase ALG2 | GT4_ALG2-like |
| Uv8b_04467 | alpha-1,2-mannosyltransferase | Glyco_transf_15 |
| Uv8b_03749 | Dolichyl-phosphate-mannose-protein | PMT_2 |
| Uv8b_03269 | Mannosyltransferase OCH1 or related enzyme | OCH1 |
| Uv8b_03119 | alpha-1,2-Mannosyltransferase | Glyco_transf_15 |
| Uv8b_04469 | alpha-1,3-Mannosyltransferase | ALG3 super family |
| Uv8b_02986 | Cryptococcal mannosyltransferase 1 | CAP59_mtransfer |
| UvI_02025670 | Dolichyl-phosphate-mannose-protein | PMT_2 |
| UvI_02035730 | Mannosyltransferase OCH1 or related enzyme | PRK08315 OCH1 |
| UvI_02008980 | alpha-1,2-Mannosyltransferase | Glyco_transf_15 |
| UvI_02020590 | Cryptococcal mannosyltransferase 1 | CAP59_mtransfer |
| UvI_02055610 | alpha-1,2-Mannosyltransferase | Glyco_transf_15 |
| UvI_02045040 | lpha-1,3/1,6-Mmannosyltransferase ALG2 | GT4_ALG2-like |
| UvI_02019520 | Mannosyltransferase OCH1 | OCH1 |
| UvI_02012570 | Dolichol-phosphate mannosyltransferase subunit 3 (DPM3) | DPM3 |
| UvI_02003470 | alpha-1,2-Mannosyltransferase | Glyco_transf_15 |
| UvI_02058330 | alpha-1,2-Mannosyltransferase ALG11 | GT4_ALG11-like |
| UvI_02029900 | Mannosyltransferase OCH1 or related enzyme | OCH1 |
| UvI_02060380 | Dolichyl-phosphate beta-D-mannosyltransferase | PLN02726 |
| Gene | Orthologous Genes | Base Number | Intron | Number of AA |
|---|---|---|---|---|
| UvALG3 | FgALG3 | 1416 | 2 | 471 |
| UvALG15 | Sc PE-2 | 1248 | 2 | 415 |
| UvALG32 | ScOCH1 | 1101 | 1 | 366 |
| Locus ID | Gene Name | Amino Acid Length/aa | Molecular Weight/kDa | Theoretical PI | Instability Index | Grand Average of Hydropathicity |
|---|---|---|---|---|---|---|
| >XP_042997901.1 | UvALG3 | 435 | 48,986.95 | 9.81 | 33.75 | 0.426 |
| >XP_042997909.1 | UvALG15 | 415 | 48,270.18 | 5.92 | 40.7 | −0.555 |
| >XP_042995325.1 | UvALG32 | 366 | 41,467.94 | 5.76 | 40.14 | −3.634 |
| Primer Name | Primer Sequence 5′-3′ |
|---|---|
| β- tubulin F | TCGTCGATTTGGAGCCTGGT |
| β- tubulin R | ATCTTGGAGATGAGCAAGGT |
| UvALG3-F | TCTAGACCTACTGCAGCATA |
| UvALG3-R | ACCAGAAGCAACGATTCTAT |
| UvALG15-F | ATTTCGCTGCTTCACGGCCT |
| UvALG15-R | GCTGAAGGAGCCATTGTAAA |
| UvALG32-F | TGATAATGACCCGATGGATT |
| UvALG32-R | ATTGTGATGATGACTCCCTT |
| Vector | Restriction Enzyme | Fragment (bp) |
|---|---|---|
| PXEHA3 | Xba I and Hind III | 975 + 9020 |
| PXEHA15 | Ecor I and Xba I | 3495 + 6887 |
| PXEHA32 | Kpn I and Hind III | 3425 + 6962 |
| Primers | Fragment (bp) |
|---|---|
| UvALG3-UF/UvALG3-UR | 771 |
| UvALG3-DF/UvALG3-DR | 969 |
| UvALG15-UF/UvALG15-UR | 1058 |
| UvALG15-DF/UvALG15-DR | 1075 |
| UvALG32-UF/UvALG32-UR | 1136 |
| UvALG32-DF/UvALG32-DR | 1131 |
| Hyg-1/Hyg-2 | 622 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Zhang, Y.; Qu, L.; Zhou, Z.; Zhai, H.; Wei, S.; Wang, Y. Functional Analysis of Mannosyltransferase-Related Genes UvALGs in Ustilaginoidea virens. Int. J. Mol. Sci. 2025, 26, 2979. https://doi.org/10.3390/ijms26072979
Wang S, Zhang Y, Qu L, Zhou Z, Zhai H, Wei S, Wang Y. Functional Analysis of Mannosyltransferase-Related Genes UvALGs in Ustilaginoidea virens. International Journal of Molecular Sciences. 2025; 26(7):2979. https://doi.org/10.3390/ijms26072979
Chicago/Turabian StyleWang, Shilong, Yating Zhang, Lili Qu, Zengran Zhou, Hongyang Zhai, Songhong Wei, and Yan Wang. 2025. "Functional Analysis of Mannosyltransferase-Related Genes UvALGs in Ustilaginoidea virens" International Journal of Molecular Sciences 26, no. 7: 2979. https://doi.org/10.3390/ijms26072979
APA StyleWang, S., Zhang, Y., Qu, L., Zhou, Z., Zhai, H., Wei, S., & Wang, Y. (2025). Functional Analysis of Mannosyltransferase-Related Genes UvALGs in Ustilaginoidea virens. International Journal of Molecular Sciences, 26(7), 2979. https://doi.org/10.3390/ijms26072979