
Interferon Regulatory Factors as a Potential Therapeutic Target for Neuroinflammation: A Focus on Alzheimer’s Disease
Abstract
1. Introduction
2. AD and Neuroinflammation
3. IRFs and Neuroinflammation in AD
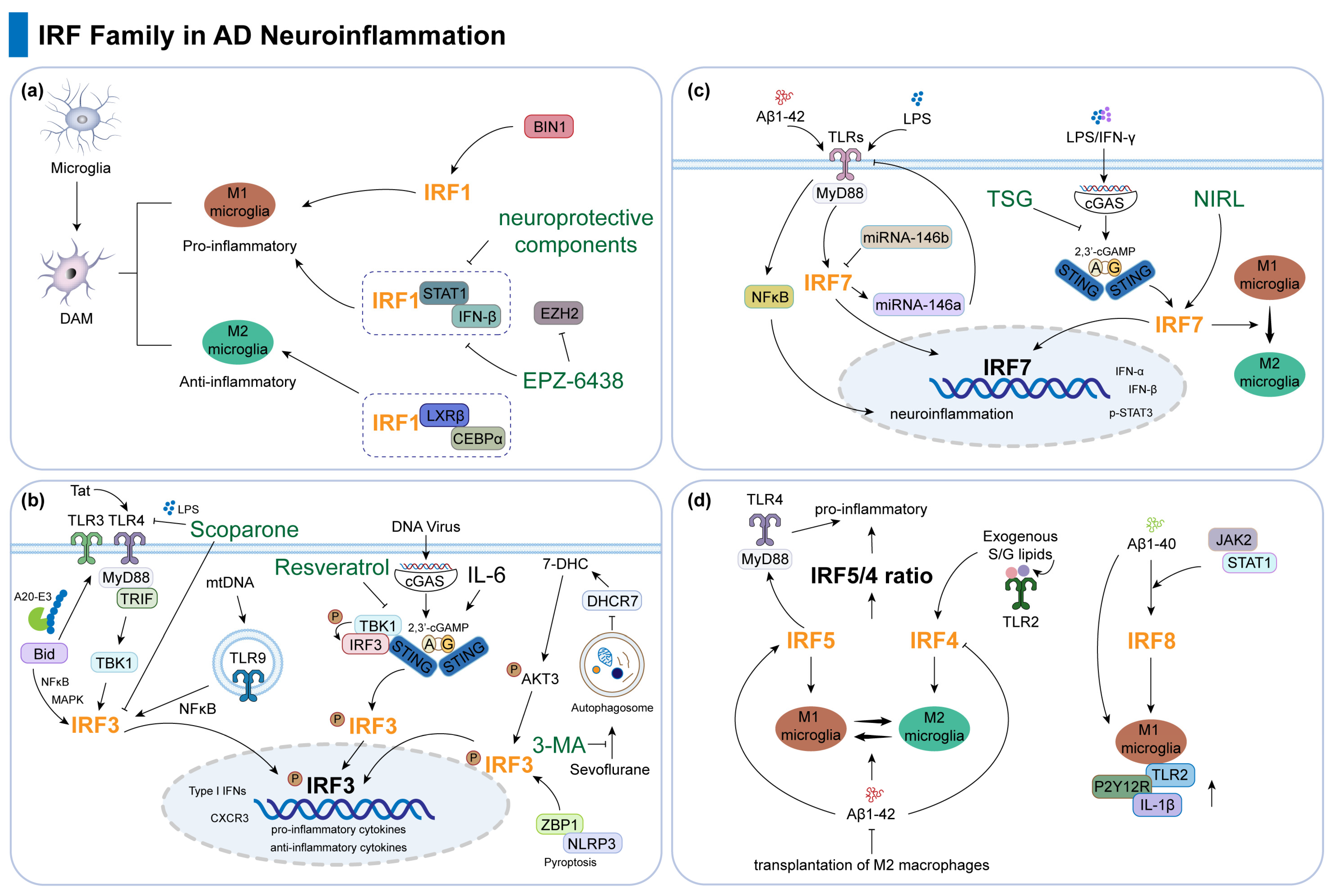
3.1. IRF1
3.2. IRF3
3.3. IRF7
3.4. Other IRFs
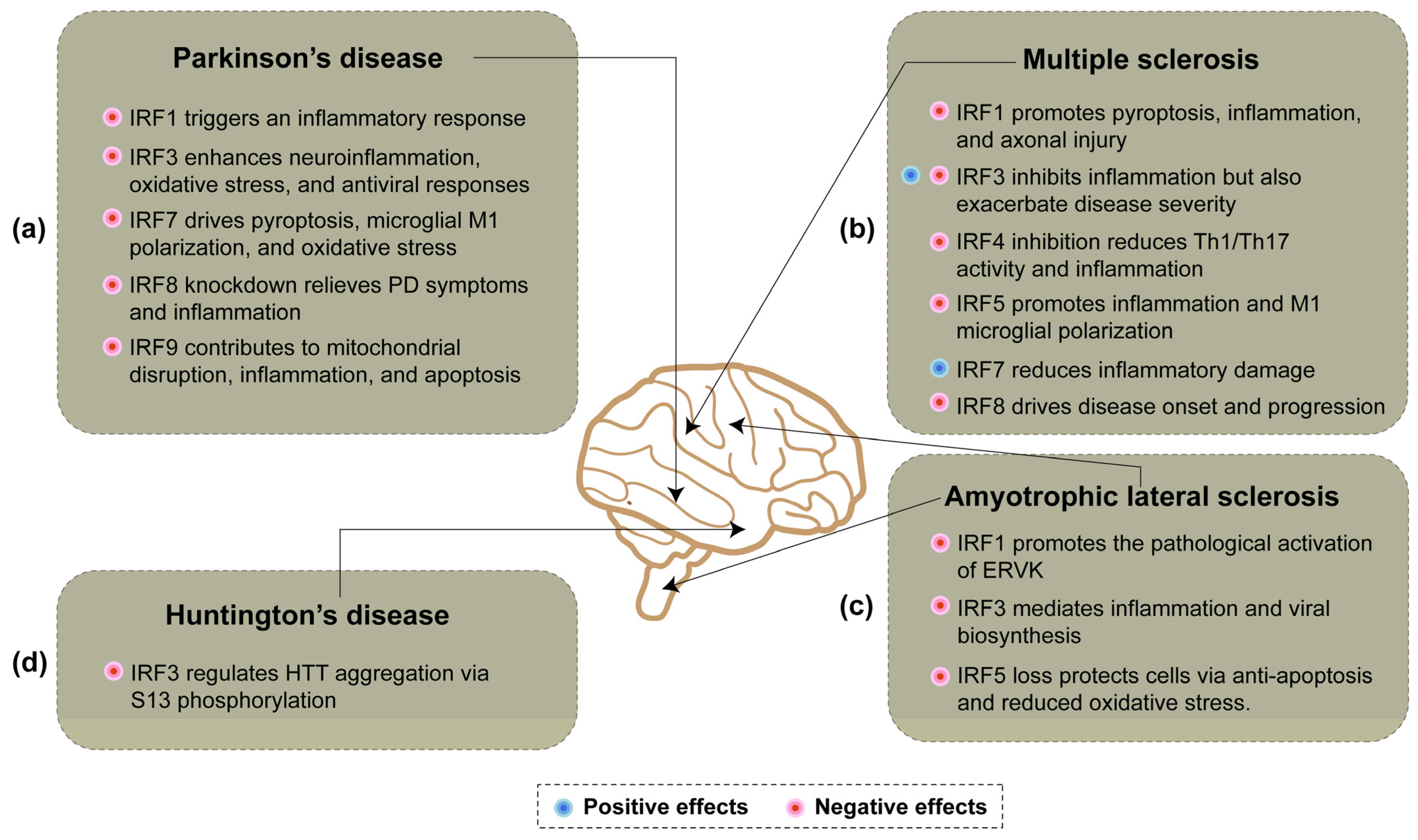
4. IRFs and Neuroinflammation in Neurodegenerative Diseases
4.1. Parkinson’s Disease
4.2. Multiple Sclerosis
4.3. Amyotrophic Lateral Sclerosis
4.4. Huntington’s Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Cataldi, R.; Sachdev, P.S.; Chowdhary, N.; Seeher, K.; Bentvelzen, A.; Moorthy, V.; Dua, T. A WHO blueprint for action to reshape dementia research. Nat. Aging 2023, 3, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Counts, N.; Chen, S.; Seligman, B.; Tortorice, D.; Vigo, D.; Bloom, D.E. Global and regional projections of the economic burden of Alzheimer’s disease and related dementias from 2019 to 2050: A value of statistical life approach. eClinicalMedicine 2022, 51, 101580. [Google Scholar] [CrossRef] [PubMed]
- Better, M.A. 2024 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2024, 20, 3708–3821. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, Y.; Zhang, N.; Xian, Y.; Tang, Y.; Ye, J.; Reza, F.; He, G.; Wen, X.; Jiang, X. The multiple roles of interferon regulatory factor family in health and disease. Signal Transduct. Target. Ther. 2024, 9, 282. [Google Scholar] [CrossRef]
- Nguyen, H.; Hiscott, J.; Pitha, P.M. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997, 8, 293–312. [Google Scholar] [CrossRef]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef]
- Amanollahi, M.; Jameie, M.; Heidari, A.; Rezaei, N. The Dialogue Between Neuroinflammation and Adult Neurogenesis: Mechanisms Involved and Alterations in Neurological Diseases. Mol. Neurobiol. 2022, 60, 923–959. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef]
- Botella Lucena, P.; Heneka, M.T. Inflammatory aspects of Alzheimer’s disease. Acta Neuropathol. 2024, 31, 148. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef]
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11, 1925. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef]
- Barczuk, J.; Siwecka, N.; Lusa, W.; Rozpedek-Kaminska, W.; Kucharska, E.; Majsterek, I. Targeting NLRP3-Mediated Neuroinflammation in Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2022, 23, 8979. [Google Scholar] [CrossRef]
- Li, Y.; Xu, H.; Wang, H.; Yang, K.; Luan, J.; Wang, S. TREM2: Potential therapeutic targeting of microglia for Alzheimer’s disease. Biomed. Pharmacother. 2023, 165, 115218. [Google Scholar] [CrossRef]
- Govindarajulu, M.; Ramesh, S.; Beasley, M.; Lynn, G.; Wallace, C.; Labeau, S.; Pathak, S.; Nadar, R.; Moore, T.; Dhanasekaran, M. Role of cGAS-Sting Signaling in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 8151. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Yu, W.S.; Lim, L.W. Exploring ER stress response in cellular aging and neuroinflammation in Alzheimer’s disease. Ageing Res. Rev. 2021, 70, 101417. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wei, Y.; Qian, Z.; Han, L. Autophagy Balances Neuroinflammation in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2023, 43, 1537–1549. [Google Scholar] [CrossRef]
- Mangalmurti, A.; Lukens, J.R. How neurons die in Alzheimer’s disease: Implications for neuroinflammation. Curr. Opin. Neurobiol. 2022, 75, 102575. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Y. Tau and neuroinflammation in Alzheimer’s disease: Interplay mechanisms and clinical translation. J. Neuroinflammation 2023, 20, 165. [Google Scholar] [CrossRef]
- Parhizkar, S.; Holtzman, D.M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 2022, 59, 101594. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Contreras, K.A.; Martinez-Diaz, J.A.; Hernandez-Aguilar, M.E.; Herrera-Covarrubias, D.; Rojas-Duran, F.; Chi-Castaneda, L.D.; Garcia-Hernandez, L.I.; Aranda-Abreu, G.E. Alterations of mRNAs and Non-coding RNAs Associated with Neuroinflammation in Alzheimer’s Disease. Mol. Neurobiol. 2024, 61, 5826–5840. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, W.; Liu, S.; Qiao, X.; Xing, Y.; Zhou, Q.; Zhang, Z. Epigenetic Regulation of Neuroinflammation in Alzheimer’s Disease. Cells 2023, 13, 79. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, H.; Liang, J.; Huang, J.; Chen, N. Exercise suppresses neuroinflammation for alleviating Alzheimer’s disease. J. Neuroinflammation 2023, 20, 76. [Google Scholar] [CrossRef]
- Grant, W.B.; Blake, S.M. Diet’s Role in Modifying Risk of Alzheimer’s Disease: History and Present Understanding. J. Alzheimer’s Dis. 2023, 96, 1353–1382. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, D.D.; Huang, S.Y.; Fang, A.P.; Li, H.B.; Zhu, H.L. Effects and mechanisms of natural products on Alzheimer’s disease. Crit. Rev. Food Sci. Nutr. 2023, 63, 3168–3188. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.L. Gut Microbiota and Dysbiosis in Alzheimer’s Disease: Implications for Pathogenesis and Treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef]
- Gao, T.; Jernigan, J.; Raza, S.A.; Dammer, E.B.; Xiao, H.; Seyfried, N.T.; Levey, A.I.; Rangaraju, S. Transcriptional regulation of homeostatic and disease-associated-microglial genes by IRF1, LXRβ, and CEBPα. Glia 2019, 67, 1958–1975. [Google Scholar] [CrossRef]
- Sudwarts, A.; Ramesha, S.; Gao, T.; Ponnusamy, M.; Wang, S.; Hansen, M.; Kozlova, A.; Bitarafan, S.; Kumar, P.; Beaulieu-Abdelahad, D.; et al. BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia. Mol. Neurodegener. 2022, 17, 33. [Google Scholar] [CrossRef]
- Arifuzzaman, S.; Das, A.; Kim, S.H.; Yoon, T.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. Selective inhibition of EZH2 by a small molecule inhibitor regulates microglial gene expression essential for inflammation. Biochem. Pharmacol. 2017, 137, 61–80. [Google Scholar] [CrossRef]
- Jantaratnotai, N.; Utaisincharoen, P.; Sanvarinda, P.; Thampithak, A.; Sanvarinda, Y. Phytoestrogens mediated anti-inflammatory effect through suppression of IRF-1 and pSTAT1 expressions in lipopolysaccharide-activated microglia. Int. Immunopharmacol. 2013, 17, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Choi, J.J.; Park, B.-K.; Yoon, S.J.; Choi, J.E.; Jin, M. Pheophytin a and chlorophyll a suppress neuroinflammatory responses in lipopolysaccharide and interferon-γ-stimulated BV2 microglia. Life Sci. 2014, 103, 59–67. [Google Scholar] [CrossRef]
- Hara, H.; Ikeda, R.; Ninomiya, M.; Kamiya, T.; Koketsu, M.; Adachi, T. Newly synthesized ’hidabeni’ chalcone derivatives potently suppress LPS-induced NO production via inhibition of STAT1, but not NF-κB, JNK, and p38, pathways in microglia. Biol. Pharm. Bull. 2014, 37, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Tang, T.M.; Lue, L.-F. Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer’s disease brains. Exp. Neurol. 2018, 309, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, S.; Fichtner, M.; Watters, O.; König, H.-G.; Prehn, J.H.M. Increased A20-E3 ubiquitin ligase interactions in bid-deficient glia attenuate TLR3- and TLR4-induced inflammation. J. Neuroinflammation 2018, 15, 130. [Google Scholar] [CrossRef]
- Niu, F.; Liao, K.; Hu, G.; Moidunny, S.; Roy, S.; Buch, S. HIV Tat-Mediated Induction of Monocyte Transmigration Across the Blood–Brain Barrier: Role of Chemokine Receptor CXCR3. Front. Cell Dev. Biol. 2021, 9, 724970. [Google Scholar] [CrossRef]
- Cho, D.-Y.; Ko, H.; Kim, J.; Kim, B.-W.; Yun, Y.-S.; Park, J.-I.; Ganesan, P.; Lee, J.-T.; Choi, D.-K. Scoparone Inhibits LPS-Simulated Inflammatory Response by Suppressing IRF3 and ERK in BV-2 Microglial Cells. Molecules 2016, 21, 1718. [Google Scholar] [CrossRef]
- Austad, S.N.; Ballinger, S.; Buford, T.W.; Carter, C.S.; Smith, D.L.; Darley-Usmar, V.; Zhang, J. Targeting whole body metabolism and mitochondrial bioenergetics in the drug development for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 511–531. [Google Scholar] [CrossRef]
- Xu, Q.; Xu, W.; Cheng, H.; Yuan, H.; Tan, X. Efficacy and mechanism of cGAMP to suppress Alzheimer’s disease by elevating TREM2. Brain Behav. Immun. 2019, 81, 495–508. [Google Scholar] [CrossRef]
- Wang, Y.; Niu, W.; Zhu, S.; Sun, J.; Lv, J.; Wang, N.; Zhang, H.; Zhang, Z.; Wang, M.; Cao, L.; et al. STING Agonist cGAMP Attenuates Sleep Deprivation-Induced Neuroinflammation and Cognitive Deficits via TREM2 Up-Regulation. Inflammation 2024, 47, 2129–2144. [Google Scholar] [CrossRef]
- Kang, N.; Shi, Y.; Song, J.; Gao, F.; Fan, M.; Jin, W.; Gao, Y.; Lv, P. Resveratrol reduces inflammatory response and detrimental effects in chronic cerebral hypoperfusion by down-regulating stimulator of interferon genes/TANK-binding kinase 1/interferon regulatory factor 3 signaling. Front. Aging Neurosci. 2022, 14, 868484. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pan, J.; Li, X.; Zhang, X.; Tian, F.; Li, M.; Wu, X.; Zhang, L.; Qin, C. Interleukin-6 deficiency reduces neuroinflammation by inhibiting the STAT3-cGAS-STING pathway in Alzheimer’s disease mice. J. Neuroinflammation 2024, 21, 282. [Google Scholar] [CrossRef]
- Hou, Q.; Yuan, J.; Li, S.; Ma, J.; Li, W.; Zhang, B.; Zhao, X.; Zhang, F.; Ma, Y.; Zheng, H.; et al. Autophagic degradation of DHCR7 activates AKT3 and promotes sevoflurane-induced hippocampal neuroinflammation in neonatal mice. Free. Radic. Biol. Med. 2024, 222, 304–316. [Google Scholar] [CrossRef]
- Guo, H.; Chen, R.; Li, P.; Yang, Q.; He, Y. ZBP1 mediates the progression of Alzheimer’s disease via pyroptosis by regulating IRF3. Mol. Cell. Biochem. 2023, 478, 2849–2860. [Google Scholar] [CrossRef]
- Romagnoli, M.; Porcellini, E.; Carbone, I.; Veerhuis, R.; Licastro, F. Impaired Innate Immunity Mechanisms in the Brain of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1126. [Google Scholar] [CrossRef]
- Zhao, G.N.; Jiang, D.S.; Li, H. Interferon regulatory factors: At the crossroads of immunity, metabolism, and disease. Biochim. Biophys. Acta. 2015, 1852, 365–378. [Google Scholar] [CrossRef]
- Fan, Z.; Zhang, X.; Zhao, S.; Zhong, S.; Li, Z.; Yan, Y.; Zhang, B.; Chen, Y. Interferon Regulatory Factor 5 Regulates the Phagocytosis of Microglia and Alleviate Alzheimer’s Pathology. J. Gerontol. A Biol. Sci. Med. Sci. 2024, 79, glae031. [Google Scholar] [CrossRef] [PubMed]
- Korvatska, O.; Kiianitsa, K.; Ratushny, A.; Matsushita, M.; Beeman, N.; Chien, W.M.; Satoh, J.I.; Dorschner, M.O.; Keene, C.D.; Bammler, T.K.; et al. Triggering Receptor Expressed on Myeloid Cell 2 R47H Exacerbates Immune Response in Alzheimer’s Disease Brain. Front. Immunol. 2020, 11, 559342. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Main, B.S.; Brody, K.M.; Zhang, M.; Taylor, J.M.; Crack, P.J. Soluble amyloid triggers a myeloid differentiation factor 88 and interferon regulatory factor 7 dependent neuronal type-1 interferon response in vitro. J. Neuroinflammation 2015, 12, 71. [Google Scholar] [CrossRef]
- Chithanathan, K.; Jurgenson, M.; Guha, M.; Yan, L.; Zarkovskaja, T.; Pook, M.; Magilnick, N.; Boldin, M.P.; Rebane, A.; Tian, L.; et al. Paradoxical attenuation of neuroinflammatory response upon LPS challenge in miR-146b deficient mice. Front. Immunol. 2022, 13, 996415. [Google Scholar] [CrossRef]
- Gao, D.; Hao, J.P.; Li, B.Y.; Zheng, C.C.; Miao, B.B.; Zhang, L.; Li, Y.L.; Li, L.; Li, X.J.; Zhang, L. Tetrahydroxy stilbene glycoside ameliorates neuroinflammation for Alzheimer’s disease via cGAS-STING. Eur. J. Pharmacol. 2023, 953, 175809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Wang, Z.; Chen, J.W.; Yuan, S.Y.; Zhao, L.; Zhong, J.Y.; Chen, J.J.; Lin, W.J.; Wu, W.S. The neuroprotective effect of near infrared light therapy in aged mice with postoperative neurocognitive disorder by upregulating IRF7. J. Affect. Disord. 2024, 349, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Yang, N.; Liu, Y.Y.; Zheng, J.; Ji, C.; Zuo, P.P. M2 Macrophage Transplantation Ameliorates Cognitive Dysfunction in Amyloid-beta-Treated Rats Through Regulation of Microglial Polarization. J. Alzheimer’s. Dis. 2016, 52, 483–495. [Google Scholar] [CrossRef]
- Mamun, A.A.; Liu, F. Role of IRF4-Mediated Inflammation: Implication in Neurodegenerative Diseases. Neurol. Neurother. Open Access. J. 2017, 2, 000107. [Google Scholar] [CrossRef]
- Mihori, S.; Nichols, F.; Provatas, A.; Matz, A.; Zhou, B.; Blesso, C.N.; Panier, H.; Daddi, L.; Zhou, Y.; Clark, R.B. Microbiome-derived bacterial lipids regulate gene expression of proinflammatory pathway inhibitors in systemic monocytes. Front. Immunol. 2024, 15, 1415565. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Zeng, Q.; Man, R.; Luo, Y.; Zeng, L.; Zhong, Y.; Lu, B.; Wang, X. IRF-8 is Involved in Amyloid-beta(1-40) (Abeta(1-40))-induced Microglial Activation: A New Implication in Alzheimer’s Disease. J. Mol. Neurosci. 2017, 63, 159–164. [Google Scholar] [CrossRef]
- Tarassishin, L.; Bauman, A.; Suh, H.-S.; Lee, S.C. Anti-Viral and Anti-Inflammatory Mechanisms of the Innate Immune Transcription Factor Interferon Regulatory Factor 3: Relevance to Human CNS Diseases. J. Neuroimmune Pharmacol. 2012, 8, 132–144. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Perry, G.; Rezaei, N. Toll-like receptors in Alzheimer’s disease. J. Neuroimmunol. 2020, 348, 577362. [Google Scholar] [CrossRef]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS–STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef]
- Ferecskó, A.S.; Smallwood, M.J.; Moore, A.; Liddle, C.; Newcombe, J.; Holley, J.; Whatmore, J.; Gutowski, N.J.; Eggleton, P. STING-Triggered CNS Inflammation in Human Neurodegenerative Diseases. Biomedicines 2023, 11, 1375. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhang, H.; Xia, M.; Gu, J.; Guo, K.; Wang, H.; Miao, C. Programmed Death of Microglia in Alzheimer’s Disease: Autophagy, Ferroptosis, and Pyroptosis. J. Prev. Alzheimer’s Dis. 2023, 10, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448.e438. [Google Scholar] [CrossRef]
- Joshi, R.; Brezani, V.; Mey, G.M.; Guixe-Muntet, S.; Ortega-Ribera, M.; Zhuang, Y.; Zivny, A.; Werneburg, S.; Gracia-Sancho, J.; Szabo, G. IRF3 regulates neuroinflammatory responses and the expression of genes associated with Alzheimer’s disease. J. Neuroinflammation 2024, 21, 212. [Google Scholar] [CrossRef]
- Yan, C.; Luo, Z.; Li, W.; Li, X.; Dallmann, R.; Kurihara, H.; Li, Y.F.; He, R.R. Disturbed Yin-Yang balance: Stress increases the susceptibility to primary and recurrent infections of herpes simplex virus type 1. Acta. Pharm. Sin. B 2020, 10, 383–398. [Google Scholar] [CrossRef]
- Valencia-Sanchez, S.; Davis, M.; Martensen, J.; Hoeffer, C.; Link, C.; Opp, M.R. Sleep-wake behavior and responses to sleep deprivation and immune challenge of protein kinase RNA-activated knockout mice. Brain Behav. Immun. 2024, 121, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Guo, Y.; Xu, X.; Zhao, L.; Shen, X.; Sun, L.; Xie, P. BoDV-1 infection induces neuroinflammation by activating the TLR4/MyD88/IRF5 signaling pathway, leading to learning and memory impairment in rats. J. Med. Virol. 2021, 93, 6163–6171. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.G.; Savitt, J.M. Parkinson’s Disease. Med. Clin. N. Am. 2019, 103, 337–350. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef]
- Herrero, M.-T.; Estrada, C.; Maatouk, L.; Vyas, S. Inflammation in Parkinson’s disease: Role of glucocorticoids. Front. Neuroanat. 2015, 9, 00032. [Google Scholar] [CrossRef]
- Yunfu, W.; Guangjian, L.; Ping, Z.; Yanpeng, S.; Xiaoxia, F.; Wei, H.; Jiang, Y.; Jingquan, H.; Songlin, W.; Hongyan, Z.; et al. PINK1 and Its Familial Parkinson’s Disease-Associated Mutation Regulate Brain Vascular Endothelial Inflammation. J. Mol. Neurosci. 2014, 53, 109–116. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, R.; Zhang, Z.; Liu, Q.; Zhang, Y.; Wang, Q.; Yuan, H. Mitochondrial Protein PINK1 Positively Regulates RLR Signaling. Front. Immunol. 2019, 10, 01069. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, X.; He, X.; Wang, Y.; Gao, S.; Ren, L.; Shi, Y. Curcumin exerts anti-inflammatory and antioxidative properties in 1-methyl-4-phenylpyridinium ion (MPP+)-stimulated mesencephalic astrocytes by interference with TLR4 and downstream signaling pathway. Cell Stress Chaperones 2016, 21, 697–705. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Wang, C.; Liu, W.; Hayashi, T.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Ikejima, T. Silibinin ameliorates depression/anxiety-like behaviors of Parkinson’s disease mouse model and is associated with attenuated STING-IRF3-IFN-β pathway activation and neuroinflammation. Physiol. Behav. 2021, 241, 113593. [Google Scholar] [CrossRef] [PubMed]
- Rui, W.J.; Li, S.; Yang, L.; Liu, Y.; Fan, Y.; Hu, Y.C.; Ma, C.M.; Wang, B.W.; Shi, J.P. Microglial AIM2 alleviates antiviral-related neuro-inflammation in mouse models of Parkinson’s disease. Glia 2022, 70, 2409–2425. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Liu, Y.; Li, J.; Chen, D.; Xu, J.; Song, J.; Chen, J.; Sun, S. Investigating the TLR4/TAK1/IRF7 axis in NLRP3-Mediated Pyroptosis in Parkinson’s Disease. Inflammation 2023, 47, 404–420. [Google Scholar] [CrossRef]
- Zhou, S.; Li, T.; Zhang, W.; Wu, J.; Hong, H.; Quan, W.; Qiao, X.; Cui, C.; Qiao, C.; Zhao, W.; et al. The cGAS-STING-interferon regulatory factor 7 pathway regulates neuroinflammation in Parkinson’s disease. Neural Regen. Res. 2025, 20, 2361–2372. [Google Scholar] [CrossRef]
- Xing, X.; Xu, F.; Wang, Y.; Liu, H. Role of the OTUB1/IRF7/NOX4 axis in oxidative stress injury and inflammatory responses in mice with Parkinson’s disease. Psychogeriatrics 2022, 23, 32–44. [Google Scholar] [CrossRef]
- Ma, L.; Mi, N.; Wang, Z.; Bao, R.; Fang, J.; Ren, Y.; Xu, X.; Zhang, H.; Tang, Y. Knockdown of IRF8 alleviates neuroinflammation through regulating microglial activation in Parkinson’s disease. J. Chem. Neuroanat. 2024, 138, 102424. [Google Scholar] [CrossRef]
- Quan, P.; Li, X.; Si, Y.; Sun, L.; Ding, F.F.; Fan, Y.; Liu, H.; Wei, C.; Li, R.; Zhao, X.; et al. Single cell analysis reveals the roles and regulatory mechanisms of type-I interferons in Parkinson’s disease. Cell Commun. Signal. 2024, 22, 212. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Zhou, F.; Li, J.; Lu, G.; Zhao, Y.; Wang, Y. MiR-20a-5p Regulates MPP+-Induced Oxidative Stress and Neuroinflammation in HT22 Cells by Targeting IRF9/NF-κB Axis. Evid. Based Complement. Altern. Med. 2021, 2021, 6621206. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Tichauer, J.E.; Arellano, G.; Acuna, E.; Gonzalez, L.F.; Kannaiyan, N.R.; Murgas, P.; Panadero-Medianero, C.; Ibanez-Vega, J.; Burgos, P.I.; Loda, E.; et al. Interferon-gamma ameliorates experimental autoimmune encephalomyelitis by inducing homeostatic adaptation of microglia. Front. Immunol. 2023, 14, 1191838. [Google Scholar] [CrossRef]
- Loda, E.; Balabanov, R. Interferon regulatory factor 1 regulation of oligodendrocyte injury and inflammatory demyelination. Rev. Neurosci. 2012, 23, 145–152. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, Y.; Liebenson, D.; Liggett, T.; Goswami, R.; Stefoski, D.; Balabanov, R. IRF-1 signaling in central nervous system glial cells regulates inflammatory demyelination. J. Neuroimmunol. 2011, 233, 147–159. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, Y.; Tao, D.; Liebenson, D.; Liggett, T.; Goswami, R.; Stefoski, D.; Balabanov, R. Central nervous system expression of interferon regulatory factor 1 regulates experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 2010, 5, 260–265. [Google Scholar] [CrossRef]
- Feng, X.; Petraglia, A.L.; Chen, M.; Byskosh, P.V.; Boos, M.D.; AT, R. Low expression of interferon-stimulated genes in active multiple sclerosis is linked to subnormal phosphorylation of STAT1. J. Neuroimmunol. 2002, 129, 205–215. [Google Scholar]
- Fortunato, G.; Calcagno, G.; Bresciamorra, V.; Salvatore, E.; Filla, A.; Capone, S.; Liguori, R.; Borelli, S.; Gentile, I.; Borrelli, F.; et al. Multiple Sclerosis and Hepatitis C Virus Infection Are Associated with Single Nucleotide Polymorphisms in Interferon Pathway Genes. J. Interferon Cytokine Res. 2008, 28, 141–152. [Google Scholar] [CrossRef]
- Annibali, V.; Umeton, R.; Palermo, A.; Severa, M.; Etna, M.P.; Giglio, S.; Romano, S.; Ferraldeschi, M.; Buscarinu, M.C.; Vecchione, A.; et al. Analysis of coding and non-coding transcriptome of peripheral B cells reveals an altered interferon response factor (IRF)-1 pathway in multiple sclerosis patients. J. Neuroimmunol. 2018, 324, 165–171. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, Y.; Tao, D.; Liebenson, D.; Liggett, T.; Goswami, R.; Clarke, R.; Stefoski, D.; Balabanov, R. Overexpression of the Dominant-Negative Form of Interferon Regulatory Factor 1 in Oligodendrocytes Protects against Experimental Autoimmune Encephalomyelitis. J. Neurosci. 2011, 31, 8329–8341. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Bojanowski, K.; Chaudhuri, R.K. Transcriptomic Analysis of Fumarate Compounds Identifies Unique Effects of Isosorbide Di-(Methyl Fumarate) on NRF2, NF-kappaB and IRF1 Pathway Genes. Pharmaceuticals 2022, 15, 461. [Google Scholar] [CrossRef] [PubMed]
- Tarassishin, L.; Loudig, O.; Bauman, A.; Shafit-Zagardo, B.; Suh, H.S.; Lee, S.C. Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory miR-155 and miR-155 *. Glia 2011, 59, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, D.C.; O’Brien, K.; Young, A.; Fonseca-Kelly, Z.; Rostami, A.; Gran, B. Interferon regulatory factor (IRF) 3 is critical for the development of experimental autoimmune encephalomyelitis. J. Neuroinflammation 2014, 11, 130. [Google Scholar] [CrossRef]
- Tarassishin, L.; Suh, H.S.; Lee, S.C. Interferon regulatory factor 3 plays an anti-inflammatory role in microglia by activating the PI3K/Akt pathway. J. Neuroinflammation 2011, 8, 187. [Google Scholar] [CrossRef]
- Salem, M.; Mony, J.T.; Lobner, M.; Khorooshi, R.; Owens, T. Interferon regulatory factor-7 modulates experimental autoimmune encephalomyelitis in mice. J. Neuroinflammation 2011, 8, 181. [Google Scholar] [CrossRef]
- Hundeshagen, A.; Hecker, M.; Paap, B.K.; Angerstein, C.; Kandulski, O.; Fatum, C.; Hartmann, C.; Koczan, D.; Thiesen, H.J.; Zettl, U.K. Elevated type I interferon-like activity in a subset of multiple sclerosis patients: Molecular basis and clinical relevance. J. Neuroinflammation 2012, 9, 140. [Google Scholar] [CrossRef]
- Esen, N.; Rainey-Barger, E.K.; Huber, A.K.; Blakely, P.K.; Irani, D.N. Type-I interferons suppress microglial production of the lymphoid chemokine, CXCL13. Glia 2014, 62, 1452–1462. [Google Scholar] [CrossRef]
- Garnier, A.; Laffont, S.; Garnier, L.; Kaba, E.; Deutsch, U.; Engelhardt, B.; Guery, J.C. CD49d/CD29-integrin controls the accumulation of plasmacytoid dendritic cells into the CNS during neuroinflammation. Eur. J. Immunol. 2019, 49, 2030–2043. [Google Scholar] [CrossRef]
- Kristjansdottir, G.; Sandling, J.K.; Bonetti, A.; Roos, I.M.; Milani, L.; Wang, C.; Gustafsdottir, S.M.; Sigurdsson, S.; Lundmark, A.; Tienari, P.J.; et al. Interferon regulatory factor 5 (IRF5) gene variants are associated with multiple sclerosis in three distinct populations. J. Med. Genet. 2008, 45, 362–369. [Google Scholar] [CrossRef]
- Shen, Y.; Zhao, J.; Yang, R.; Yang, H.; Guo, M.; Ji, B.; Du, G.; Li, L. Panobinostat Attenuates Experimental Autoimmune Encephalomyelitis in Mice via Suppressing Oxidative Stress-Related Neuroinflammation and Mitochondrial Dysfunction. Int. J. Mol. Sci. 2024, 25, 12035. [Google Scholar] [CrossRef]
- Yoshida, Y.; Yoshimi, R.; Yoshii, H.; Kim, D.; Dey, A.; Xiong, H.; Munasinghe, J.; Yazawa, I.; O’Donovan, M.J.; Maximova, O.A.; et al. The transcription factor IRF8 activates integrin-mediated TGF-beta signaling and promotes neuroinflammation. Immunity 2014, 40, 187–198. [Google Scholar] [CrossRef]
- Garcia-Santibanez, R.; Burford, M.; Bucelli, R.C. Hereditary Motor Neuropathies and Amyotrophic Lateral Sclerosis: A Molecular and Clinical Update. Curr. Neurol. Neurosci. Rep. 2018, 18, 93. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Kawakami, H. Optineurin and amyotrophic lateral sclerosis. Geriatr. Gerontol. Int. 2012, 13, 528–532. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Irie, T.; Kawabata, R.; Yoshida, A.; Maruyama, H.; Kawakami, H. Optineurin with amyotrophic lateral sclerosis-related mutations abrogates inhibition of interferon regulatory factor-3 activation. Neurosci. Lett. 2011, 505, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Weil, R.; Laplantine, E.; Génin, P. Regulation of TBK1 activity by Optineurin contributes to cell cycle-dependent expression of the interferon pathway. Cytokine Growth Factor Rev. 2016, 29, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Slowicka, K.; Vereecke, L.; Mc Guire, C.; Sze, M.; Maelfait, J.; Kolpe, A.; Saelens, X.; Beyaert, R.; van Loo, G. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-κB signaling. Eur. J. Immunol. 2016, 46, 971–980. [Google Scholar] [CrossRef]
- de Majo, M.; Topp, S.D.; Smith, B.N.; Nishimura, A.L.; Chen, H.-J.; Gkazi, A.S.; Miller, J.; Wong, C.H.; Vance, C.; Baas, F.; et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol. Aging 2018, 71, 266.e1–266.e10. [Google Scholar] [CrossRef]
- Weinreich, M.; Shepheard, S.R.; Verber, N.; Wyles, M.; Heath, P.R.; Highley, J.R.; Kirby, J.; Shaw, P.J. Neuropathological characterization of a novel TANK binding kinase (TBK1) gene loss of function mutation associated with amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2019, 46, 279–291. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, W.; Huang, B.; Chen, X.; Huang, C. UBQLN2 Promotes the Production of Type I Interferon via the TBK1-IRF3 Pathway. Cells 2020, 9, 1205. [Google Scholar] [CrossRef]
- Miller, M.S.; Rialdi, A.; Ho, J.S.Y.; Tilove, M.; Martinez-Gil, L.; Moshkina, N.P.; Peralta, Z.; Noel, J.; Melegari, C.; Maestre, A.M.; et al. Senataxin suppresses the antiviral transcriptional response and controls viral biogenesis. Nat. Immunol. 2015, 16, 485–494. [Google Scholar] [CrossRef]
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N.; Ross, S.R. NF-κB and IRF1 Induce Endogenous Retrovirus K Expression via Interferon-Stimulated Response Elements in Its 5′ Long Terminal Repeat. J. Virol. 2016, 90, 9338–9349. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, L.; Liu, C.; Duan, W.; Zhang, S.; Li, Z.; Yi, L.; Guo, M.; Bi, Y.; Li, C.; et al. IRF5 knockdown reverses TDP-related phenotypes partially by increasing TBK1 expression. Brain Res. 2023, 1798, 148155. [Google Scholar] [CrossRef]
- Du, G.; Dong, W.; Yang, Q.; Yu, X.; Ma, J.; Gu, W.; Huang, Y. Altered Gut Microbiota Related to Inflammatory Responses in Patients With Huntington’s Disease. Front. Immunol. 2020, 11, 603594. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Cariulo, C.; Martufi, P.; Verani, M.; Toledo-Sherman, L.; Lee, R.; Dominguez, C.; Petricca, L.; Caricasole, A. IKBKB reduces huntingtin aggregation by phosphorylating serine 13 via a non-canonical IKK pathway. Life Sci. Alliance 2023, 6, e202302006. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| IRFs | Phenotype | References |
|---|---|---|
| IRF1 | IRF1 modulates pro-inflammatory and anti-inflammatory DAM. | [29] |
| IRF1 transcriptionally regulates microglia together with LXRβ and CEBPα. | [29] | |
| IRF1 mediates inflammation-related processes of BIN1 and EZH2 inhibitor. | [30,31] | |
| Anti-inflammatory agents suppress the IRF1-related signaling pathway. | [32,33,34] | |
| IRF3 | IRF3 mRNA is highly elevated in AD brains. | [35] |
| Bid, Tat, mtDNA positively and Scoparone negatively regulate TLR signaling to affect the pro-neuroinflammatory response of IRF3. | [36,37,38,39] | |
| cGAS-STING-IRF3 pathway in AD: cGAMP and IL-6 inhibit neuroinflammation, resveratrol protects cognition. | [40,41,42,43] | |
| Autophagy, pyroptosis, and IRF3 in AD: aggregation clearance, alleviation of inflammation and cognitive impairment by 3-MA and ZBP1 silencing. | [44,45] | |
| IRF7 | IRF7 mRNA is reduced in AD brains but upregulated in microglia during CNS injury. | [46,47,48] |
| TREM2 R47H variant increases AD risk and neuroinflammation by enhancing IRF7 activation and type I interferon responses. | [49] | |
| IRF7 knockdown suppresses Aβ-induced IFN-α/β and p-STAT3 via TLR-Myd88 signaling, exacerbating AD pathogenesis. | [50] | |
| miR-146b deficiency increases IRF7 expression, which upregulates miR-146a, inhibits TLR4, and reduces NF-κB activation and neuroinflammation. | [51] | |
| TSG reduces IRF7 and neuroinflammation via cGAS-STING inhibition, with potential AD benefits. | [52] | |
| NIRL upregulates IRF7, promoting microglia phenotype shift, reducing brain damage and improving cognitive function. | [53] | |
| IRF4 and IRF5 | IRF5 promotes pro-inflammatory M1 microglial polarization and Aβ-driven neuroinflammation in AD, while IRF4 enhances anti-inflammatory M2 polarization and neuroprotection. | [54] |
| Modulating the IRF5/4 ratio alleviates pathology via immune regulation and Aβ clearance, supported by microbiome-derived lipids and therapeutic interventions. | [48,55,56] | |
| IRF8 | IRF8 is upregulated in AD brains, associated with microglial activation markers, and is involved in TREM2-related AD pathogenesis. | [57] |
| In AD transgenic Tg2576 mice, Aβ1-40 upregulates IRF8 via the JAK2/STAT1 pathway, driving microglial activation; silencing IRF8 reduces this effect. | [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, X.; Diao, W.; Wang, H.; Yin, X.; Qian, W. Interferon Regulatory Factors as a Potential Therapeutic Target for Neuroinflammation: A Focus on Alzheimer’s Disease. Int. J. Mol. Sci. 2025, 26, 2906. https://doi.org/10.3390/ijms26072906
Fan X, Diao W, Wang H, Yin X, Qian W. Interferon Regulatory Factors as a Potential Therapeutic Target for Neuroinflammation: A Focus on Alzheimer’s Disease. International Journal of Molecular Sciences. 2025; 26(7):2906. https://doi.org/10.3390/ijms26072906
Chicago/Turabian StyleFan, Xing, Weikang Diao, Hao Wang, Xiaomin Yin, and Wei Qian. 2025. "Interferon Regulatory Factors as a Potential Therapeutic Target for Neuroinflammation: A Focus on Alzheimer’s Disease" International Journal of Molecular Sciences 26, no. 7: 2906. https://doi.org/10.3390/ijms26072906
APA StyleFan, X., Diao, W., Wang, H., Yin, X., & Qian, W. (2025). Interferon Regulatory Factors as a Potential Therapeutic Target for Neuroinflammation: A Focus on Alzheimer’s Disease. International Journal of Molecular Sciences, 26(7), 2906. https://doi.org/10.3390/ijms26072906