
Differential Temperature-Induced Responses in Immortalized Oral and Skin Keratinocytes


Abstract
1. Introduction
2. Results
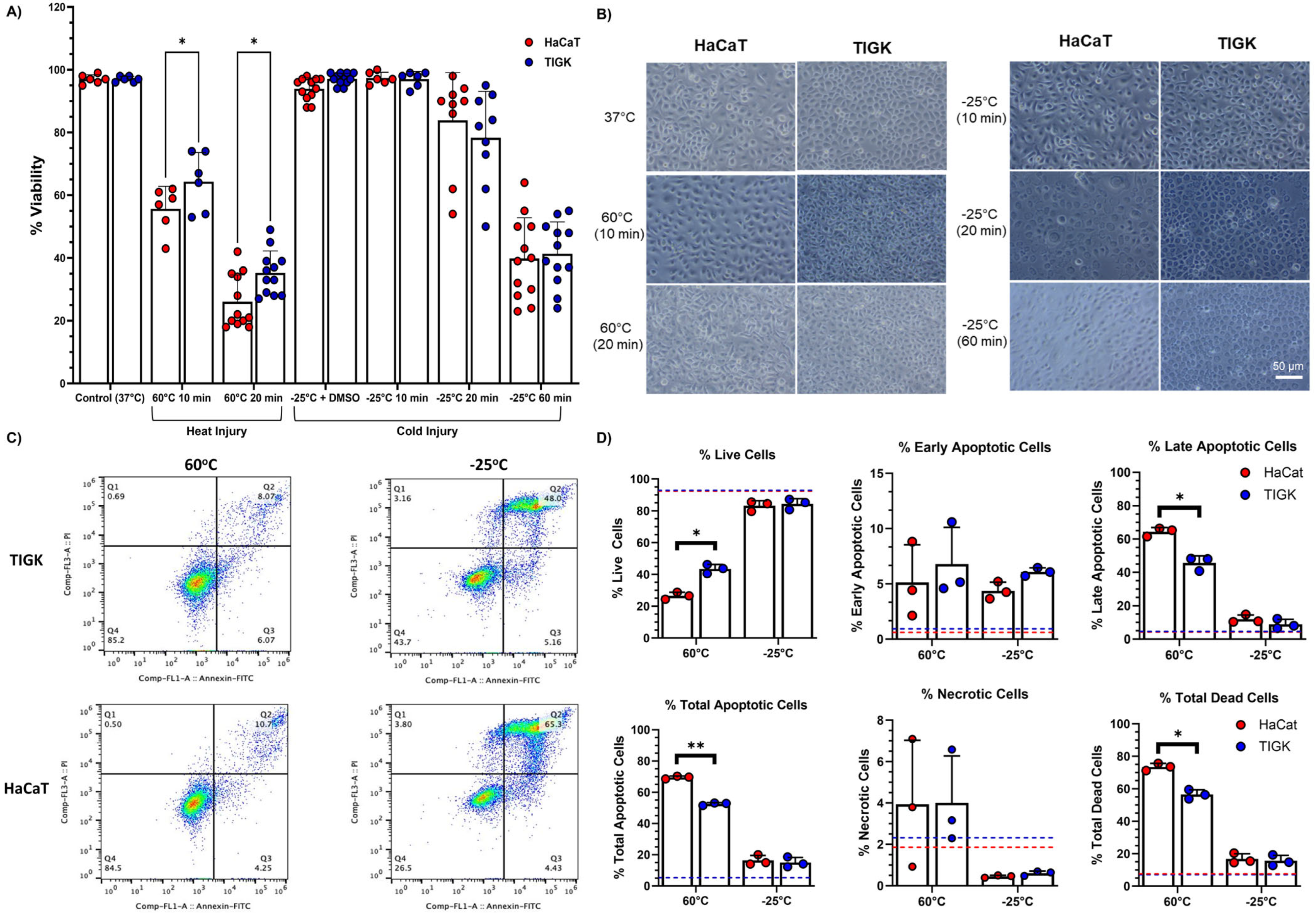
2.1. TIGKs Exhibit Enhanced Resistance to Heat Injury
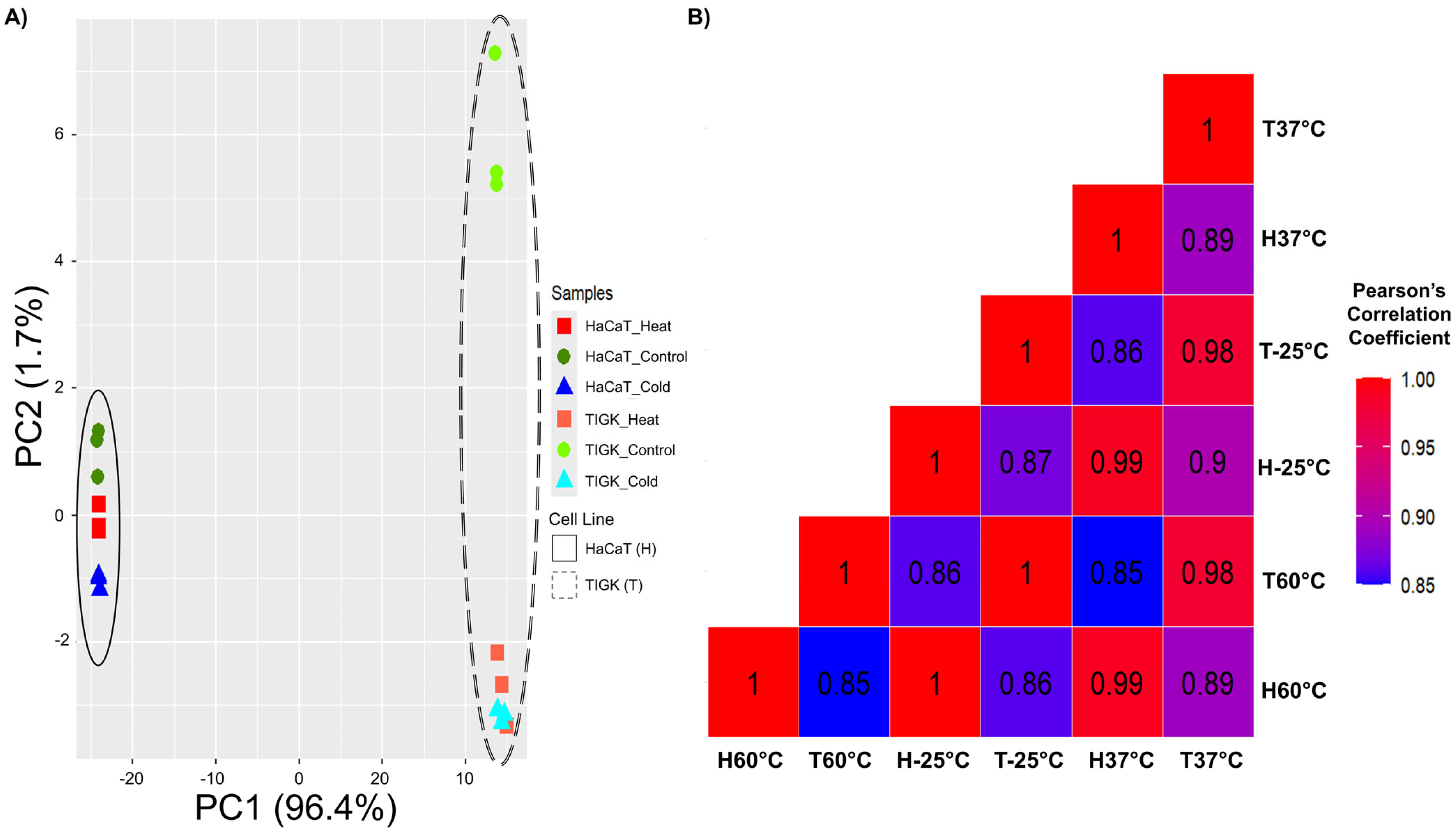
2.2. HaCaTs and TIGKs Cluster Both by Cell Type and Injury
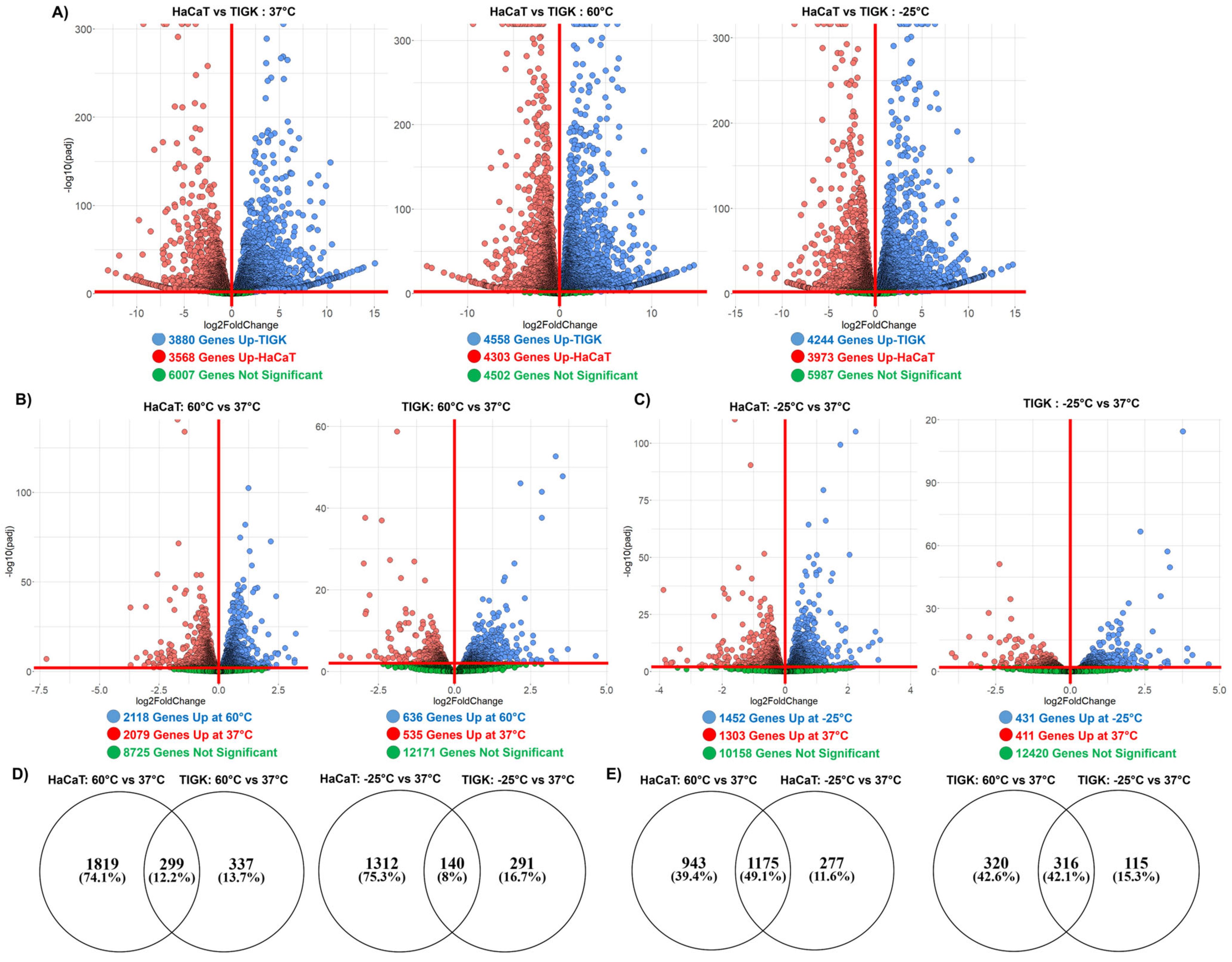
2.3. HaCaTs and TIGKs Exhibit Transcriptomic Differences at Baseline and in Response to Temperature-Induced Injury
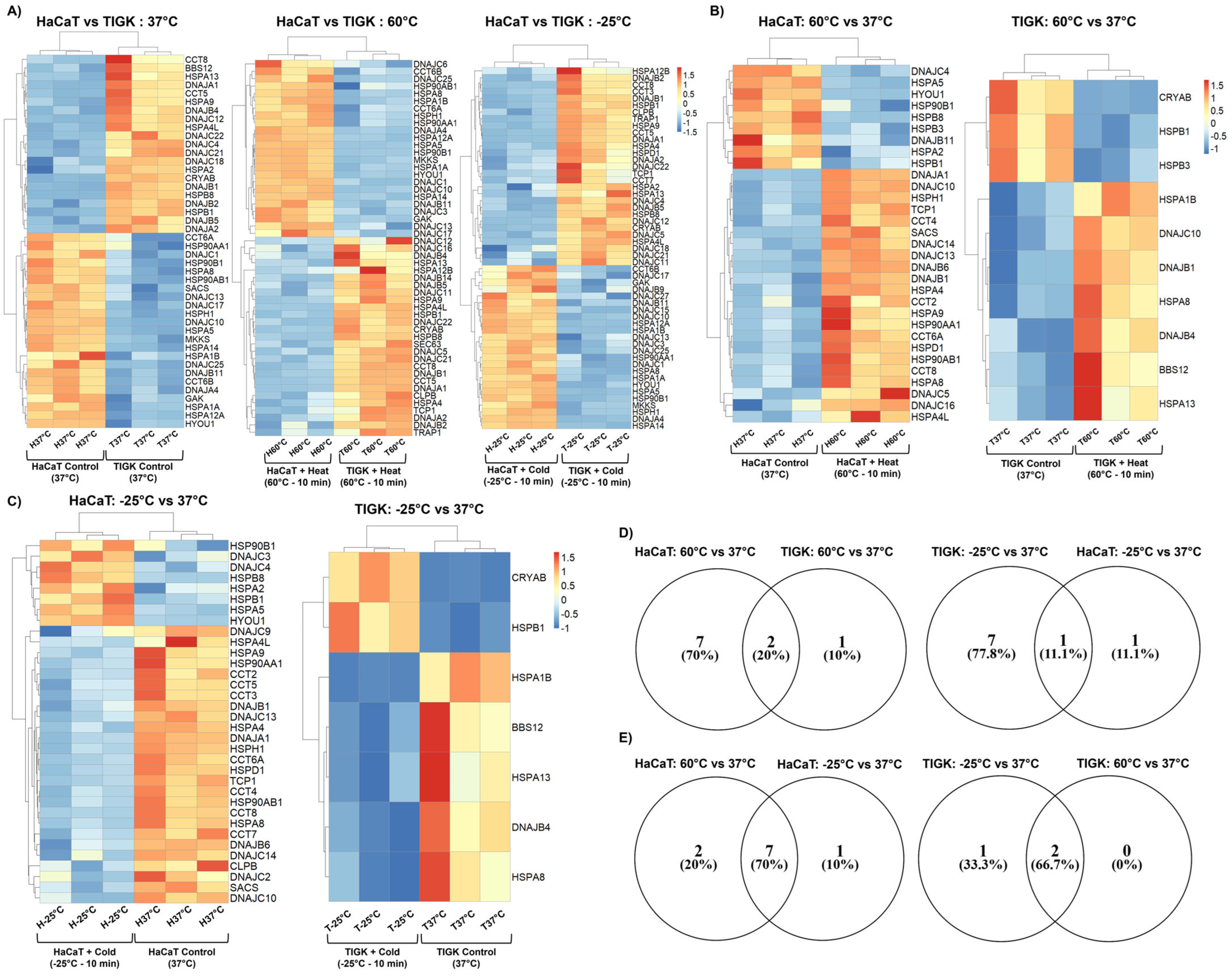
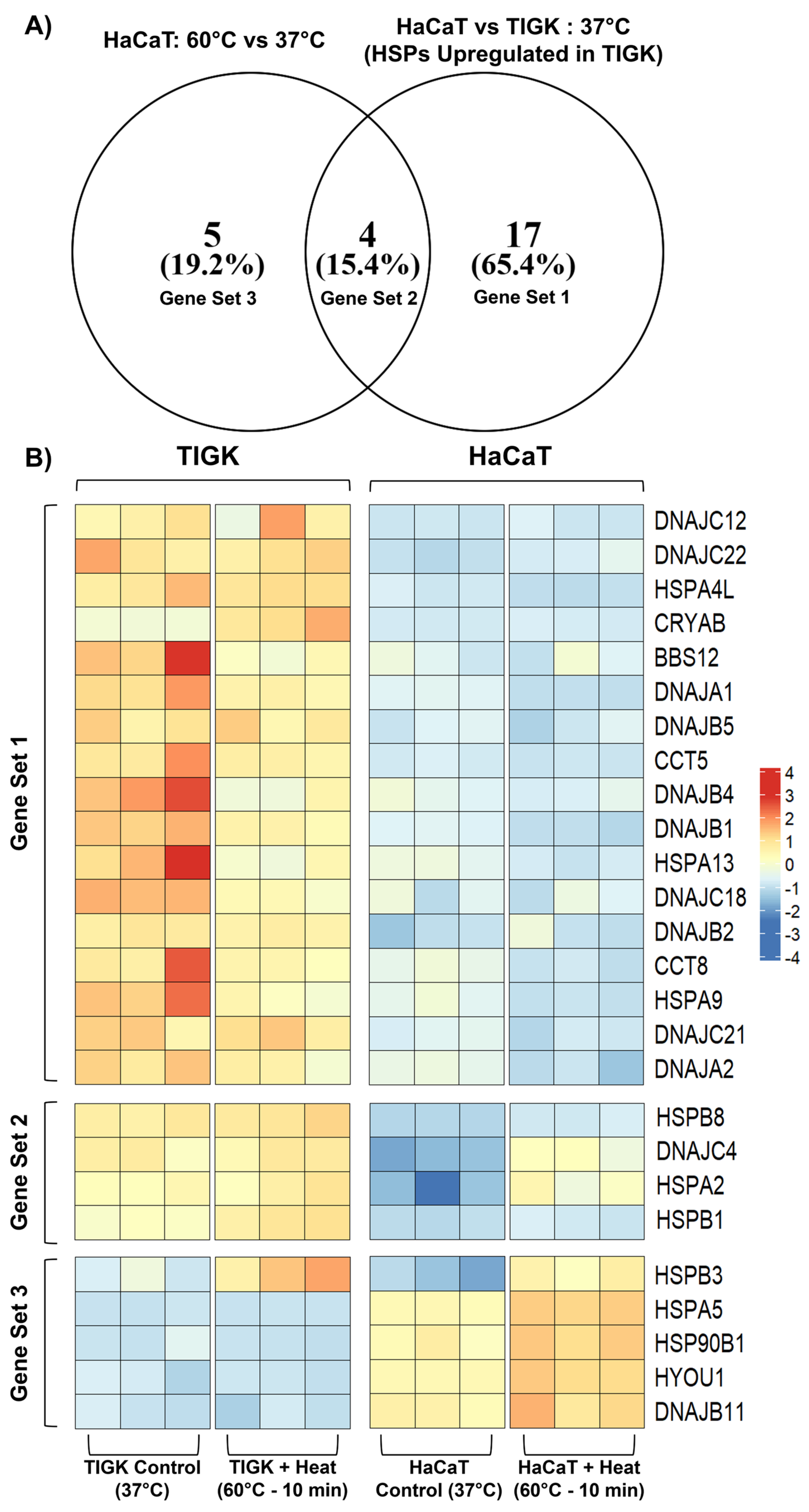
2.4. HaCaTs and TIGKs Exhibit Differential Expression of Heat Shock Proteins
2.5. Constitutively Expressed Heat Shock Proteins in Uninjured TIGKs May Enhance Their Resistance to Temperature Injury
3. Discussion
4. Materials and Methods
4.1. Skin and Oral Keratinocyte Culture
4.2. Skin and Oral Keratinocyte Temperature-Induced Injury and Viability
4.3. Assessment of Cellular Apoptosis
4.4. RNA Extraction, Library Preparation, and Data Processing for mRNA-Sequencing
4.5. Comparison of HaCaTs and TIGKs to Primary Skin and Oral Keratinocytes
4.6. Gene Ontology and Reactome Pathway Enrichment Analysis of DEGs
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muñoz-Garcia, A.; Ro, J.; Brown, J.C.; Williams, J.B. Cutaneous water loss and sphingolipids in the stratum corneum of house sparrows, Passer domesticus L. from desert and mesic environments as determined by reversed phase high-performance liquid chromatography coupled with atmospheric pressure photospray ionization mass spectrometry. J. Exp. Biol. 2008, 211 Pt 3, 447–458. [Google Scholar] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell. Biol. 2012, 212, 1–115. [Google Scholar]
- Sadler, K.E.; Moehring, F.; Stucky, C.L. Keratinocytes contribute to normal cold and heat sensation. Elife 2020, 9, e58625. [Google Scholar]
- Matylevitch, N.P.; Schuschereba, S.T.; Mata, J.R.; Gilligan, G.R.; Lawlor, D.F.; Goodwin, C.W.; Bowman, P.D. Apoptosis and accidental cell death in cultured human keratinocytes after thermal injury. Am. J. Pathol. 1998, 153, 567–577. [Google Scholar]
- Holland, D.B.; Roberts, S.G.; Wood, E.J.; Cunliffe, W.J. Cold shock induces the synthesis of stress proteins in human keratinocytes. J. Investig. Dermatol. 1993, 101, 196–199. [Google Scholar]
- Szpaderska, A.M.; Zuckerman, J.D.; DiPietro, L.A. Differential injury responses in oral mucosal and cutaneous wounds. J. Dent. Res. 2003, 82, 621–626. [Google Scholar]
- Szpaderska, A.M.; Zuckerman, J.D.; DiPietro, L.A. Distinct patterns of angiogenesis in oral and skin wounds. J. Dent. Res. 2005, 84, 309–314. [Google Scholar]
- Turabelidze, A.; Guo, S.; Chung, A.Y.; Chen, L.; Dai, Y.; Marucha, P.T.; DiPietro, L.A. Intrinsic differences between oral and skin keratinocytes. PLoS ONE 2014, 9, e101480. [Google Scholar]
- Warburton, G.; Nares, S.; Angelov, N.; Brahim, J.S.; Dionne, R.A.; Wahl, S.M. Transcriptional events in a clinical model of oral mucosal tissue injury and repair. Wound Repair Regen. 2005, 13, 19–26. [Google Scholar]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10, eaap8798. [Google Scholar]
- Mak, K.; Manji, A.; Gallant-Behm, C.; Wiebe, C.; Hart, D.A.; Larjava, H.; Häkkinen, L. Scarless healing of oral mucosa is characterized by faster resolution of inflammation and control of myofibroblast action compared to skin wounds in the red Duroc pig model. J. Dermatol. Sci. 2009, 56, 168–180. [Google Scholar] [CrossRef]
- Wong, J.W.; Gallant-Behm, C.; Wiebe, C.; Mak, K.; Hart, D.A.; Larjava, H.; Häkkinen, L. Wound healing in oral mucosa results in reduced scar formation as compared with skin: Evidence from the red Duroc pig model and humans. Wound Repair Regen. 2009, 17, 717–729. [Google Scholar] [CrossRef]
- Chen, L.; Arbieva, Z.H.; Guo, S.; Marucha, P.T.; Mustoe, T.A.; DiPietro, L.A. Positional differences in the wound transcriptome of skin and oral mucosa. BMC Genom. 2010, 11, 471. [Google Scholar] [CrossRef]
- Leonardo, T.R.; Shi, J.; Chen, D.; Trivedi, H.M.; Chen, L. Differential Expression and Function of Bicellular Tight Junctions in Skin and Oral Wound Healing. Int. J. Mol. Sci. 2020, 21, 2966. [Google Scholar] [CrossRef]
- Leonardo, T.R.; Chen, L.; Schrementi, M.E.; Shi, J.; Marucha, P.T.; Glass, K.; DiPietro, L.A. Transcriptional changes in human palate and skin healing. Wound Repair Regen. 2023, 31, 156–170. [Google Scholar] [CrossRef]
- Gillis, P.; Savla, U.; Volpert, O.V.; Jimenez, B.; Waters, C.M.; Panos, R.J.; Bouck, N.P. Keratinocyte growth factor induces angiogenesis and protects endothelial barrier function. J. Cell Sci. 1999, 112 Pt 12, 2049–2057. [Google Scholar] [CrossRef]
- Machesney, M.; Tidman, N.; Waseem, A.; Kirby, L.; Leigh, I. Activated keratinocytes in the epidermis of hypertrophic scars. Am. J. Pathol. 1998, 152, 1133–1141. [Google Scholar]
- Jiang, Y.; Tsoi, L.C.; Billi, A.C.; Ward, N.L.; Harms, P.W.; Zeng, C.; Maverakis, E.; Kahlenberg, J.M.; Gudjonsson, J.E. Cytokinocytes: The diverse contribution of keratinocytes to immune responses in skin. JCI Insight 2020, 5, e142067. [Google Scholar] [CrossRef]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef]
- Scieglinska, D.; Krawczyk, Z.; Sojka, D.R.; Gogler-Pigłowska, A. Heat shock proteins in the physiology and pathophysiology of epidermal keratinocytes. Cell Stress Chaperones 2019, 24, 1027–1044. [Google Scholar] [CrossRef]
- Moritz, A.R.; Henriques, F.C. Studies of Thermal Injury: II. The Relative Importance of Time and Surface Temperature in the Causation of Cutaneous Burns. Am. J. Pathol. 1947, 23, 695–720. [Google Scholar]
- Doppegieter, M.; van Leeuwen, T.G.; van Weert, A.; Aalders, M.C.G.; Bakker, E. Subminute thermal damage to cell types present in the skin. Int. J. Hyperth. 2024, 41, 2354435. [Google Scholar]
- Lindquist, S. The heat-shock response. Annu. Rev. Biochem. 1986, 55, 1151–1191. [Google Scholar]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef]
- Murshid, A.; Prince, T.L.; Lang, B.; Calderwood, S.K. Role of Heat Shock Factors in Stress-Induced Transcription. Methods Mol. Biol. 2018, 1709, 23–34. [Google Scholar]
- Maytin, E.V.; Wimberly, J.M.; Anderson, R.R. Thermotolerance and the heat shock response in normal human keratinocytes in culture. J. Investig. Dermatol. 1990, 95, 635–642. [Google Scholar]
- Bowman, P.D.; Schuschereba, S.T.; Lawlor, D.F.; Gilligan, G.R.; Mata, J.R.; DeBaere, D.R. Survival of human epidermal keratinocytes after short-duration high temperature: Synthesis of HSP70 and IL-8. Am. J. Physiol. 1997, 272 Pt 1, C1988–C1994. [Google Scholar]
- Sakamoto, H.; Mashima, T.; Yamamoto, K.; Tsuruo, T. Modulation of heat-shock protein 27 (Hsp27) anti-apoptotic activity by methylglyoxal modification. J. Biol. Chem. 2002, 277, 45770–45775. [Google Scholar]
- Al-Fageeh, M.B.; Smales, C.M. Control and regulation of the cellular responses to cold shock: The responses in yeast and mammalian systems. Biochem. J. 2006, 397, 247–259. [Google Scholar]
- Cedraz, H.; Gromboni, J.G.G.; Garcia, A.A.P.J.; Farias Filho, R.V.; Souza, T.M.; Oliveira, E.R.; Oliveira, E.B.; Nascimento, C.S.D.; Meneghetti, C.; Wenceslau, A.A. Heat stress induces expression of HSP genes in genetically divergent chickens. PLoS ONE 2017, 12, e0186083. [Google Scholar]
- Bastaki, N.K.; Albarjas, T.A.; Almoosa, F.A.; Al-Adsani, A.M. Chronic heat stress induces the expression of HSP genes in the retina of chickens (Gallus gallus). Front. Genet. 2023, 14, 1085590. [Google Scholar] [CrossRef] [PubMed]
- Rennekampff, H.O.; Alharbi, Z. Burn Injury: Mechanisms of Keratinocyte Cell Death. Med. Sci. 2021, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.H.; Rose, L.F.; Fletcher, J.L.; Wu, J.C.; Leung, K.P.; Chan, R.K. Antecedent thermal injury worsens split-thickness skin graft quality: A clinically relevant porcine model of full-thickness burn, excision and grafting. Burns 2017, 43, 223–231. [Google Scholar] [PubMed]
- Isozaki, S.; Tanaka, H.; Horioka, K.; Konishi, H.; Kashima, S.; Takauji, S.; Fujiya, M.; Druid, H. Hypoxia-induced nuclear translocation of β-catenin in the healing process of frostbite. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166385. [Google Scholar] [CrossRef]
- Basit, H.; Wallen, T.J.; Dudley, C. Frostbite. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Scieglinska, D.; Piglowski, W.; Chekan, M.; Mazurek, A.; Krawczyk, Z. Differential expression of HSPA1 and HSPA2 proteins in human tissues; tissue microarray-based immunohistochemical study. Histochem. Cell Biol. 2011, 135, 337–350. [Google Scholar]
- Echchgadda, I.; Roth, C.C.; Cerna, C.Z.; Wilmink, G.J. Temporal gene expression kinetics for human keratinocytes exposed to hyperthermic stress. Cells 2013, 2, 224–243. [Google Scholar] [CrossRef]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef]
- Filipczak, P.T.; Piglowski, W.; Glowala-Kosinska, M.; Krawczyk, Z.; Scieglinska, D. HSPA2 overexpression protects V79 fibroblasts against bortezomib-induced apoptosis. Biochem. Cell Biol. 2012, 90, 224–231. [Google Scholar]
- Shen, J.; Li, M.; Min, L. HSPB8 promotes cancer cell growth by activating the ERK-CREB pathway and is indicative of a poor prognosis in gastric cancer patients. Oncol. Rep. 2018, 39, 2978–2986. [Google Scholar]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Algothmi, K.M.; Mahasneh, Z.M.H.; Abdelnour, S.A.; Khalaf, Q.A.W.; Noreldin, A.E.; Barkat, R.A.; Khalifa, N.E.; Khafaga, A.F.; Tellez-Isaias, G.; Alqhtani, A.H.; et al. Protective impacts of mitochondria enhancers against thermal stress in poultry. Poult. Sci. 2024, 103, 103218. [Google Scholar]
- Lu, J.; Li, H.; Yu, D.; Zhao, P.; Liu, Y. Heat stress inhibits the proliferation and differentiation of myoblasts and is associated with damage to mitochondria. Front. Cell Dev. Biol. 2023, 11, 1171506. [Google Scholar]
- Wang, S.; Cheng, F.; Ji, Q.; Song, M.; Wu, Z.; Zhang, Y.; Ji, Z.; Feng, H.; Belmonte, J.C.I.; Zhou, Q.; et al. Hyperthermia differentially affects specific human stem cells and their differentiated derivatives. Protein Cell 2022, 13, 615–622. [Google Scholar] [PubMed]
- Zan, G.X.; Qin, Y.C.; Xie, W.W.; Gao, C.Q.; Yan, H.C.; Wang, X.Q.; Zhou, J.Y. Heat stress disrupts intestinal stem cell migration and differentiation along the crypt-villus axis through FAK signaling. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119431. [Google Scholar]
- Gao, D.; Critser, J.K. Mechanisms of cryoinjury in living cells. Ilar J. 2000, 41, 187–196. [Google Scholar]
- Mazur, P. Freezing of living cells: Mechanisms and implications. Am. J. Physiol. 1984, 247 Pt 1, C125–C142. [Google Scholar]
- Read, D.E.; Reed Herbert, K.; Gorman, A.M. Heat shock enhances NGF-induced neurite elongation which is not mediated by Hsp25 in PC12 cells. Brain Res. 2008, 1221, 14–23. [Google Scholar]
- Kudo, T.A.; Kanetaka, H.; Mochizuki, K.; Tominami, K.; Nunome, S.; Abe, G.; Kosukegawa, H.; Abe, T.; Mori, H.; Mori, K.; et al. Induction of neurite outgrowth in PC12 cells treated with temperature-controlled repeated thermal stimulation. PLoS ONE 2015, 10, e0124024. [Google Scholar]
- Hossain, M.E.; Matsuzaki, K.; Katakura, M.; Sugimoto, N.; Mamun, A.A.; Islam, R.; Hashimoto, M.; Shido, O. Direct exposure to mild heat promotes proliferation and neuronal differentiation of neural stem/progenitor cells in vitro. PLoS ONE 2017, 12, e0190356. [Google Scholar]
- Wan, Q.; Song, D.; Li, H.; He, M.L. Stress proteins: The biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal Transduct. Target Ther. 2020, 5, 125. [Google Scholar]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [PubMed]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [PubMed]
- Carretero, M.; Escámez, M.J.; García, M.; Duarte, B.; Holguín, A.; Retamosa, L.; Jorcano, J.L.; del Río, M.; Larcher, F. In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. J. Investig. Dermatol. 2008, 128, 223–236. [Google Scholar]
- Liu, Y.; Xiong, W.; Wang, C.W.; Shi, J.P.; Shi, Z.Q.; Zhou, J.D. Resveratrol promotes skin wound healing by regulating the miR-212/CASP8 axis. Lab Investig. 2021, 101, 1363–1370. [Google Scholar]
- Ranzato, E.; Patrone, M.; Mazzucco, L.; Burlando, B. Platelet lysate stimulates wound repair of HaCaT keratinocytes. Br. J. Dermatol. 2008, 159, 537–545. [Google Scholar]
- Long, M.; Rojo de la Vega, M.; Wen, Q.; Bharara, M.; Jiang, T.; Zhang, R.; Zhou, S.; Wong, P.K.; Wondrak, G.T.; Zheng, H.; et al. An Essential Role of NRF2 in Diabetic Wound Healing. Diabetes 2016, 65, 780–793. [Google Scholar]
- Moffatt-Jauregui, C.E.; Robinson, B.; de Moya, A.V.; Brockman, R.D.; Roman, A.V.; Cash, M.N.; Culp, D.J.; Lamont, R.J. Establishment and characterization of a telomerase immortalized human gingival epithelial cell line. J. Periodontal Res. 2013, 48, 713–721. [Google Scholar]
- Colombo, I.; Sangiovanni, E.; Maggio, R.; Mattozzi, C.; Zava, S.; Corbett, Y.; Fumagalli, M.; Carlino, C.; Corsetto, P.A.; Scaccabarozzi, D.; et al. HaCaT Cells as a Reliable In Vitro Differentiation Model to Dissect the Inflammatory/Repair Response of Human Keratinocytes. Mediat. Inflamm. 2017, 2017, 7435621. [Google Scholar]
- Schürer, N.; Köhne, A.; Schliep, V.; Barlag, K.; Goerz, G. Lipid composition and synthesis of HaCaT cells, an immortalized human keratinocyte line, in comparison with normal human adult keratinocytes. Exp. Dermatol. 1993, 2, 179–185. [Google Scholar]
- Smith, C.J.; Parkinson, E.K.; Yang, J.; Pratten, J.; O’Toole, E.A.; Caley, M.P.; Braun, K.M. Investigating wound healing characteristics of gingival and skin keratinocytes in organotypic cultures. J. Dent. 2022, 125, 104251. [Google Scholar]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [PubMed]
- Shukla, L.; Lee, S.A.; Du, M.R.M.; Karnezis, T.; Ritchie, M.E.; Shayan, R. A transcriptomic dataset evaluating the effect of radiotherapy injury on cells of skin and soft tissue. Data Brief 2022, 41, 107828. [Google Scholar]
- Liu, Y.; Sun, Y.; Yang, J.; Wu, D.; Yu, S.; Liu, J.; Hu, T.; Luo, J.; Zhou, H. DNMT1-targeting remodeling global DNA hypomethylation for enhanced tumor suppression and circumvented toxicity in oral squamous cell carcinoma. Mol. Cancer 2024, 23, 104. [Google Scholar]
- Uchiyama, A.; Nayak, S.; Graf, R.; Cross, M.; Hasneen, K.; Gutkind, J.S.; Brooks, S.R.; Morasso, M.I. SOX2 Epidermal Overexpression Promotes Cutaneous Wound Healing via Activation of EGFR/MEK/ERK Signaling Mediated by EGFR Ligands. J. Investig. Dermatol. 2019, 139, 1809–1820.e8. [Google Scholar]
- Boukamp, P.; Popp, S.; Bleuel, K.; Tomakidi, E.; Bürkle, A.; Fusenig, N.E. Tumorigenic conversion of immortal human skin keratinocytes (HaCaT) by elevated temperature. Oncogene 1999, 18, 5638–5645. [Google Scholar]
- hTERT TIGKs. Available online: https://www.atcc.org/products/crl-3397 (accessed on 16 March 2025).
- Romanovsky, A.A. Skin temperature: its role in thermoregulation. Acta Physiol. 2014, 210, 498–507. [Google Scholar]
- Denda, M.; Sokabe, T.; Fukumi-Tominaga, T.; Tominaga, M. Effects of skin surface temperature on epidermal permeability barrier homeostasis. J. Investig. Dermatol. 2007, 127, 654–659. [Google Scholar]
- Xiao, R.; Xu, X.Z.S. Temperature Sensation: From Molecular Thermosensors to Neural Circuits and Coding Principles. Annu. Rev. Physiol. 2021, 83, 205–230. [Google Scholar]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Seal, R.L.; Braschi, B.; Gray, K.; Jones, T.E.M.; Tweedie, S.; Haim-Vilmovsky, L.; Bruford, E.A. Genenames.org: The HGNC resources in 2023. Nucleic Acids Res. 2023, 51, D1003–D1009. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Bruford, E.A.; Braschi, B.; Denny, P.; Jones, T.E.M.; Seal, R.L.; Tweedie, S. Guidelines for human gene nomenclature. Nat. Genet. 2020, 52, 754–758. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2016. [Google Scholar]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007–2015. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 19 August 2024).
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (37 °C) | Heat-Induced Injury (60 °C) | Cold-Induced Injury (−25 °C) | |
|---|---|---|---|
| Upregulated in HaCaTs | 3568 | 4303 | 3973 |
| Upregulated in TIGKs | 3880 | 4558 | 4244 |
| Not Significant | 6007 | 4502 | 5098 |
| HaCaTs | TIGKs | |||
|---|---|---|---|---|
| Heat-Induced Injury Response (60 °C vs. 37 °C) | Cold-Induced Injury Response (−25 °C vs. 37 °C) | Heat-Induced Injury Response (60 °C vs. 37 °C) | Cold-Induced Injury Response (−25 °C vs. 37 °C) | |
| Upregulated | 2118 | 1452 | 636 | 431 |
| Downregulated | 2079 | 1303 | 535 | 411 |
| Not Significant | 8725 | 10,158 | 12,171 | 12,420 |
| Control (37 °C) | Heat-Induced Injury (60 °C) | Cold-Induced Injury (−25 °C) | |
|---|---|---|---|
| Upregulated in HaCaTs | 23 | 24 | 29 |
| Upregulated in TIGKs | 21 | 27 | 24 |
| HaCaTs | TIGKs | |||
|---|---|---|---|---|
| Heat-Induced Injury Response (60 °C vs. 37 °C) | Cold-Induced Injury Response (−25 °C vs. 37 °C) | Heat-Induced Injury Response (60 °C vs. 37 °C) | Cold-Induced Injury Response (−25 °C vs. 37 °C) | |
| Upregulated | 9 | 8 | 3 | 2 |
| Downregulated | 22 | 26 | 7 | 5 |
| Gene Set | Gene Set Meaning | HSPs |
|---|---|---|
| 1 | HSPs activated in HaCaTs at −25 °C relative to 37 °C | DNAJC12, DNAJC22, HSPA4L, CRYAB, BBS12, DNAJA1, DNAJB5, CCT5, DNAJB4, DNAJB1, HSPA13, DNAJC18, DNAJB2, CCT8, HSPA9, DNAJC21, DNAJA2 |
| 2 | HSPs activated in HaCaTs at −25 °C relative to 37 °C and differentially upregulated in TIGKs relative to HaCaTs at 37 °C | HSPB8, DNAJC4, HSPA2, HSPB1 |
| 3 | HSPs differentially upregulated in TIGKs relative to HaCaTs at 37 °C | HSPB3, HSPA5, HSP90B1, HYOU1, DNAJB11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, C.; Yuan, H.; Chen, A.K.; DiPietro, L.A.; Chen, L. Differential Temperature-Induced Responses in Immortalized Oral and Skin Keratinocytes. Int. J. Mol. Sci. 2025, 26, 2851. https://doi.org/10.3390/ijms26072851
Han C, Yuan H, Chen AK, DiPietro LA, Chen L. Differential Temperature-Induced Responses in Immortalized Oral and Skin Keratinocytes. International Journal of Molecular Sciences. 2025; 26(7):2851. https://doi.org/10.3390/ijms26072851
Chicago/Turabian StyleHan, Chen, Heidi Yuan, Amy K. Chen, Luisa A. DiPietro, and Lin Chen. 2025. "Differential Temperature-Induced Responses in Immortalized Oral and Skin Keratinocytes" International Journal of Molecular Sciences 26, no. 7: 2851. https://doi.org/10.3390/ijms26072851
APA StyleHan, C., Yuan, H., Chen, A. K., DiPietro, L. A., & Chen, L. (2025). Differential Temperature-Induced Responses in Immortalized Oral and Skin Keratinocytes. International Journal of Molecular Sciences, 26(7), 2851. https://doi.org/10.3390/ijms26072851