1. Introduction
Alzheimer disease (AD) is the most prevalent neurodegenerative condition among the elderly. In comparison to diseases such as cancer, heart disease, and stroke, it has become one of the leading causes of death in individuals over 65 years of age [
1]. It is widely accepted that AD arises from a combination of pathological processes, including β-amyloid (Aβ) abnormality, microtubule-associated protein tau (Tau) hyperphosphorylation, neuroinflammation, neurotransmitter imbalance, and neuronal damage [
2]. The upstream regulators of these pathological features have not been fully clarified. Since the most widely adopted treatments aimed at reducing the burden of Aβ have not proven effective, it is urgent to find a new treatment for AD. The ideal therapeutic strategy should not only eliminate the pathological features of the disease but also facilitate some functional recovery [
3]. Emerging evidence has shown that autophagy system disorders are the key pathological features of AD [
4]. Therefore, one of the new strategies is to find small molecules that can promote neurogenesis and regulate autophagy.
Nerve growth factor (NGF) is the first recognized neurotrophic factor and is considered a potential candidate drug for AD treatment [
5]. Preclinical evidence highlights the critical role of NGF in neuroprotection, resilience, and cognitive function in models of neuronal injury and aging [
6,
7]. The PC12 cell bioassay system has been established as a reliable method for investigating potential drugs with NGF-mimic activity. Following NGF treatment, PC12 cells cease dividing and start to differentiate, similar to neurons. However, NGF faces challenges in crossing the blood–brain barrier (BBB) due to its high polarity and large molecular weight, which limits its therapeutic potential. Consequently, developing small molecules with NGF-mimic activity is a promising approach for AD treatment.
Adult neurogenesis has been found in some areas of the brain, especially in the hippocampus, during the life of various mammals [
8]. Blocking the adult hippocampal neurogenesis aggravates cognitive impairment in the AD mouse model. However, targeting neurogenesis may improve cognitive dysfunction in AD [
9]. Wnt signaling plays a major role in regulating adult hippocampal neurogenesis [
10], whereas the phosphatidylinositol 3-kinase (PI3K) pathway is implicated in neuroregeneration [
11]. Discovering small molecules that can regulate these two signaling pathways and promote neurogenesis is a new idea for the treatment of AD.
Additionally, autophagy plays a crucial regulatory role in neurodegenerative diseases. Mutations in genes associated with autophagy and lysosomal function contribute to the development of various neurodegenerative diseases [
12]. Notably, abnormalities in the lysosome and autophagy system within neurons represent an early prominent feature of AD [
13,
14]. Chaperone-mediated autophagy (CMA) is a lysosomal-dependent selective degradation pathway involved in the pathogenesis of cancer and neurodegenerative diseases. A remarkable aspect of CMA is its ability to selectively degrade proteins based on a recognition motif present in their amino acid sequence [
15]. This specificity enables the elimination of particular proteins (substrates that contain the Lys-Phe-Glu-Arg-Gln (KFERQ) motif) without affecting adjacent proteins [
16]. One of the key proteins in CMA is heat-shock cognate protein 70 (Hsc70), a molecular chaperone from the family of heat-shock proteins (HSP) [
17]. It has been reported that Hsc70 has diverse intracellular functions, including binding to aggregated and misfolded peptides as well as facilitating the degradation of target proteins such as amyloid precursor protein (APP), hexokinase 2 (HK2), and pyruvate kinase M2 (PKM2) through the ubiquitin-proteasome system (UPS) and CMA [
18,
19,
20]. Meanwhile, Hsc70 may help prevent the accumulation of phosphorylated Tau (p-Tau) protein [
21]. Consequently, identifying compounds that activate the CMA pathway of Hsc70 to clear misfolded proteins is anticipated to be a promising strategy for preventing or treating AD.
To investigate the activity and mechanisms of compounds for treating AD, selecting an appropriate animal model is critical. Commonly used AD models include aging animals, transgenic animals (such as APP/PS1 double transgenic mice, 3×Tg mice, and 5×FAD mice), and exogenously induced (such as Aβ, D-galactose, and high-fat diet (HFD)-induced AD mice) models. Among these, the HFD-induced AD mouse model is less invasive, effectively simulates AD symptoms, and accelerates pathology under natural conditions. The metabolic defects caused by HFD contribute to AD risk through mechanisms involving neuroinflammation and amyloidosis [
22] and usually lead to gut microbiota disorder. For this study, HFD-induced AD mice were utilized.
Previous studies identified several neuritogenic compounds, termed gentisides, from the traditional Chinese medicine
Gentiana rigescens Franch in PC12 cells. Structure–activity relationship (SAR) analyses of these gentisides led to the synthesis of hundreds of alkyl benzoate derivatives [
23]. Activity-based protein profiling (ABPP) identified Hsc70 as the target protein for one of the leading compounds [
24]. However, the poor water solubility and low bioavailability of the leading compound necessitated the development of more derivatives with improved properties. From these derivatives, a new lead compound, TBG096, was identified. This report demonstrates that TBG096 exerts NGF-mimic activity and ameliorates memory dysfunction in AD mouse models by promoting neurogenesis and regulation of the Hsc70/HK2/PKM2/LAMP2A signaling pathway.
3. Discussion
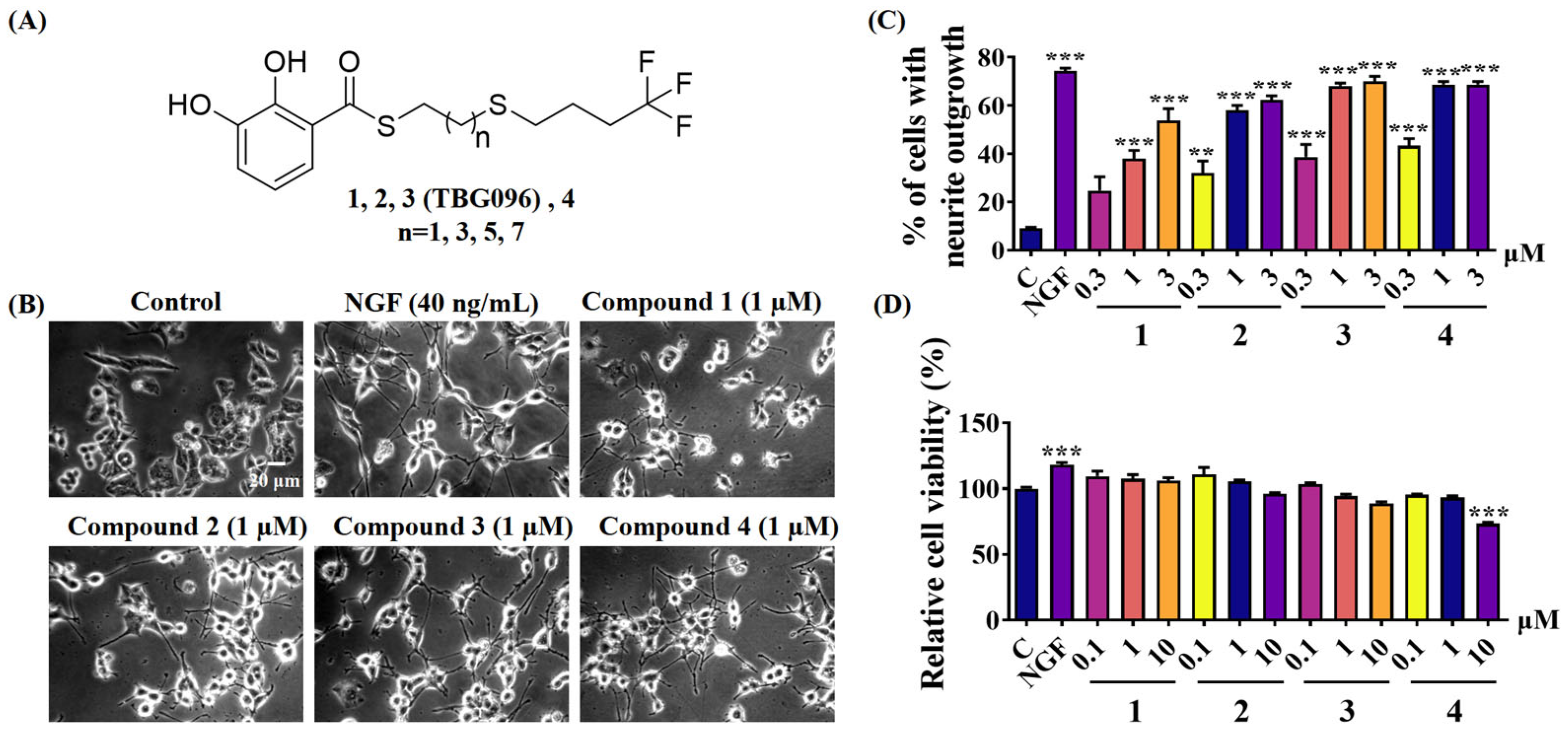
Previous studies identified gentiside derivatives with novel NGF-mimic effects. However, these compounds exhibited low bioavailability and poor ability to cross the BBB. To address these limitations, this study introduced structural modifications, including alterations to alkyl chain length and substitution of the terminal methyl group’s hydrogen atom with fluorine (
Figures S1 and S2). These modifications led to the development of thiobenzoate compounds with improved water solubility (
Figure 1), and TBG096 was discovered as a lead compound with NGF-mimic activity and favorable safety (
Figure 1 and
Figure S3).
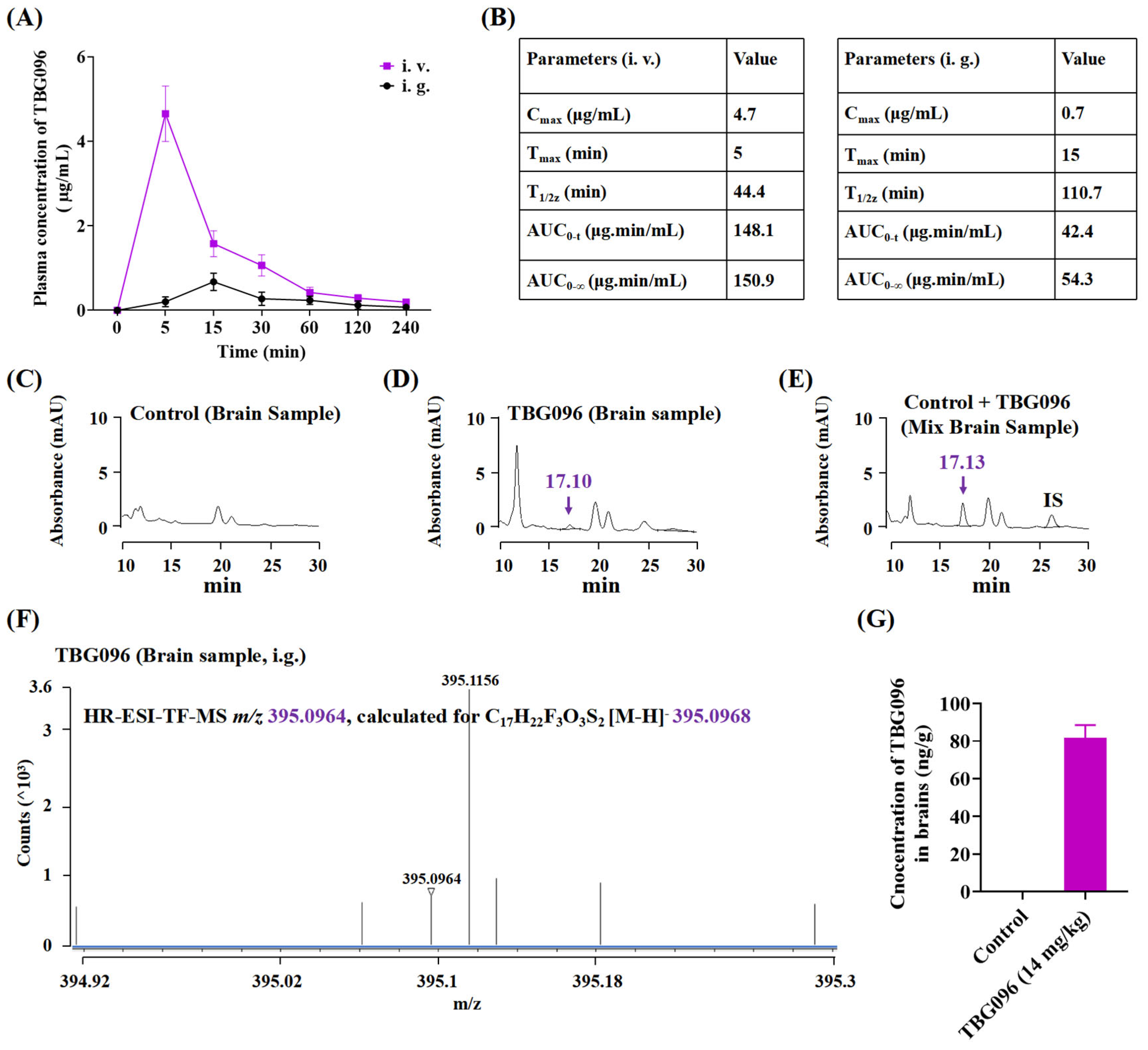
As a potential therapeutic candidate for neurodegenerative diseases, TBG096 required good bioavailability and BBB permeability. Pharmacokinetic studies assessed its brain tissue concentration, while HPLC and HR ESI-TOF-MS analysis confirmed that TBG096 exhibited favorable bioavailability and successfully crossed the BBB (
Figure 2 and
Figure S4). NGF cannot penetrate the BBB to produce a therapy effect due to high molecular weight and polarity [
28]. TBG096 and Met can avoid this limitation, pass through BBB [
29], and arrive at the brain tissue to exert anti-AD effects. However, Met mainly acts on the liver and intestine [
30]. Interestingly, the oral bioavailability and the half-life of TBG096 and metformin are 36% and 1.85 h as well as 55% and 1.92 h [
31,
32], respectively. However, TBG096 has anti-AD effects at low concentrations of 0.1 and 5 mg/kg. These findings suggest that TBG096 is a highly promising candidate drug for anti-AD treatment.
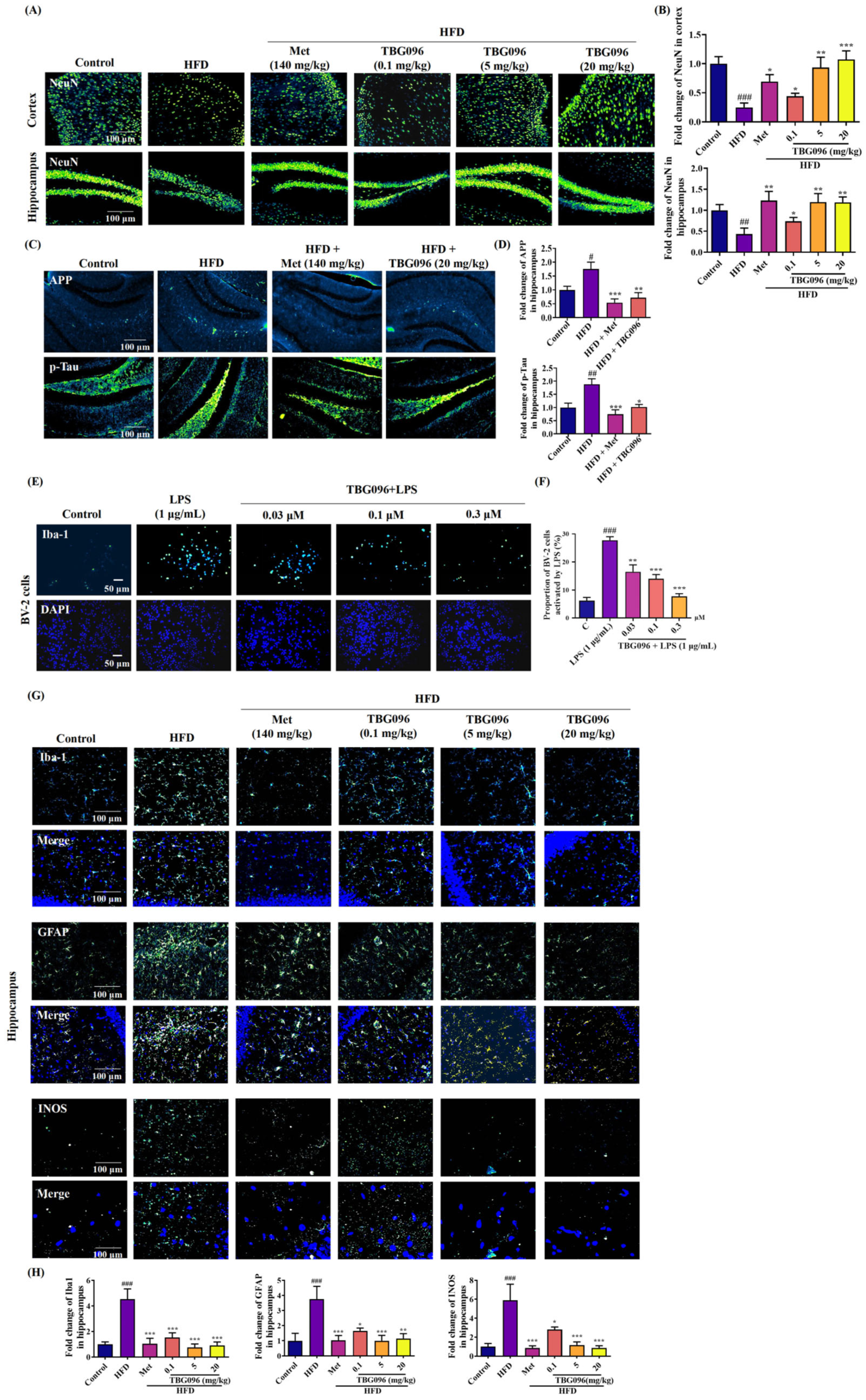
Key pathological features of AD include the loss of neurons [
33]. To evaluate the effects of TBG096 on neuronal integrity, immunofluorescence staining was performed on the cerebral cortex and hippocampus of AD mice. The analysis revealed marked reductions in mature neurons, increased APP, and phosphorylated Tau proteins due to long-term HFD feeding, which were notably reversed following TBG096 treatment, indicating that TBG096 has a neuroprotective effect (
Figure 3). Neuroinflammation is another major contributor to AD pathology, driven primarily by astrocytes and microglia [
34]. The effects of TBG096 on microglial and astrocytic activity as well as INOS levels were assessed (
Figure 3).
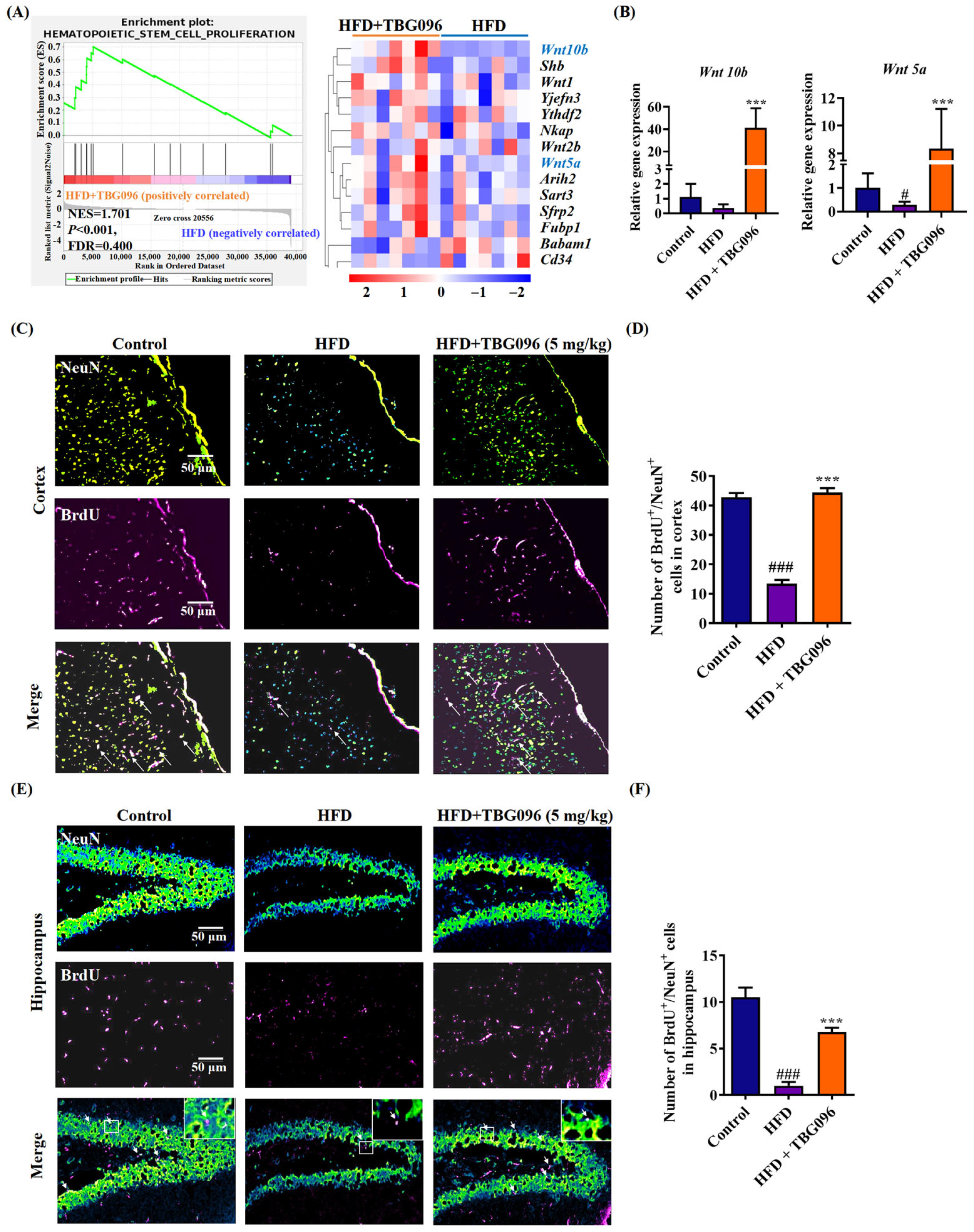
Reducing Aβ deposition has been a primary therapeutic focus for AD, but evidence supporting its ability to halt disease progression remains inconclusive. Recently, neuronal regeneration has been gradually accepted, and emerging evidence has reported that blocking the adult hippocampal neurogenesis (AHN) exacerbated cognitive impairment in AD mice, which exhibit typical AD pathological features such as Aβ aggregation; similar changes in AHN were also found in AD patients [
35]. Furthermore, combining neurogenesis with brain-derived neurotrophic factor (BDNF) has been shown to replicate the cognitive benefits of exercise in AD models [
3]. To explore the role of neurogenesis in the anti-AD effects of TBG096, the sequencing analysis of transcriptome and immunofluorescence staining of newborn neurons in the hippocampus and cortex of HFD mice was evaluated (
Figure 4 and
Figure S5). These experiments show that Wnt signaling is involved in the function of TBG096. Interestingly, treatment with TBG096 increased the number of newborn neurons in both regions. Although neurogenesis is traditionally associated with the hippocampus, studies have reported potential neurogenic activity in the cortex of adult non-human primates [
36]. Neurogenic activity observed in the cerebral cortex of TBG096-treated mice warrants further investigation to determine its extent and possible migratory behavior within the brain. These results confirmed that TBG096 promotes neurogenesis and mitigates neuroinflammation, contributing to its anti-AD effects.
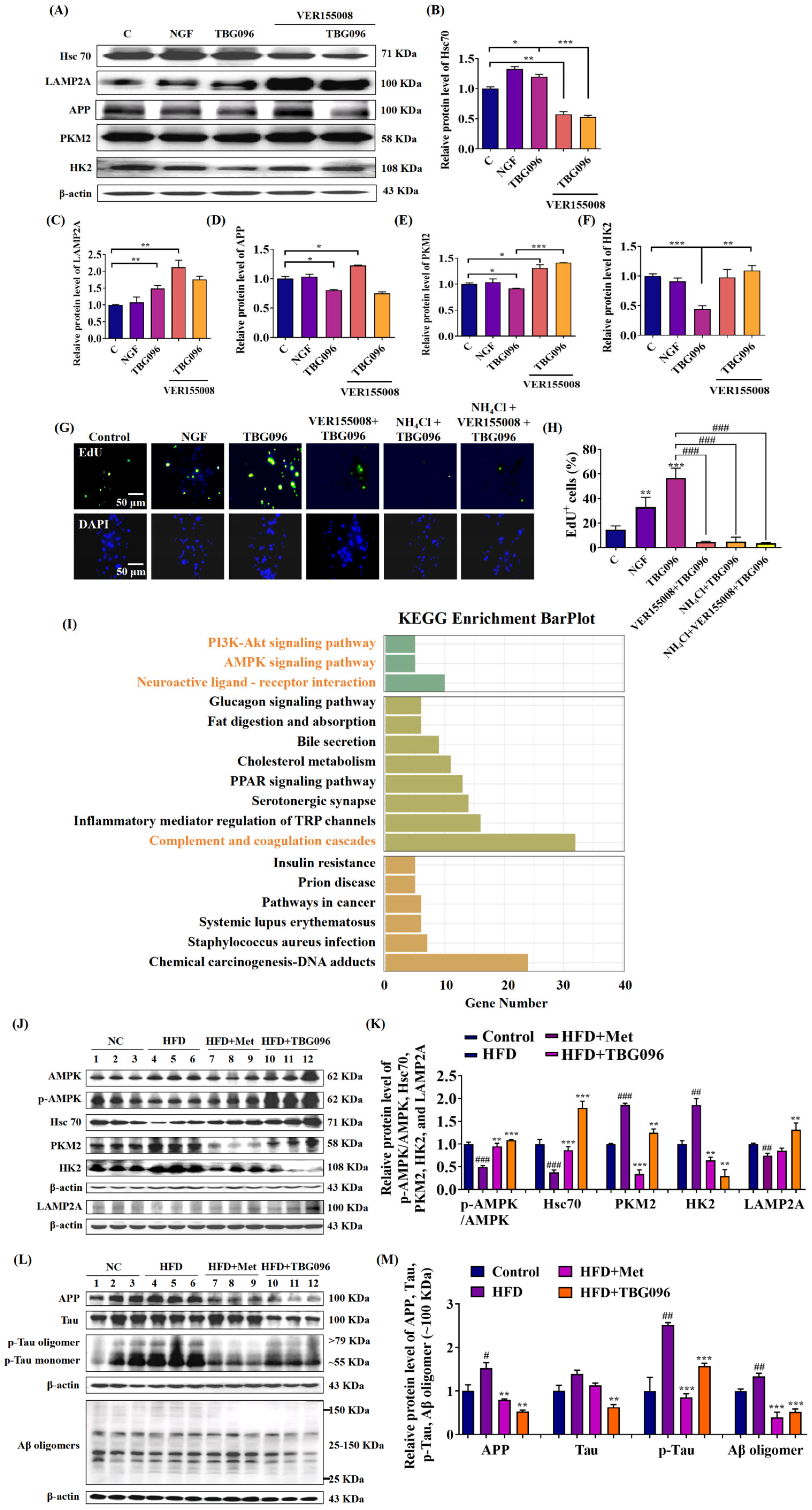
CMA is a selective autophagic process that degrades cytosolic proteins containing KFERQ motifs through lysosomal pathways [
16]. To indicate whether CMA participates in the NGF mimic and anti-AD effects of TBG096, the inhibitor experiment and Western blot analysis were conducted. The results in
Figure 5,
Figures S6 and S7 clarify that TBG096 exerted NGF-mimic activity in PC12 cells and anti-AD effect by activating Hsc70 and regulation downstream of CMA. The APP protein as one of the substrates of CMA is responsible for generating Aβ peptides, which subsequently aggregate to form senile plaques in the brain [
20]. Concurrently, the Tau protein, critical for stabilizing microtubules, undergoes hyperphosphorylation in AD, contributing to neuroinflammation, synaptic dysfunction, and cognitive decline [
37]. Thus, the effects of TBG096 on APP, Aβ, Tau, and p-Tau levels were evaluated. Significant reductions in these proteins after TBG096 treatment, as shown in
Figure 5 and
Figure S7, suggest that CMA may take part in the clearance of Aβ and p-Tau in AD mice of TBG096. In the present study, we only clarify that TBG096 produces anti-AD effects via promoting neurogenesis and modification of CMA. Whether CMA is involved in the neurogenesis of TBG096 needs more evidence to confirm.
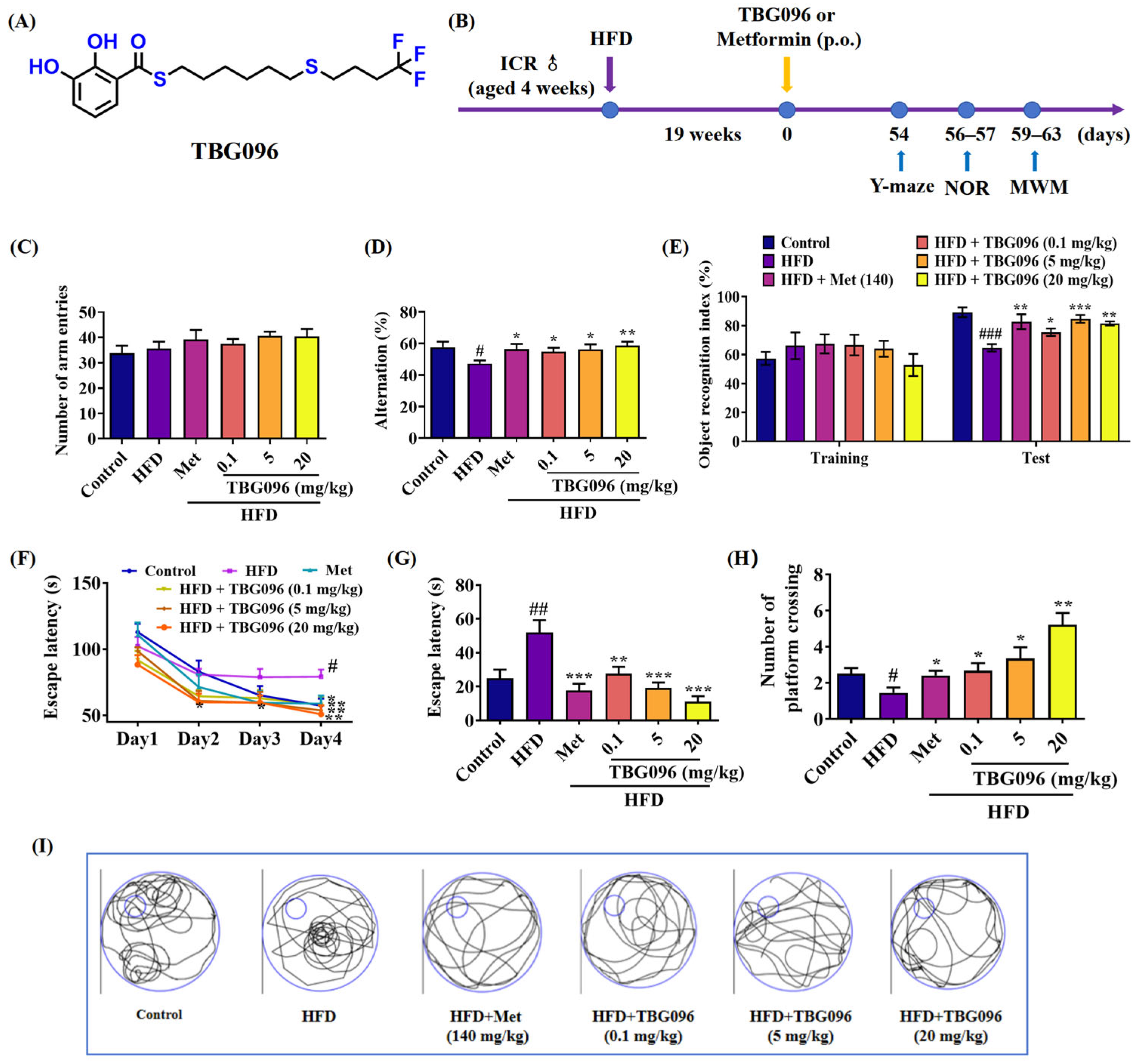
TBG096 was evaluated in the HFD-induced AD mice—selected for its close resemblance to human AD pathology. The behavioral assessments in
Figure 6 and
Figure S8 revealed that TBG096 at all doses of 0.1, 5, and 20 mg/kg exerted anti-AD effects in this model in a dose-dependent manner, and the optimal dose for TBG096 was 20 mg/kg, with comparable efficacy to Met at a concentration of 140 mg/kg. These results align with findings for other compounds such as CuB and 2H-GPS, which utilize different mechanisms to exert anti-AD effects [
38,
39]. CuB primarily acts through neurogenesis and neuroprotection, while 2H-GPS regulates the Wnt signaling pathway. In contrast, TBG096 exerts NGF-mimic activity in PC12 cells and addresses memory dysfunction in AD by promoting neurogenesis and regulation of the Hsc70 signaling pathway.
Additionally, we discovered a compound similar to TBG096 modulating the abundance of gut microbiota in previous studies [
40]. Hence, we examined TBG096′s impact on gut microbiota in HFD-induced AD mice. In HFD mice, the abundance of the beneficial gut bacteria
Ligilactobacillus was significantly decreased but improved with TBG096 (5 and 20 mg/kg) or Met (140 mg/kg) treatment (
Figure S9A). The principal coordinate analysis (PCoA) showed HFD altered gut microbiota composition, which TBG096 reversed towards normal levels, with 20 mg/kg closest to the control group (
Figure S9B). The α diversity analysis revealed significant differences between TBG096 (0.1 mg/kg) and HFD groups, with TBG096′s goods coverage near the control group (
Figure S9C). Phylum composition analysis showed the abundance of
Firmicutes was decreased and
Bacteroidota increased in the HFD group, but this was reversed with TBG096 (20 mg/kg) and Met (140 mg/kg) treatment towards normal levels (
Figure S9D). Thus, TBG096 may reshape gut microbiota by enhancing
Ligilactobacillus and normalizing
Firmicutes and
Bacteroidota, which are dominant in the intestinal microbial community. Clinical studies have shown that gut microbiota is related to the risk of AD [
41]. Changes in gut microbiota can increase the risk of AD by promoting neuroinflammation. In subsequent research, we will conduct an in-depth study on the relationship between specific gut microbiota modulated by TBG096 and neuroinflammation.
In conclusion, this study confirms TBG096 as a promising candidate drug for AD. This molecule has good bioavailability and can cross the BBB. TBG096 exerted NGF-mimic activity by Hsc70 and produced anti-AD effects in HFD-induced AD mice via promoting neurogenesis (
Figure 7). In the future, we will examine the potential limitations of TBG096, such as its long-term safety, impact on non-neuronal cells, and synergistic side effects with other AD treatment strategies. Meanwhile, we will focus on identifying the binding targets and binding sites of TBG096 and verify whether these proteins are indeed therapeutic targets responsible for the anti-AD effects of TBG096.
4. Materials and Methods
4.1. General
Silica gel (200–300 mesh, Yantai Research Institute of Chemical Industry, Yantai, China) and reversed-phase C18 (octadecylsilyl, ODS) silica gel (Cosmosil 75 C18-OPN, Nacalai Tesque, Kyoto, Japan) were used for column chromatography. Precoated silica gel (0.25 mm and RP-18 plate (0.25 mm; Merck KGaA, Darmstadt, Germany)) was used for TLC. The preparative HPLC was performed on an HPLC system equipped with ELITE P-230 pumps (Dalian Elite Inc., Dalian, China). NMR spectra were recorded on a Bruker AV III-500 spectrometer (Bruker, Karlsruhe, Germany). High-resolution (HR) ESI-TOF-MS were recorded on an Agilent 6224A LC/MS (Agilent Technologies Inc., Beijing, China).
4.2. Reagents and Antibodies
The sources of chemicals used in experiments are as follows: DMSO (#D4540), NGF (#N2513), LPS (#L2880), metformin (#317240), and BrdU (#B5002) were purchased from Sigma-Aldrich (St. Louis, MO, USA); VER155008 (#HY-10941) was purchased from MedChemExpress (Shanghai, China); NH4Cl (#A116372) was purchased from Aladdin Biochemical Technology (Shanghai, China). The following antibodies were used in this study: HK2 (#22029-1-AP), PKM2 (#15822-1-AP), APP (#60342), and Tau (#66499-1-Ig) from Protein Tech (Chicago, IL, USA); LAMP2A(#ET1601-24) from HuaAn Biotechnology (Hangzhou, China); Aβ (#8243S), AMPK (#2532), and p-AMPK (#2535) from Cell Signaling Technology (Boston, MA, USA); NeuN (#ab177487), BrdU (#ab8152), Hsc70 (#ab51052), p-Tau (phospho S396) (#ab32057), Iba1 (#ab5076), GFAP (#ab7260), INOS (#ab178945), and goat anti-rabbit IgG H&L (Alexa Fluor® 488, #ab6702) from Abcam (Cambridge, UK); rabbit anti-mouse IgG (Cy3 conjugate) from Millipore (Merck KGaA, Darmstadt, Germany); and β-actin (#CW0096M) and secondary antibody horseradish peroxidase [HRP]-linked anti-rabbit and anti-mouse IgGs (#CW0103) from CoWin Biotech Company (Beijing, China).
4.3. Synthesis and Purification of TBG096
The synthesis of TBG096 (compound 3) was used as an example, and the synthesis and purification procedures of TBG096 were undertaken as follows. At first, 1,6-Hexanedithiol (3.01 g, 20.00 mmol) was dissolved in anhydrous N,N-dimethylformamide (DMF) (50 mL). After that, 60% NaH (1.8 g, 45 mmol) was added at 0 °C, kept stirring for 30 min, and warmed up to room temperature for another 30 min. Subsequently, 1-bromo-4,4,4-trifluorobutane (3.0 mL, 20.00 mmol) and tetrabutylammonium iodide (TBAI) (740 mg, 2.00 mmol) were added in at 0 °C. Then, the mixture was stirred and warmed up to room temperature overnight. The reaction was stopped by adding 500 mL of EtOAc. The mixture was washed with 1 N HCl solution, water, and saturated aqueous solution of NaHCO3 and NaCl, successively. The organic phase was dried over Na2SO4, filtered, and then concentrated. Next, the product of the first step was dissolved in anhydrous CH2Cl2 (50 mL), and DMAP (2.44 g, 20.0 mmol), EDC·HCl (3.82 g, 20.0 mmol), and 2,3-dihydroxybenzoic acid (1.54 g, 10.00 mmol) were added into this mixture. The mixture was stirred overnight at room temperature and then concentrated under vacuum. The crude mixture was purified by silica gel open column (n-hexane: EtOAc = 80:1) and ODS open column (Methanol: H2O = 80:20) and then purified by HPLC (SP-ODS-A (20 × 250 mm), MeOH/H2O = 87:13 in 40 min, flow rate: 8 mL/min, detected at 254 nm) to obtain a pure compound 3 (TBG096, tR = 28 min, 5 injections, 249 mg, yield 6.3%).
The structure of TBG096 was determined by
1H NMR,
13C NMR, and HR ESI-MS data.
1H NMR (500 MHz, CDCl
3):
δ = 11.24 (1H, s), 7.41 (1H, dd,
J = 1.4, 8.0 Hz), 7.11 (1H, dd,
J = 1.4, 8.0 Hz), 6.82 (1H, t,
J = 8.0 Hz), 5.67 (1H, s), 3.07 (2H, t,
J = 7.3 Hz), 2.55 (4H, m), 2.22 (2H, m), 1.85 (2H, m), 1.70 (2H, m), 1.60 (2H, m), 1.45 (4H, m).
13C NMR (125 MHz, CDCl
3):
δ = 197.9, 146.8, 145.4, 128.3 (q,
1J (C, F) = 276.3 Hz), 120.1, 119.9, 119.5, 77.4, 32.6 (q,
2J (C, F) = 28.6 Hz), 31.9, 31.0, 29.5, 29.3, 28.8, 28.5, 28.4, 22.0 (q,
3J (C, F) = 2.6 Hz). HR ESI-TOF-MS
m/z 419.0933, calcd. for C
17H
23F
3O
3S
2Na [M + Na]
+ 419.0933. The synthesis and purification procedures of compounds
1,
2, and
4 and other details are presented in the
Supporting Information (Sections S1.1 and S1.2; Figures S1 and S2).
4.4. Cell Culture and Activity Evaluations for Neurogenesis and Anti-Inflammation
PC12 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM medium (Dulbecco’s Modified Eagle Medium, CellMax, Lanzhou, China), supplemented with 10% HS (Horse Serum, Solarbio, Beijing, China), 7.5% FBS (Foetal Bovine Serum, CellMax, Lanzhou, China), and 1% penicillin/streptomycin (Solarbio, Beijing, China).
The neurogenic activity of samples was evaluated according to the methods described in our previous study [
24]. Briefly, approximately 5 × 10
4 cells were seeded in each well of a 24-well microplate and were cultured under a humidified atmosphere with 5% CO
2 at 37 °C for 24 h. Then, the medium in each well was replaced with 1 mL of serum-free DMEM containing 40 ng/mL NGF as a positive control, 0.5% DMSO as a negative control, and the test sample after 24 h, respectively. About 100–200 cells of each well were counted from a randomly selected area and repeated thrice after 48 h. A cell-bearing neurite outgrowth longer than the diameter of the cell body was identified as a positive cell. The activity was presented as the percentage of positive cells in the selected area.
BV-2 cells were obtained from Nanjing CoBioer Biotechnology Ltd., Co., Nanjing, China, and cultured in RPMI 1640 medium (Roswell Park Memorial Institute 1640 medium, Gibco, Grand Island, NY, USA) supplemented with 10% FBS and 1% penicillin/streptomycin. The anti-inflammatory activity of samples in BV-2 cells was determined as follows. Approximately 5 × 104 cells were seeded in each well of a 24-well microplate and were cultured under a humidified atmosphere with 5% CO2 at 37 °C for 24 h. Then, the medium was replaced by 500 µL RPMI 1640 medium containing 0.5% DMSO or samples at different concentrations for 2 h, and 500 µL medium containing LPS (1 µg/mL) was added and incubated for 24 h. Next, the medium of each well was sucked out, and each well was washed with phosphate-buffered saline (PBS) three times and fixed with 4% paraformaldehyde (500 µL/well) for 20 min at room temperature. The fixative was discarded, washed with PBS, and blocked for 60 min. After removing the blocking solution, the primary antibody (Anti-Iba1 rabbit monoclonal antibody) was diluted 1000 times, and 200 µL of diluted primary antibody was added to each well and then incubated at 4 °C overnight. The next day, the wells were washed with PBS, and goat anti-rabbit IgG (250 µL/well) diluted 1000 times was added and incubated at room temperature for 1 h. After that, the cell nucleus was stained with diluted DAPI (SouthernBiotech, Birmingham, AL, USA) for 10 min, and the fluorescence image of BV-2 cells was observed under the fluorescence inverted microscope. Three regions were randomly selected to be observed and recorded with pictures. Image J software version 1.8.0 (National Institutes of Health, Bethesda, MD, USA) was finally used to analyze the fluorescence density of BV-2 cells.
4.5. Methyl Thiazolyl Tetrazolium (MTT) Assay
PC12 cells were firstly inoculated into a 24-well plate and cultured for 24 h. Following this, samples at different doses were added and cultured for 24 h. Subsequently, the 0.5 mL serum-free medium containing MTT (0.2 mg/mL) of each well was used to replace the original medium and incubated for 2 h. After removing the solution, the purple crystal was dissolved in 200 μL of DMSO, and the absorbance was measured at 570 nm. The cell survival rate for each group was calculated based on the control group.
4.6. Inhibitor Experiment
In the inhibitor experiment of NGF-mimic activity, approximately 5 × 104 PC12 cells were seeded into each well of a 24-well plate and incubated for 24 h. After that, the medium in each well was replaced with 500 μL of serum-free DMEM or the serum-free DMEM containing VER155008, the inhibitor of Hsc70 protein, incubated in a CO2 incubator for 30 min. Subsequently, 500 μL of serum-free DMEM containing different samples was added and cultured, respectively. The PC12 cells with neurite outgrowth were recorded after 48 h. For the Western blot analysis, the experiment included five groups, namely the control (C) group, the NGF (40 ng/mL) group, the TBG096 (1 µM) group, the VER155008 (3 µM) group, and the VER155008 (3 µM) + TBG096 (1 µM) group. Afterwards, approximately 2 × 106 cells per dish were seeded into ten 6 cm Petri dishes with 5 mL of DMEM medium and incubated for 24 h. After giving different treatments with samples, the cells continued to culture for 4 h. Protein extraction was conducted to prepare for the subsequent Western blot analysis.
In the inhibitor experiment of EdU cell proliferation activity, PC12 cells were seeded into each well of a 24-well plate as above for 24 h. Then, the medium in each well was replaced with 500 μL of serum-free EM containing VER155008 (inhibitor of Hsc70, 3 µM), or NH
4Cl (inhibitor of lysosome, 250 µM) for 30 min according to the literature [
24,
42], and 500 μL of serum-free DMEM containing different samples was added and cultured for 24 h, respectively. After that, PC12 cells were treated with EdU for 2 h according to the operation steps of the BeyoClick™ EdU-488 cell proliferation detection kit (#C0071S, Beyotime Biotechnology, Shanghai, China). After cell labeling, the supernatant was removed. Then, PC12 cells were fixed, washed, and blocked, and the click reaction solution was added at room temperature for 30 min. After that, DAPI staining was performed for 10 min. Finally, PC12 cells were detected by fluorescence microscope (Olympus IX53, Tokyo, Japan).
4.7. Design of Animal Experiments
In experiments with the HFD-induced AD mouse model, a total of 60 Institute of Cancer Research (ICR) male mice aged 4 weeks from Zhejiang Academy of Medical Sciences (Hangzhou, China) were used for animal experiments (permit number: ZJU20230121). The animals were provided free access to a chow diet and water. After one week of acclimatization, ten mice were fed a normal chow diet, whereas the remaining 60 mice were fed the HFD (24.2% crude protein, 42.1% carbohydrate, and 25.4% fat; Biotechnology HD Co., Ltd., Beijing, China). The HFD and normal diet were alternated every two days to maintain freshness. Weekly measurements were taken for water intake, food intake, and body weight. In the eighteenth week, the mice underwent Y-maze and NOR tests. The Morris water maze test was conducted during the nineteenth week. After confirming the successful establishment of the AD model characterized by cognitive impairment, oral administration of treatments commenced. The mice were randomly divided into the following groups: (i) control group, (ii) HFD-only group, (iii) HFD + Met 140 mg/kg group, (iv) HFD + TBG096 0.1 mg/kg group, (v) HFD + TBG096 5 mg/kg group, and (vi) HFD + TBG096 20 mg/kg group. Each group had 10 mice. Soybean oil and MilliQ water were used as vehicles to dissolve TBG096 and Met. The drugs were orally administrated to mice once a day according to the different doses of TBG096 and Met, and this animal experiment was conducted for three months.
In order to investigate the effect of TBG096 on promoting neuronal regeneration, animal experiments were carried out according to the method previously reported [
38]. Fifteen 4-week-old ICR male mice purchased from Zhejiang Academy of Medical Sciences (Hangzhou, China) were randomly divided into three groups, namely the (i) normal control group, (ii) HFD group, and (iii) HFD + TBG096 5 mg/kg group, and given a normal diet or high-fat diet for two months to construct the AD mouse model, respectively. After confirming the successful construction of the AD model, TBG096 was administered once a day at a dose of 5 mg/kg for 1 month, and soybean oil was used as a vehicle. BrdU was then injected into the mice at a dose of 100 mg/kg in four injections over two days, each 12 h apart. Administration of TBG096 was continued for 28 days. At the end of animal experiments, the mice were anesthetized and cardiac perfusion stopped. The brains of mice were taken, fixed, and sectioned, and immunostaining was performed with NeuN and BrdU antibodies.
In the acute toxicity experiment of mice, a total of 40 ICR male mice aged 8 weeks from Zhejiang Academy of Medical Sciences (Hangzhou, China) were used. A standard experimental scheme was adopted according to the requirements of Zhejiang University Animal Experiment Ethics Committee (animal use license number: SYXK(Zhe)2019-0002, permit number: ZJU20230219). The mice were randomly divided into the following groups: (i) control group, (ii) TBG096 100 mg/kg group, (iii) TBG096 300 mg/kg group, and (iv) TBG096 600 mg/kg group. Each group had 10 mice. Soybean oil was used as a vehicle to dissolve TBG096. The body weight, food intake, and water consumption of mice in each group were recorded every day for two weeks. Blood was collected for biochemical index analysis.
4.8. The Pharmacokinetic Analysis of TBG096
In the pharmacokinetic analysis, a total of 23 female SD rats aged 8 weeks from Zhejiang Academy of Medical Sciences (Hangzhou, China) (permit number: ZJU20240191) were used. A total of 15 rats were randomly divided into the following groups: (i) control group, (ii) TBG096 (14 mg/kg) oral administration group, and (iii) TBG096 (14 mg/kg) intravenous administration group. TBG096 was administered orally (dissolved in soybean oil) or intravenously (dissolved in Milli-Q water containing 2% Tween 80 and 5% ethanol) at 14 mg/kg after 12 h of fasting. Then, the blood from the posterior ocular venous plexus of rats was taken at 5, 15, 30, 60, 90, 120, 180, and 240 min after administration. A total of eight rats were randomly divided into two groups for taking brain samples: (i) control group and (ii) TBG096 (14 mg/kg) oral administration group.
The collected blood was placed in a centrifuge tube (containing 20 µL heparin sodium solution) and centrifuged at 3000 rpm for 10 min to obtain a supernatant (plasma). Brain samples were collected after the oral administration of 14 mg/kg TBG096 and heart perfusion with PBS. Then, 10 µL of 10% formic acid and 10 µL of internal standard (10 ng/µL) were added to 200 µL of plasma (compound 2 was used as internal standard) or 0.2 g of brain sample (compound 4 was used as internal standard) and vortexed for 10 s and then left at −80 °C overnight. Next, 600 µL or 3 mL of extraction solvent (isopropanol) was added to plasma or brain samples, vortexed for 5 min, left at −30 °C for 2 h, and then centrifuged at 12,000 rpm for 15 min. Next, the upper organic phase was transferred to another tube, centrifuged, and concentrated to dryness. The residue was dissolved in 100 µL of mobile phase, vortexed for 0.5 min, and centrifuged at 12,000 rpm for 3 min, and the supernatant was then prepared for HPLC analysis (Supersil Phenyl 5 µm (4.6 × 200 mm), elution with MeCN/H2O containing 0.1% formic acid (plasma samples: 57% MeCN/H2O, brain samples: 56% MeCN/H2O) for 30 min, flow rate: 1 mL/min, sample size: 20 µL, detected at 284 nm) and HR ESI-TOF-MS analysis.
4.9. Animal Behavioral Tests
4.9.1. Y-Maze Test
The Y-maze test is used to evaluate working memory, and the process was referenced in another report [
39]. In brief, the Y-maze consists of three black plastic arms (A, B, and C) with the same length (30 cm), width (10 cm), height (15 cm), and 120° relative to adjacent arms. Each mouse is placed facing the wall of arm A and allowed to explore the maze freely for 5 min. The behavior of mice is recorded with a video recorder, and the data are stored in ANY-MAZE software version 6.35 (Stoelting, Chicago, IL, USA). The total number of entries made by the mice into each arm—considered as either whole or half-body entries—is recorded. An alternation is defined as a mouse moving through all three arms without returning to the initial arm on the third visit. Alternation: number of alternations/(number of arm entries − 2) × 100%.
4.9.2. Novel Object Recognition (NOR) Test
The NOR test is utilized to assess the working memory capabilities of mice. The NOR apparatus consists of an open, black plastic box, two identical objects, and one novel object. The activity of the mice within the open field is captured and recorded using a video recorder, with data subsequently stored in ANY-MAZE software. The test is conducted over a span of two days, incorporating habituation, training, and testing stages. During the habituation phase, the mice are placed in the box devoid of any objects, allowing them to explore freely for a duration of five min. Following a 24 h interval, the NOR test is administered, which consists of both the training and testing phases. In the training phase, each mouse is positioned in the central area of the box, where two identical objects are placed equidistant from the mouse. Each training session lasts for five minutes. After a one-hour break, one of the identical objects is replaced with a new, distinct object, and the exploration duration for each mouse is recorded. Exploration is defined by the time a mouse spends touching or sniffing an object. The exploration times during both the training and testing phases are documented via video recording, and the data are organized within ANY-MAZE software. The object recognition index was defined as follows: (time spent on the new object)/(time spent on the old object + time spent on the new object) × 100%. The data are calculated by ANY-MAZE software.
4.9.3. Morris Water Maze (MWM) Test
The MWM test is used to measure spatial memory and long-term memory. The whole MWM equipment includes a circular water tank (diameter 1.5 m, depth 50 cm) with warm water (22 ± 1 °C) 35 cm deep, a platform (diameter 14 cm, immersed 1 cm in water), a camera, and a computer for data analysis. The MWM test is conducted over five 5 days: 4 days for training and 1 day for testing. The pool is divided into four quadrants, and at least three quadrants are tested at a time. In the training stage, the mouse is placed on the wall of one of the four quadrants and allowed to swim and find the platform hidden underwater within 120 s. If the mouse cannot find the platform, the experimenter will guide it and let it stay on the platform for 10 s. The experiment lasts for 4 days. On the fifth day of the test phase, the platform is removed, and each mouse is placed in a similar starting position and allowed to move freely for 90 s. The swimming trajectories are recorded and stored in the computer. The time taken by each mouse to reach its destination (escape delay) and the number of times the mouse passes through the platform are recorded by the camera, respectively, and analyzed using ANY-MAZE software.
4.10. Immunostaining of Brain Tissues
At the end of animal experiments, three mice in each group were taken for heart perfusion with cold PBS and 4% paraformaldehyde. After that, the brains of mice were fixed with 4% paraformaldehyde for 24 h and gradually dehydrated with 15% and 30% sucrose solutions for 48 h, respectively. The fixed and embedded brain sample was placed in a cryostat and cut into 20 µm thick slices with a cryostat (Thermo Fisher, Waltham, MA, USA), and brain slices in the hippocampus and cortex were collected. Finally, the brain slices were placed in antifreeze (glycerol/ethylene/PBS = 3:3:4) and kept in a refrigerator at −30 °C until immunostaining. The brain slices were fixed on a glass slide and hydrated with PBS for 5 min. Then, blocking buffer was added to the brain slices and incubated at room temperature for 1 h. Brain slices were washed twice with PBS and incubated with anti-NeuN, anti-BrdU, anti-Iba1, anti-GFAP, anti-INOS, anti-APP, or anti-p-Tau at 4 °C for 24 h, respectively. Subsequently, the brain slices were incubated with secondary antibody (goat anti-rabbit IgG H&L (Alexa Fluor® 488) or rabbit anti-mouse IgG (Cy3 conjugate)) at room temperature for 2 h after washing with PBS three times. Finally, a cover glass was used to lightly cover the slices stained with DAPI. The slides loaded with brain sections were observed with an upright two-photon confocal microscope (Olympus BX61, Tokyo, Japan). Biomarker proteins in the cerebral cortex and hippocampus were digitized by Image J software (National Institutes of Health, Bethesda, MD, USA).
4.11. Western Blot Analysis
Approximately 50 mg of cerebral cortex from brain tissues was homogenized in RIPA lysis buffer containing 1% protease inhibitor and 1% phosphatase inhibitor. Then, the sample was centrifuged at 12,000 rpm at 4 °C for 10 min, and the supernatant was transferred to a new tube. Protein concentrations were determined using the BCA kit, and 30 µg of protein from each sample was denatured by heating in water at 100 °C for 10 min. Subsequently, approximately 30 µg of protein from each sample was loaded into each well. After conducting gel electrophoresis at 80 V for 15 min and then at 120 V for 60 min, the proteins were transferred to the polyvinylidene fluoride (PVDF) membrane for 90 min. Then, the PVDF membrane was blocked with 5% skimmed milk at room temperature for 60 min. The membrane was incubated with a diluted primary antibody at 4 °C overnight. After washing the membrane three times with TBST buffer (1× TBS buffer containing 0.05% (v/v) tween 20), it was incubated with the secondary antibody for 1 h at room temperature. Finally, the strip was developed using chemiluminescence ECL reagent, and the blot density was quantified with ImageJ (National Institutes of Health, Bethesda, MD, USA).
4.12. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted and quantified from 50 mg of the cerebral cortex using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA), respectively. Subsequently, 2.5 µg of RNA from each sample was reverse-transcribed using a HiFi-MMLV cDNA kit (Cowin Biotechnology Company, Beijing, China). Transcripts were quantified by PCR analysis performed with a CFX 96 Touch (Bio-Rad, Hercules, CA, USA) and SYBR Premixed EX Taq (Takara, Otsu, Japan). PCR was performed under the following conditions: 95 °C for 2 min, followed by 40 cycles, 94 °C for 15 s, 60 °C for 25 s, and 72 °C for 10 s. All results were normalized to the level of 18S RNA, and the relative mRNA transcription levels were estimated by 2−ΔΔCT. The number of animal samples in each group was five, and all samples were run in triplicate, and their average values were calculated. The mouse primers of Wnt10b (NM_011718.2), Wnt5a (NM_001256224.2), and 18S RNA (NR_003278.3) were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China), and their sequences are as follows: Wnt10b-sense: 5′-AATGCCATGGAACGTCAGGC-3′; antisense: 5′-CTCCAGAGTTGCGGTTGTGG-3′. Wnt5a-sense: 5′-AGACCTCAGAGGGGATGGAC-3′; antisense: 5′-TCTCCGTGCACTTCTTGCAT-3′. 18S-sense: 5′-TAACCCGTTGAACCCCATT-3; antisense: 5′-CCATCCAATCGGTAGTAGCG-3′.
4.13. RNA Sequencing Analysis
After extracting total RNA (5 µg) from cerebral cortex samples, mRNA was purified using Dynabeads Oligo(dT) (Thermo Fisher, Waltham, MA, USA), involving two purification steps. The purified mRNA was then cleaved into smaller fragments at 94 °C for 5–7 min. These fragments were reverse-transcribed into cDNA using SuperScript™ II Reverse Transcriptase (Invitrogen, cat. 1896649, Carlsbad, CA, USA). The resulting cDNA served as a template for synthesizing U-labeled second-strand DNAs with E. coli DNA polymerase I, RNase H from NEB, and dUTP solution (Thermo Fisher, cat. R0133, Waltham, MA, USA). The ends of these DNA strands were blunted and ligated with dual-index adapters, followed by size selection using AMPure XP beads. The U-labeled second-strand DNAs were treated with a heat-labile UDG enzyme from NEB and then underwent PCR amplification. The PCR protocol was performed as follows: 95 °C for 3 min, followed by eight cycles of denaturation at 98 °C for 15 s, 60 °C for 15 s, and 72 °C for 5 min. The final cDNA libraries had an average insert size of 300 ± 50 bp. Finally, the Illumina NovaSeq™ 6000 system (Illumina, San Diego, CA, USA) was utilized to perform 2 × 150 bp paired-end sequencing (PE150), following the manufacturer’s protocol. This analysis was conducted by LC-Bio Technology Co., Ltd. in Hangzhou, China.
4.14. Biostatistical Analysis
The statistical analysis was carried out by using GraphPad Prism biostatistics (GraphPad Prism 9.0, San Diego, CA, USA). The significant differences between control and treatment groups were analyzed by one-way ANOVA or unpaired t-test. All the data were expressed by mean ± SEM, and p < 0.05 indicated statistical significance.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}