
Mechanotransduction and Skeletal Muscle Atrophy: The Interplay Between Focal Adhesions and Oxidative Stress


Abstract
1. Introduction
2. Mechanotransduction in Skeletal Muscle

{kind=link}
{kind=link}
{kind=link}
| Mechanosensory Protein | Function | Mechanisms | References |
|---|---|---|---|
| Neuronal Nitric Oxide Synthase (nNOS) | Regulates blood flow, insulin-induced glucose uptake, satellite-cell activation, and muscle regeneration. | Localizes to the sarcolemma, interacts with the dystrophin–glycoprotein complex, and produces nitric oxide (NO), modulating vasodilation and glucose uptake. Disruption leads to impaired repair and increased inflammation. | [38,39,40,41,42,43] |
| Perlecan | Acts as a mechanosensory protein regulating metabolism, muscle growth, and repair. | Binds ECM components, interacts with growth factors, and modulates hypertrophy/atrophy pathways. Promotes nNOS delocalization during atrophy, enhancing protein degradation. | [44,45,46,47] |
| Focal-Adhesion Complex | Links actin cytoskeleton to the ECM, transmitting mechanical signals for gene expression and cellular adaptation. | Composed of integrins, talin, vinculin, and FAK, mediating biochemical responses to mechanical stimuli. Dysfunction leads to disrupted force transmission and impaired regeneration. | [48,49,50] |
| Dystrophin–Glycoprotein Complex (DGC) | Maintains sarcolemma integrity and transmits contractile forces. | Anchors actin cytoskeleton to the ECM, interacting with dystroglycans and sarcoglycans. Loss of function leads to sarcolemma instability. | [51,52,53] |
| Dysferlin | Facilitates membrane repair, which is critical for muscle recovery post injury. | Involved in vesicle fusion and resealing, interacting with caveolin-3 and annexins. Deficiency results in impaired repair and chronic inflammation. | [54,55,56,57] |
| Integrins | Mediates cell–ECM adhesion and transmits mechanical signals. | Binds ECM proteins like fibronectin/collagen and activates intracellular signaling via FAK. Contributes to stress fiber formation and mechanosensing. | [58,59,60,61,62] |
| Talin | Links integrins to the actin cytoskeleton and supports focal adhesion signaling. | Binds integrin cytoplasmic tails, recruiting vinculin and other focal adhesion proteins. Impairment weakens focal-adhesion strength, leading to atrophy. | [36,63,64,65] |
| Vinculin | Reinforces cell–ECM adhesion and regulates cell motility. | Interacts with talin and actin, stabilizing focal adhesions and modulating actin dynamics. Essential for maintaining structural integrity during stress. | [66,67,68,69,70] |
| Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) (YAP/TAZ) | Transcriptional co-activators modulate gene expression via mechanical signals. | Translocate to the nucleus under mechanical stimuli, interacting with transcription factors. Critical for stress adaptation, dysregulation promotes atrophy. | [24,71,72,73,74] |
| Piezo 1/2 | Mechanosensitive ion channels regulate the cellular response to mechanical stimuli. | Open under force to mediate ion influx, triggering downstream signaling pathways. Hyperactivation disrupts homeostasis and promotes oxidative stress. | [22,75,76,77] |
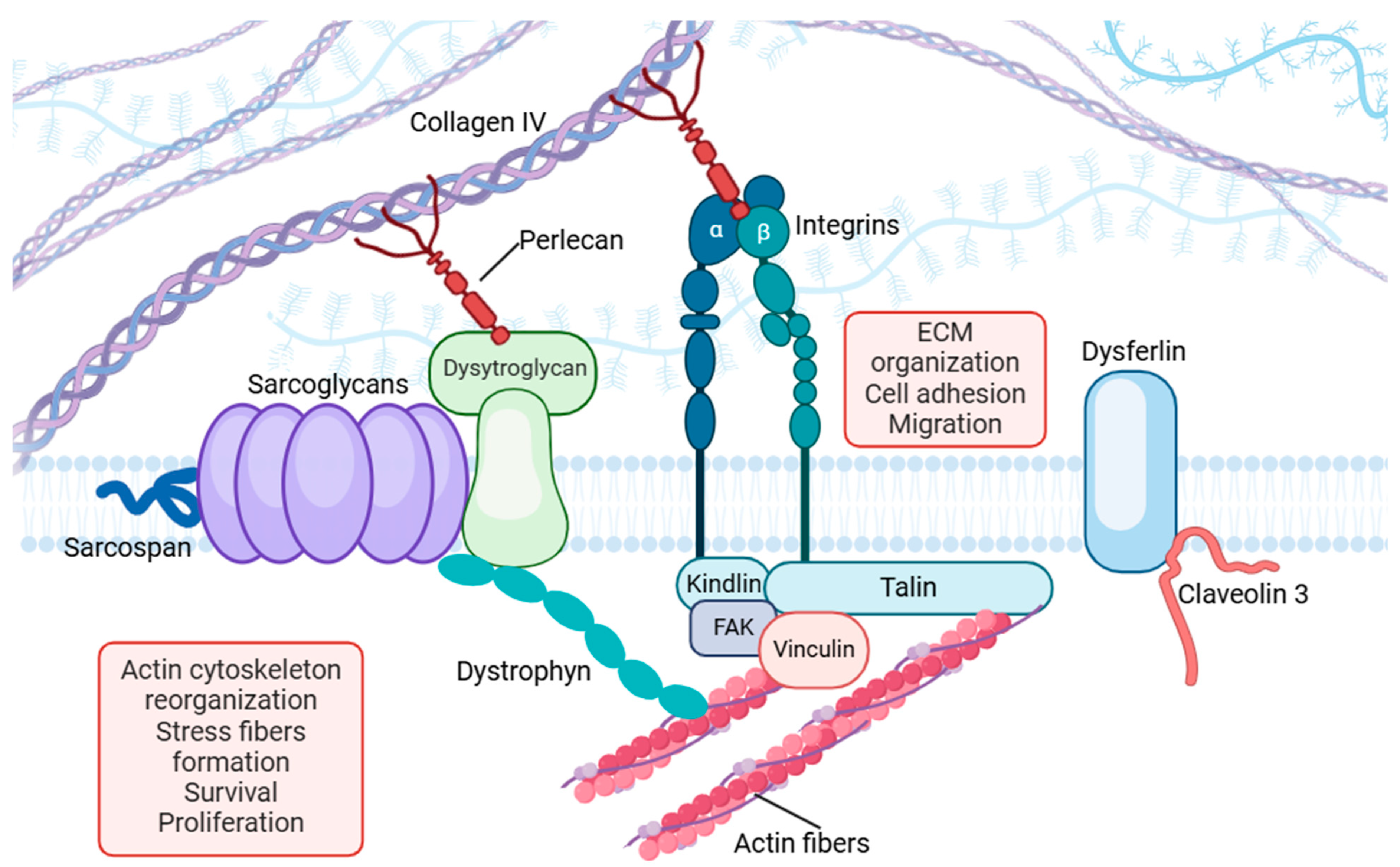
3. Focal Adhesions as Mechanosensors
3.1. Key Molecular Components in Focal Adhesions
3.1.1. Integrins
3.1.2. Focal Adhesion Kinase (FAK)
3.1.3. Talin and Vinculin
3.1.4. Dysferlin
3.1.5. Perlecan
3.2. Functional Integration of Focal Adhesion Components
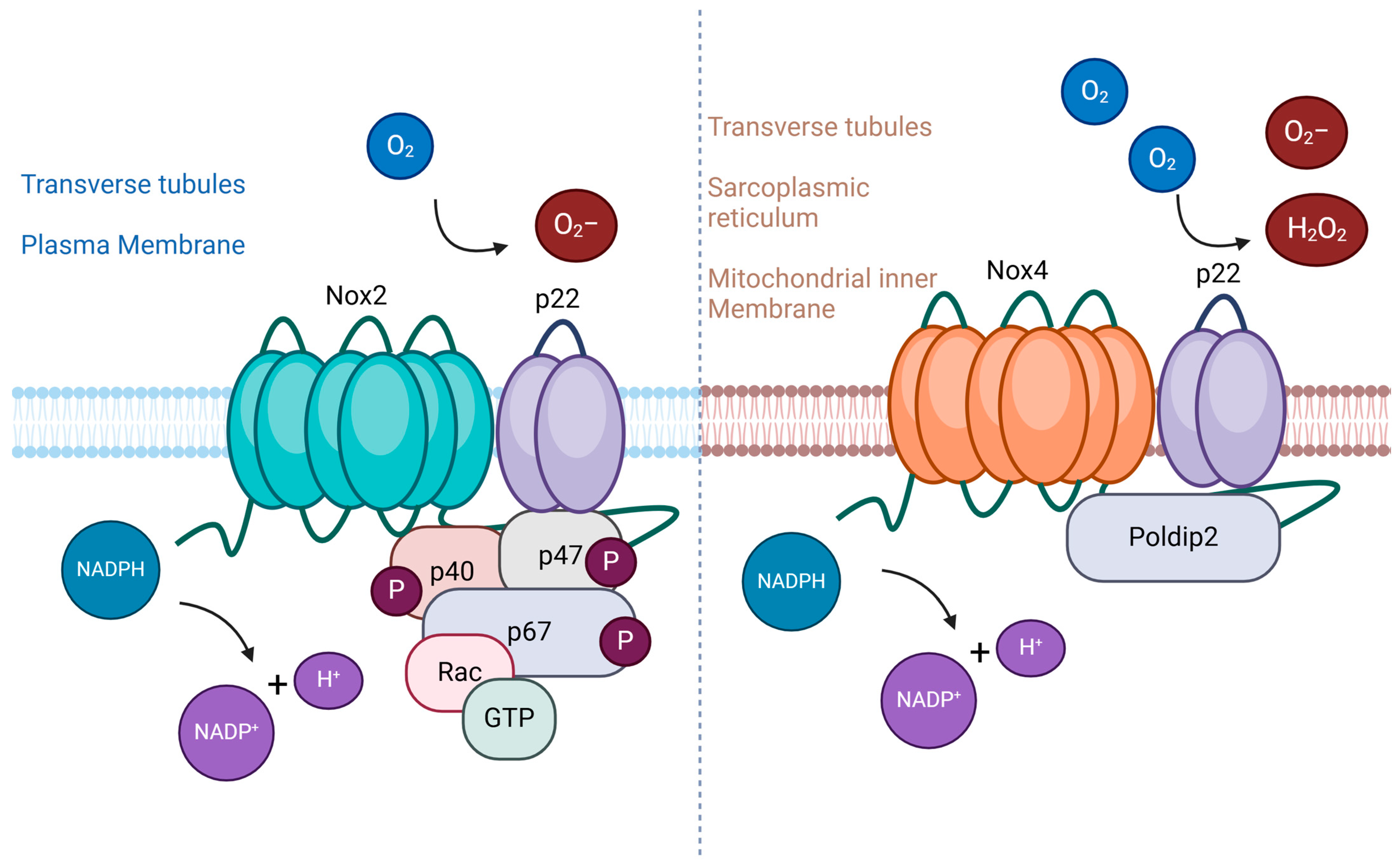
4. Oxidative Stress in Muscle Atrophy
4.1. NADPH Oxidase Family as Key Mediator
4.2. Therapeutic Strategies Targeting Oxidative Stress and NOX2
4.2.1. Antioxidants
4.2.2. Anti-Inflammatory Agents
4.2.3. Controlled Physical Activity and Multimodal Therapies
4.2.4. Heat-Shock Proteins
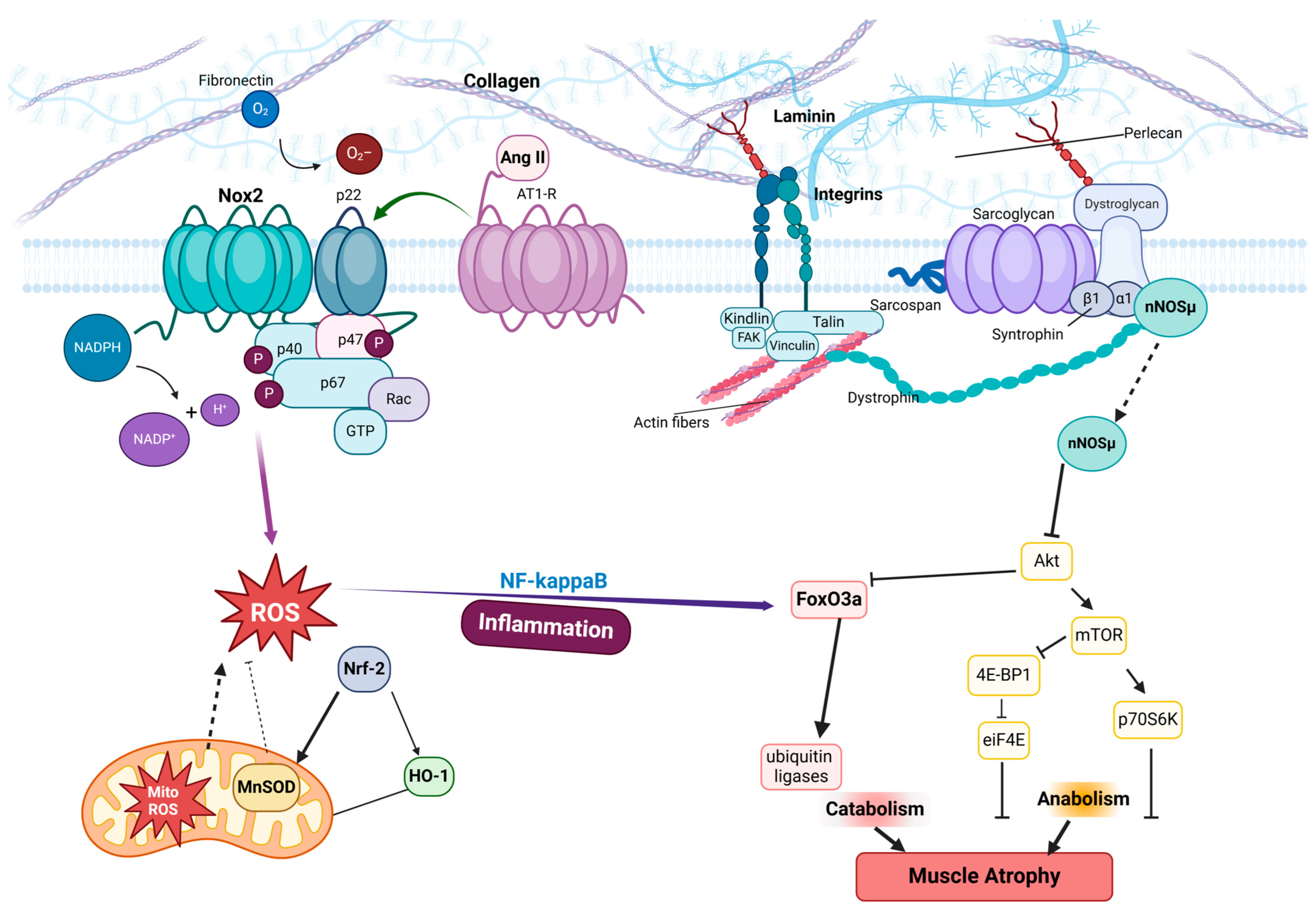
5. Interplay Between Focal Adhesions and Mechanical Unloading
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Okimura, Y. Branched chain amino acids and muscle atrophy protection. Branched Chain Amino Acids Clin. Nutr. 2015, 2, 49–63. [Google Scholar]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin–proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar]
- Gao, Y.; Arfat, Y.; Wang, H.; Goswami, N. Muscle atrophy induced by mechanical unloading: Mechanisms and potential countermeasures. Front. Physiol. 2018, 9, 235. [Google Scholar]
- Tascher, G.; Brioche, T.; Maes, P.; Chopard, A.l.; O’gorman, D.; Gauquelin-Koch, G.; Blanc, S.p.; Bertile, F. Proteome-wide adaptations of mouse skeletal muscles during a full month in space. J. Proteome Res. 2017, 16, 2623–2638. [Google Scholar] [PubMed]
- Teodori, L.; Costa, A.; Campanella, L.; Albertini, M.C. Skeletal muscle atrophy in simulated microgravity might be triggered by immune-related microRNAs. Front. Physiol. 2019, 9, 1926. [Google Scholar]
- Lawler, J.M.; Hord, J.M.; Ryan, P.; Holly, D.; Janini Gomes, M.; Rodriguez, D.; Guzzoni, V.; Garcia-Villatoro, E.; Green, C.; Lee, Y.; et al. Nox2 Inhibition Regulates Stress Response and Mitigates Skeletal Muscle Fiber Atrophy during Simulated Microgravity. Int. J. Mol. Sci. 2021, 22, 3252. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.T.; Yang, X.; Tian, R.; Li, Y.H.; Wang, C.Y.; Fan, Y.B.; Sun, L.W. Cells respond to space microgravity through cytoskeleton reorganization. FASEB J. 2022, 36, e22114. [Google Scholar]
- Evans, W.J. Skeletal muscle loss: Cachexia, sarcopenia, and inactivity. Am. J. Clin. Nutr. 2010, 91, 1123S–1127S. [Google Scholar]
- Fitts, R.H.; Riley, D.R.; Widrick, J.J. Functional and structural adaptations of skeletal muscle to microgravity. J. Exp. Biol. 2001, 204, 3201–3208. [Google Scholar]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Models Mech. 2013, 6, 25–39. [Google Scholar]
- Gordon, B.S.; Kelleher, A.R.; Kimball, S.R. Regulation of muscle protein synthesis and the effects of catabolic states. Int. J. Biochem. Cell Biol. 2013, 45, 2147–2157. [Google Scholar]
- Bashan, N.; Kovsan, J.; Kachko, I.; Ovadia, H.; Rudich, A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol. Rev. 2009, 89, 44. [Google Scholar]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [PubMed]
- Burkholder, T.J. Mechanotransduction in skeletal muscle. Front. Biosci. J. Virtual Libr. 2007, 12, 174. [Google Scholar]
- Baumgarten, C.M. Origin of Mechanotransduction: Stretch-Activated ion Channels. Madame Curie Bioscience Database; 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6374/ (accessed on 17 March 2025).
- Plopper, G.E.; McNamee, H.P.; Dike, L.E.; Bojanowski, K.; Ingber, D.E. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol. Biol. Cell 1995, 6, 1349–1365. [Google Scholar] [PubMed]
- Ramirez, M.P.; Anderson, M.J.; Kelly, M.D.; Sundby, L.J.; Hagerty, A.R.; Wenthe, S.J.; Odde, D.J.; Ervasti, J.M.; Gordon, W.R. Dystrophin missense mutations alter focal adhesion tension and mechanotransduction. Proc. Natl. Acad. Sci. USA 2022, 119, e2205536119. [Google Scholar]
- Rando, T.A. The dystrophin–glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve 2001, 24, 1575–1594. [Google Scholar]
- Yang, X.; Ren, H.; Guo, X.; Hu, C.; Fu, J. The Expressions and Mechanisms of Sarcomeric Proteins in Cancers. Dis. Markers 2020, 2020, 16. [Google Scholar]
- Chen, Y.; Ju, L.; Rushdi, M.; Ge, C.; Zhu, C. Receptor-mediated cell mechanosensing. Mol. Biol. Cell 2017, 28, 3134–3155. [Google Scholar]
- Quach, N.L.; Biressi, S.; Reichardt, L.F.; Keller, C.; Rando, T.A. Focal adhesion kinase signaling regulates the expression of caveolin 3 and β1 integrin, genes essential for normal myoblast fusion. Mol. Biol. Cell 2009, 20, 3422–3435. [Google Scholar]
- Lewis, A.H.; Cui, A.F.; McDonald, M.F.; Grandl, J. Transduction of Repetitive Mechanical Stimuli by Piezo1 and Piezo2 Ion Channels. Cell Rep. 2017, 19, 2572–2585. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 1–40. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Jiang, C.; Wen, Y.; Kuroda, K.; Hannon, K.; Rudnicki, M.A.; Kuang, S. Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis. Model. Mech. 2014, 7, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, Y.; Downey, G.P.; Klip, A. Stimulation of glucose transport in L6 muscle cells by long-term intermittent stretch—Relaxation. FEBS Lett. 1992, 301, 94–98. [Google Scholar]
- Yu, K.-T.; Czech, M. The type I insulin-like growth factor receptor mediates the rapid effects of multiplication-stimulating activity on membrane transport systems in rat soleus muscle. J. Biol. Chem. 1984, 259, 3090–3095. [Google Scholar] [PubMed]
- Ihlemann, J.; Ploug, T.; Hellsten, Y.; Galbo, H. Effect of stimulation frequency on contraction-induced glucose transport in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E862–E867. [Google Scholar]
- Sakamoto, K.; Aschenbach, W.G.; Hirshman, M.F.; Goodyear, L.J. Akt signaling in skeletal muscle: Regulation by exercise and passive stretch. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1081–E1088. [Google Scholar]
- Powers, S.K.; Lennon, S.L. Analysis of cellular responses to free radicals: Focus on exercise and skeletal muscle. Proc. Nutr. Soc. 1999, 58, 1025–1033. [Google Scholar]
- Reid, M.B. Invited Review: Redox modulation of skeletal muscle contraction: What we know and what we don’t. J. Appl. Physiol. 2001, 90, 724–731. [Google Scholar]
- Cheema, U.; Brown, R.; Mudera, V.; Yang, S.Y.; McGrouther, G.; Goldspink, G. Mechanical signals and IGF-I gene splicing in vitro in relation to development of skeletal muscle. J. Cell. Physiol. 2005, 202, 67–75. [Google Scholar] [CrossRef]
- Musi, N.; Yu, H.; Goodyear, L. AMP-activated protein kinase regulation and action in skeletal muscle during exercise. Biochem. Soc. Trans. 2003, 31, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, R.; Hattori, A.; Ikeuchi, Y.; Anderson, J.E.; Allen, R.E. Release of hepatocyte growth factor from mechanically stretched skeletal muscle satellite cells and role of pH and nitric oxide. Mol. Biol. Cell 2002, 13, 2909–2918. [Google Scholar] [PubMed]
- Palmer, R.M.; Reeds, P.J.; Atkinson, T.; Smith, R.H. The influence of changes in tension on protein synthesis and prostaglandin release in isolated rabbit muscles. Biochem. J. 1983, 214, 1011–1014. [Google Scholar] [CrossRef]
- Lu, F.; Zhu, L.; Bromberger, T.; Yang, J.; Yang, Q.; Liu, J.; Plow, E.F.; Moser, M.; Qin, J. Mechanism of integrin activation by talin and its cooperation with kindlin. Nat. Commun. 2022, 13, 2362. [Google Scholar] [CrossRef] [PubMed]
- Roca-Cusachs, P.; Del Rio, A.; Puklin-Faucher, E.; Gauthier, N.C.; Biais, N.; Sheetz, M.P. Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc. Natl. Acad. Sci. USA 2013, 110, E1361–E1370. [Google Scholar] [CrossRef]
- Kobzik, L.; Reid, M.B.; Bredt, D.S.; Stamler, J.S. Nitric oxide in skeletal muscle. Nature 1994, 372, 546–548. [Google Scholar]
- Froehner, S.C.; Reed, S.M.; Anderson, K.N.; Huang, P.L.; Percival, J.M. Loss of nNOS inhibits compensatory muscle hypertrophy and exacerbates inflammation and eccentric contraction-induced damage in mdx mice. Hum. Mol. Genet. 2015, 24, 492–505. [Google Scholar] [CrossRef]
- Lawler, J.M.; Kunst, M.; Hord, J.M.; Lee, Y.; Joshi, K.; Botchlett, R.E.; Ramirez, A.; Martinez, D.A. EUK-134 ameliorates nNOSμ translocation and skeletal muscle fiber atrophy during short-term mechanical unloading. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R470–R482. [Google Scholar] [CrossRef]
- Reid, M. Role of nitric oxide in skeletal muscle: Synthesis, distribution and functional importance. Acta Physiol. Scand. 1998, 162, 401–409. [Google Scholar] [CrossRef]
- Bahadoran, Z.; Mirmiran, P.; Ghasemi, A. Role of nitric oxide in insulin secretion and glucose metabolism. Trends Endocrinol. Metab. 2020, 31, 118–130. [Google Scholar] [CrossRef]
- Anderson, J.E. A role for nitric oxide in muscle repair: Nitric oxide–mediated activation of muscle satellite cells. Mol. Biol. Cell 2000, 11, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Guilak, F.; Hayes, A.J.; Melrose, J. Perlecan in pericellular mechanosensory cell-matrix communication, extracellular matrix stabilisation and mechanoregulation of load-bearing connective tissues. Int. J. Mol. Sci. 2021, 22, 2716. [Google Scholar] [CrossRef]
- Hayes, A.J.; Farrugia, B.L.; Biose, I.J.; Bix, G.J.; Melrose, J. Perlecan, a multi-functional, cell-instructive, matrix-stabilizing proteoglycan with roles in tissue development has relevance to connective tissue repair and regeneration. Front. Cell Dev. Biol. 2022, 10, 856261. [Google Scholar] [CrossRef]
- Melrose, J. Perlecan, a modular instructive proteoglycan with diverse functional properties. Int. J. Biochem. Cell Biol. 2020, 128, 105849. [Google Scholar] [CrossRef] [PubMed]
- Arikawa-Hirasawa, E. Impact of the heparan sulfate proteoglycan perlecan on human disease and health. Am. J. Physiol. Cell Physiol. 2022, 322, C1117–C1122. [Google Scholar] [CrossRef]
- Wu, C. Focal adhesion: A focal point in current cell biology and molecular medicine. Cell Adhes. Migr. 2007, 1, 13–18. [Google Scholar] [CrossRef]
- Kuo, J.C. Mechanotransduction at focal adhesions: Integrating cytoskeletal mechanics in migrating cells. J. Cell. Mol. Med. 2013, 17, 704–712. [Google Scholar] [CrossRef]
- Malik-Sheriff, R.S.; Imtiaz, S.; Grecco, H.E.; Zamir, E. Diverse patterns of molecular changes in the mechano-responsiveness of focal adhesions. Sci. Rep. 2018, 8, 2187. [Google Scholar] [CrossRef]
- Acharyya, S.; Butchbach, M.E.; Sahenk, Z.; Wang, H.; Saji, M.; Carathers, M.; Ringel, M.D.; Skipworth, R.J.; Fearon, K.C.; Hollingsworth, M.A. Dystrophin glycoprotein complex dysfunction: A regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005, 8, 421–432. [Google Scholar] [CrossRef]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [PubMed]
- Wilson, D.G.S.; Tinker, A.; Iskratsch, T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun. Biol. 2022, 5, 1022. [Google Scholar]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar]
- Chiu, Y.-H.; Hornsey, M.A.; Klinge, L.; Jørgensen, L.H.; Laval, S.H.; Charlton, R.; Barresi, R.; Straub, V.; Lochmüller, H.; Bushby, K. Attenuated muscle regeneration is a key factor in dysferlin-deficient muscular dystrophy. Hum. Mol. Genet. 2009, 18, 1976–1989. [Google Scholar]
- Roche, J.A.; Lovering, R.M.; Bloch, R.J. Impaired recovery of dysferlin-null skeletal muscle after contraction-induced injury in vivo. Neuroreport 2008, 19, 1579–1584. [Google Scholar]
- Defour, A.; Van der Meulen, J.H.; Bhat, R.; Bigot, A.; Bashir, R.; Nagaraju, K.; Jaiswal, J.K. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. 2014, 5, e1306. [Google Scholar] [PubMed]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [PubMed]
- Vachon, P.H. Integrin signaling, cell survival, and anoikis: Distinctions, differences, and differentiation. J. Signal Transduct. 2011, 2011, 738137. [Google Scholar]
- Giancotti, F.G. Integrin signaling: Specificity and control of cell survival and cell cycle progression. Curr. Opin. Cell Biol. 1997, 9, 691–700. [Google Scholar]
- Riquelme, M.A.; Gu, S.; Hua, R.; Jiang, J.X. Mechanotransduction via the coordinated actions of integrins, PI3K signaling and Connexin hemichannels. Bone Res. 2021, 9, 8. [Google Scholar]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, A.P.; Burridge, K. Molecular mechanisms for focal adhesion assembly through regulation of protein–protein interactions. Structure 1996, 4, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Otani, Y.; Guo, Y.; Yan, J.; Goult, B.T.; Howe, A.K. The focal adhesion protein talin is a mechanically gated A-kinase anchoring protein. Proc. Natl. Acad. Sci. USA 2024, 121, e2314947121. [Google Scholar] [CrossRef]
- Zhu, L.; Plow, E.F.; Qin, J. Initiation of focal adhesion assembly by talin and kindlin: A dynamic view. Protein Sci. 2021, 30, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Carisey, A.; Tsang, R.; Greiner, A.M.; Nijenhuis, N.; Heath, N.; Nazgiewicz, A.; Kemkemer, R.; Derby, B.; Spatz, J.; Ballestrem, C. Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner. Curr. Biol. 2013, 23, 271–281. [Google Scholar] [CrossRef]
- Goldmann, W.H. Role of vinculin in cellular mechanotransduction. Cell Biol. Int. 2016, 40, 241–256. [Google Scholar] [CrossRef]
- Rahman, A.; Carey, S.P.; Kraning-Rush, C.M.; Goldblatt, Z.E.; Bordeleau, F.; Lampi, M.C.; Lin, D.Y.; García, A.J.; Reinhart-King, C.A. Vinculin regulates directionality and cell polarity in two-and three-dimensional matrix and three-dimensional microtrack migration. Mol. Biol. Cell 2016, 27, 1431–1441. [Google Scholar] [CrossRef]
- Rosowski, K.A.; Boltyanskiy, R.; Xiang, Y.; Van den Dries, K.; Schwartz, M.A.; Dufresne, E.R. Vinculin and the mechanical response of adherent fibroblasts to matrix deformation. Sci. Rep. 2018, 8, 17967. [Google Scholar] [CrossRef]
- Lee, H.T.; Sharek, L.; O’Brien, E.T.; Urbina, F.L.; Gupton, S.L.; Superfine, R.; Burridge, K.; Campbell, S.L. Vinculin and metavinculin exhibit distinct effects on focal adhesion properties, cell migration, and mechanotransduction. PLoS ONE 2019, 14, e0221962. [Google Scholar] [CrossRef]
- Cai, X.; Wang, K.-C.; Meng, Z. Mechanoregulation of YAP and TAZ in cellular homeostasis and disease progression. Front. Cell Dev. Biol. 2021, 9, 673599. [Google Scholar]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, I.; McCollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Liu, H.; Hu, J.; Zheng, Q.; Feng, X.; Zhan, F.; Wang, X.; Xu, G.; Hua, F. Piezo1 channels as force sensors in mechanical force-related chronic inflammation. Front. Immunol. 2022, 13, 816149. [Google Scholar] [CrossRef]
- Qin, L.; He, T.; Chen, S.; Yang, D.; Yi, W.; Cao, H.; Xiao, G. Roles of mechanosensitive channel Piezo1/2 proteins in skeleton and other tissues. Bone Res. 2021, 9, 44. [Google Scholar] [CrossRef]
- Zhou, T.; Gao, B.; Fan, Y.; Liu, Y.; Feng, S.; Cong, Q.; Zhang, X.; Zhou, Y.; Yadav, P.S.; Lin, J. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT-YAP1-ß-catenin. eLife 2020, 9, e52779. [Google Scholar] [CrossRef] [PubMed]
- Graham, Z.A.; Gallagher, P.M.; Cardozo, C.P. Focal adhesion kinase and its role in skeletal muscle. J. Muscle Res. Cell Motil. 2015, 36, 305–315. [Google Scholar] [PubMed]
- Burridge, K.; Chrzanowska-Wodnicka, M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996, 12, 463–519. [Google Scholar]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular mechanotransduction: From tension to function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Ross, T.D.; Coon, B.G.; Yun, S.; Baeyens, N.; Tanaka, K.; Ouyang, M.; Schwartz, M.A. Integrins in mechanotransduction. Curr. Opin. Cell Biol. 2013, 25, 613–618. [Google Scholar] [CrossRef]
- Bozyczko, D.; Decker, C.; Muschler, J.; Horwitz, A.F. Integrin on developing and adult skeletal muscle. Exp. Cell Res. 1989, 183, 72–91. [Google Scholar] [PubMed]
- Disatnik, M.-H.l.n.; Rando, T.A. Integrin-mediated muscle cell spreading: The role of protein kinase C in outside-in and inside-out signaling and evidence of integrin cross-talk. J. Biol. Chem. 1999, 274, 32486–32492. [Google Scholar]
- Ugarte, G.; Santander, C.; Brandan, E. Syndecan-4 and β1 integrin are regulated by electrical activity in skeletal muscle: Implications for cell adhesion. Matrix Biol. 2010, 29, 383–392. [Google Scholar]
- Sztretye, M.; Singlár, Z.; Ganbat, N.; Al-Gaadi, D.; Szabó, K.; Köhler, Z.M.; Dux, L.; Keller-Pintér, A.; Csernoch, L.; Szentesi, P. Unravelling the effects of syndecan-4 knockdown on skeletal muscle functions. Int. J. Mol. Sci. 2023, 24, 6933. [Google Scholar] [CrossRef] [PubMed]
- Rønning, S.B.; Carlson, C.R.; Stang, E.; Kolset, S.O.; Hollung, K.; Pedersen, M.E. Syndecan-4 regulates muscle differentiation and is internalized from the plasma membrane during myogenesis. PLoS ONE 2015, 10, e0129288. [Google Scholar]
- Pegoraro, E.; Cepollaro, F.; Prandini, P.; Marin, A.; Fanin, M.; Trevisan, C.P.; El-Messlemani, A.H.; Tarone, G.; Engvall, E.; Hoffman, E.P. Integrin α7β1 in muscular dystrophy/myopathy of unknown etiology. Am. J. Pathol. 2002, 160, 2135–2143. [Google Scholar] [PubMed]
- Rooney, J.E.; Welser, J.V.; Dechert, M.A.; Flintoff-Dye, N.L.; Kaufman, S.J.; Burkin, D.J. Severe muscular dystrophy in mice that lack dystrophin and α7 integrin. J. Cell Sci. 2006, 119, 2185–2195. [Google Scholar]
- Bugiardini, E.; Nunes, A.M.; Oliveira-Santos, A.; Dagda, M.; Fontelonga, T.M.; Barraza-Flores, P.; Pittman, A.M.; Morrow, J.M.; Parton, M.; Houlden, H. Integrin α7 Mutations Are Associated With Adult-Onset Cardiac Dysfunction in Humans and Mice. J. Am. Heart Assoc. 2022, 11, e026494. [Google Scholar]
- Sarathy, A.; Wuebbles, R.D.; Fontelonga, T.M.; Tarchione, A.R.; Griner, L.A.M.; Heredia, D.J.; Nunes, A.M.; Duan, S.; Brewer, P.D.; Van Ry, T. SU9516 increases α7β1 integrin and ameliorates disease progression in the mdx mouse model of duchenne muscular dystrophy. Mol. Ther. 2017, 25, 1395–1407. [Google Scholar]
- Song, Y.; Qin, X.; Wang, H.; Miao, R.; Zhang, Y.; Miao, C.; Wang, Z. Effects of integrin α5β1 on the proliferation and migration of human aortic vascular smooth muscle cells. Mol. Med. Rep. 2016, 13, 1147–1155. [Google Scholar]
- Roy, J.; Tran, P.K.; Religa, P.; Kazi, M.; Henderson, B.; Lundmark, K.; Hedin, U. Fibronectin promotes cell cycle entry in smooth muscle cells in primary culture. Exp. Cell Res. 2002, 273, 169–177. [Google Scholar] [PubMed]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1692, 103–119. [Google Scholar]
- Pardo, J.V.; Siliciano, J.; Craig, S.W. A vinculin-containing cortical lattice in skeletal muscle: Transverse lattice elements (“costameres”) mark sites of attachment between myofibrils and sarcolemma. Proc. Natl. Acad. Sci. USA 1983, 80, 1008–1012. [Google Scholar] [PubMed]
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 2000, 150, 1209–1214. [Google Scholar] [CrossRef]
- Straub, V.; Bittner, R.E.; Léger, J.J.; Voit, T. Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J. Cell Biol. 1992, 119, 1183–1191. [Google Scholar] [PubMed]
- Bisht, B.; Goel, H.; Dey, C. Focal adhesion kinase regulates insulin resistance in skeletal muscle. Diabetologia 2007, 50, 1058–1069. [Google Scholar]
- Chen, S.; He, T.; Zhong, Y.; Chen, M.; Yao, Q.; Chen, D.; Shao, Z.; Xiao, G. Roles of focal adhesion proteins in skeleton and diseases. Acta Pharm. Sin. B 2023, 13, 998–1013. [Google Scholar]
- Kumar, A.; Murphy, R.; Robinson, P.; Wei, L.; BORIE, A.M. Cyclic mechanical strain inhibits skeletal myogenesis through activation of focal adhesion kinase, Rac-1 GTPase, and nf-kB transcription factor. FASEB J. 2004, 18, 1524–1535. [Google Scholar]
- Durieux, A.C.; Desplanches, D.; Freyssenet, D.; Flueck, M. Mechanotransduction in striated muscle via focal adhesion kinase. Biochem. Soc. Trans. 2007, 35, 1312–1313. [Google Scholar]
- Lechado, A.i.T.; Vitadello, M.; Traini, L.; Namuduri, A.V.; Gastaldello, S.; Gorza, L. Sarcolemmal loss of active nNOS (Nos1) is an oxidative stress-dependent, early event driving disuse atrophy. J. Pathol. 2018, 246, 433–446. [Google Scholar]
- Brooks, N.E.; Myburgh, K.H. Skeletal muscle wasting with disuse atrophy is multi-dimensional: The response and interaction of myonuclei, satellite cells and signaling pathways. Front. Physiol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.L.; Muñoz-Cánoves, P. Regulation and dysregulation of fibrosis in skeletal muscle. Exp. Cell Res. 2010, 316, 3050–3058. [Google Scholar]
- Yamashita, Y.; Nakada, S.; Yoshihara, T.; Nara, T.; Furuya, N.; Miida, T.; Hattori, N.; Arikawa-Hirasawa, E. Perlecan, a heparan sulfate proteoglycan, regulates systemic metabolism with dynamic changes in adipose tissue and skeletal muscle. Sci. Rep. 2018, 8, 7766. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ichikawa, N.; Kosaki, K.; Yamada, Y.; Sasaki, T.; Sakai, L.Y.; Kurosawa, H.; Hattori, N.; Arikawa-Hirasawa, E. Perlecan deficiency causes muscle hypertrophy, a decrease in myostatin expression, and changes in muscle fiber composition. Matrix Biol. 2010, 29, 461–470. [Google Scholar] [CrossRef]
- Nakada, S.; Yamashita, Y.; Machida, S.; Miyagoe-Suzuki, Y.; Arikawa-Hirasawa, E. Perlecan Facilitates Neuronal Nitric Oxide Synthase Delocalization in Denervation-Induced Muscle Atrophy. Cells 2020, 9, 2524. [Google Scholar] [CrossRef]
- Arikawa-Hirasawa, E.; Nakada, S.; Yamashita, Y.; Hattori, N. The role of perlecan in nnos mediated mechanotransduction in skeletal muscle. J. Neurol. Sci. 2017, 381, 268. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Boppart, M.D.; Mahmassani, Z.S. Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, 629–641. [Google Scholar]
- Gao, M.; Gao, W.; Papadimitriou, J.; Zhang, C.; Gao, J.; Zheng, M. Exosomes—The enigmatic regulators of bone homeostasis. Bone Res. 2018, 6, 36. [Google Scholar]
- Sartori, R.; Romanello, V.; Sandri, M. Mechanisms of muscle atrophy and hypertrophy: Implications in health and disease. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Indo, H.P.; Yen, H.-C.; Nakanishi, I.; Matsumoto, K.-i.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [PubMed]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2007, 35, 411–429. [Google Scholar]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [PubMed]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.-S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef]
- Powers, S.K. Can antioxidants protect against disuse muscle atrophy? Sports Med. 2014, 44, 155–165. [Google Scholar]
- Powers, S.K.; Kavazis, A.N.; DeRuisseau, K.C. Mechanisms of disuse muscle atrophy: Role of oxidative stress. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R337–R344. [Google Scholar] [CrossRef]
- Powers, S.K.; Kavazis, A.N.; McClung, J.M. Oxidative stress and disuse muscle atrophy. J. Appl. Physiol. 2007, 102, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Pannell, B.K. Redox characterization of functioning skeletal muscle. Front. Physiol. 2015, 6, 338. [Google Scholar]
- He, F.; Li, J.; Liu, Z.; Chuang, C.-C.; Yang, W.; Zuo, L. Redox mechanism of reactive oxygen species in exercise. Front. Physiol. 2016, 7, 486. [Google Scholar]
- Anik, M.I.; Mahmud, N.; Masud, A.A.; Khan, M.I.; Islam, M.N.; Uddin, S.; Hossain, M.K. Role of Reactive Oxygen Species in Aging and Age-Related Diseases: A Review. ACS Appl. Bio Mater. 2022, 5, 4028–4054. [Google Scholar]
- Kavazis, A.N.; Talbert, E.E.; Smuder, A.J.; Hudson, M.B.; Nelson, W.B.; Powers, S.K. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic. Biol. Med. 2009, 46, 842–850. [Google Scholar] [CrossRef]
- Powers, S.K.; Hudson, M.B.; Nelson, W.B.; Talbert, E.E.; Min, K.; Szeto, H.H.; Kavazis, A.N.; Smuder, A.J. Mitochondrial-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 2011, 39, 1749. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Smuder, A.J.; Kwon, O.-s.; Kavazis, A.N.; Szeto, H.H.; Powers, S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 2011, 111, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.M.; Van Gammeren, D.; Whidden, M.A.; Falk, D.J.; Kavazis, A.N.; Hudson, M.B.; Gayan-Ramirez, G.; Decramer, M.; DeRuisseau, K.C.; Powers, S.K. Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit. Care Med. 2009, 37, 1373. [Google Scholar] [CrossRef]
- Peterson, J.M.; Bakkar, N.; Guttridge, D.C. NF-κB signaling in skeletal muscle health and disease. Curr. Top. Dev. Biol. 2011, 96, 85–119. [Google Scholar]
- Powers, S.K.; Smuder, A.; Judge, A. Oxidative stress and disuse muscle atrophy: Cause or consequence? Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 240. [Google Scholar] [CrossRef] [PubMed]
- Whidden, M.A.; McClung, J.M.; Falk, D.J.; Hudson, M.B.; Smuder, A.J.; Nelson, W.B.; Powers, S.K. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J. Appl. Physiol. 2009, 106, 385–394. [Google Scholar] [CrossRef]
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox control of skeletal muscle regeneration. Antioxid. Redox Signal. 2017, 27, 276–310. [Google Scholar]
- Hord, J.M.; Garcia, M.M.; Farris, K.R.; Guzzoni, V.; Lee, Y.; Lawler, M.S.; Lawler, J.M. Nox2 signaling and muscle fiber remodeling are attenuated by losartan administration during skeletal muscle unloading. Physiol. Rep. 2021, 9, e14606. [Google Scholar] [CrossRef]
- Kondo, H.; Nakagaki, I.; Sasaki, S.; Hori, S.; Itokawa, Y. Mechanism of oxidative stress in skeletal muscle atrophied by immobilization. Am. J. Physiol. Endocrinol. Metab. 1993, 265, E839–E844. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxidative Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]
- Powers, S.K.; Smuder, A.J.; Criswell, D.S. Mechanistic links between oxidative stress and disuse muscle atrophy. Antioxid. Redox Signal. 2011, 15, 2519–2528. [Google Scholar]
- Lawler, J.M.; Song, W.; Demaree, S.R. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radic. Biol. Med. 2004, 35, 9–16. [Google Scholar]
- Zhang, X.; Trevino, M.B.; Wang, M.; Gardell, S.J.; Ayala, J.E.; Han, X.; Kelly, D.P.; Goodpaster, B.H.; Vega, R.B.; Coen, P.M. Impaired mitochondrial energetics characterize poor early recovery of muscle mass following hind limb unloading in old mice. J. Gerontol. Ser. 2018, 73, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, S.; Pan, X.; Dai, X.; Pan, G.; Li, Z.; Mai, X.; Tian, Y.; Zhang, S.; Liu, B. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J. Cachexia Sarcopenia Muscle 2021, 12, 746–768. [Google Scholar]
- Rajasekaran, N.S.; Shelar, S.B.; Jones, D.P.; Hoidal, J.R. Reductive stress impairs myogenic differentiation. Redox Biol. 2020, 34, 101492. [Google Scholar]
- Barbieri, E.; Sestili, P. Reactive oxygen species in skeletal muscle signaling. J. Signal Transduct. 2012, 2012, 17. [Google Scholar]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD (P) H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Chen, S.; Meng, X.-F.; Zhang, C. Role of NADPH oxidase-mediated reactive oxygen species in podocyte injury. BioMed Res. Int. 2013, 2013, 7. [Google Scholar]
- Khairallah, R.J.; Shi, G.; Sbrana, F.; Prosser, B.L.; Borroto, C.; Mazaitis, M.J.; Hoffman, E.P.; Mahurkar, A.; Sachs, F.; Sun, Y. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci. Signal. 2012, 5, 56. [Google Scholar]
- Ward, C.W.; Prosser, B.L.; Lederer, W.J. Mechanical stretch-induced activation of ROS/RNS signaling in striated muscle. Antioxid. Redox Signal. 2014, 20, 929–936. [Google Scholar] [PubMed]
- Specht, K.S.; Kant, S.; Addington, A.K.; McMillan, R.P.; Hulver, M.W.; Learnard, H.; Campbell, M.; Donnelly, S.R.; Caliz, A.D.; Pei, Y. Nox4 mediates skeletal muscle metabolic responses to exercise. Mol. Metab. 2021, 45, 101160. [Google Scholar] [PubMed]
- Shenkman, B.S.; Nemirovskaya, T.L.; Lomonosova, Y.N. No-dependent signaling pathways in unloaded skeletal muscle. Front. Physiol. 2015, 6, 298. [Google Scholar]
- Sukhanov, S.; Yoshida, T.; Tabony, A.M.; Higashi, Y.; Galvez, S.; Delafontaine, P.; Semprun-Prieto, L. Angiotensin II, oxidative stress and skeletal muscle wasting. Am. J. Med. Sci. 2011, 342, 143–147. [Google Scholar]
- Ushio-Fukai, M. Vascular signaling through G protein coupled receptors-new concepts. Curr. Opin. Nephrol. Hypertens. 2009, 18, 153. [Google Scholar]
- Adhihetty, P.J.; Uguccioni, G.; Leick, L.; Hidalgo, J.; Pilegaard, H.; Hood, D.A. The role of PGC-1α on mitochondrial function and apoptotic susceptibility in muscle. Am. J. Physiol. Cell Physiol. 2009, 297, C217–C225. [Google Scholar]
- Talbert, E.E.; Smuder, A.J.; Min, K.; Kwon, O.S.; Szeto, H.H.; Powers, S.K. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J. Appl. Physiol. 2013, 115, 529–538. [Google Scholar]
- Javadov, S.; Jang, S.; Rodriguez-Reyes, N.; Rodriguez-Zayas, A.E.; Hernandez, J.S.; Krainz, T.; Wipf, P.; Frontera, W. Mitochondria-targeted antioxidant preserves contractile properties and mitochondrial function of skeletal muscle in aged rats. Oncotarget 2015, 6, 39469. [Google Scholar]
- Vays, V.B.; Eldarov, C.M.; Vangely, I.M.; Kolosova, N.G.; Bakeeva, L.E.; Skulachev, V.P. Antioxidant SkQ1 delays sarcopenia-associated damage of mitochondrial ultrastructure. Aging 2014, 6, 140. [Google Scholar]
- Lawler, J.M.; Hord, J.M.; Lee, Y.; Joshi, K.; Kim, J.H. Redox Regulation of Caveolin-3 and MMP-9 in the Diaphragm of mdx Mice. FASEB J. 2011, 25, 1. [Google Scholar]
- Lawler, J.M.; Song, W.; Kwak, H.B. Differential response of heat shock proteins to hindlimb unloading and reloading in the soleus. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2006, 33, 200–207. [Google Scholar]
- Huang, Z.; Fang, Q.; Ma, W.; Zhang, Q.; Qiu, J.; Gu, X.; Yang, H.; Sun, H. Skeletal muscle atrophy was alleviated by salidroside through suppressing oxidative stress and inflammation during denervation. Front. Pharmacol. 2019, 10, 997. [Google Scholar]
- Lawler, J.M.; Garcia-Villatoro, E.L.; Guzzoni, V.; Hord, J.M.; Botchlett, R.; Holly, D.; Lawler, M.S.; Janini Gomes, M.; Ryan, P.; Rodriguez, D.; et al. Effect of combined fish oil & Curcumin on murine skeletal muscle morphology and stress response proteins during mechanical unloading. Nutr. Res. 2019, 65, 17–28. [Google Scholar]
- Sakellariou, G.K.; Lightfoot, A.P.; Earl, K.E.; Stofanko, M.; McDonagh, B. Redox homeostasis and age-related deficits in neuromuscular integrity and function. J. Cachexia Sarcopenia Muscle 2017, 8, 881–906. [Google Scholar]
- Nakanishi, T.; Tsujii, M.; Asano, T.; Iino, T.; Sudo, A. Protective effect of edaravone against oxidative stress in C2C12 myoblast and impairment of skeletal muscle regeneration exposed to ischemic injury in Ob/ob mice. Front. Physiol. 2020, 10, 1596. [Google Scholar]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [PubMed]
- Chen, Z.; Li, T.; Tang, H.-B.; Lu, Z.-W.; Chen, Z.-Y.; Zhao, Z.-H.; Yang, X.-L.; Zhao, L.-L.; Dang, M.-J.; Li, Y. Edaravone Dexborneol provides neuroprotective effect by inhibiting neurotoxic activation of astrocytes through inhibiting NF-κB signaling in cortical ischemia. Brain Res. Bull. 2024, 218, 111097. [Google Scholar]
- Casagrande, D.; Waib, P.H.; Júnior, A.A.J. Mechanisms of action and effects of the administration of Coenzyme Q10 on metabolic syndrome. J. Nutr. Intermed. Metab. 2018, 13, 26–32. [Google Scholar]
- Inoue, R.; Miura, M.; Yanai, S.; Nishimune, H. Coenzyme Q10 supplementation improves the motor function of middle-aged mice by restoring the neuronal activity of the motor cortex. Sci. Rep. 2023, 13, 4323. [Google Scholar]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxidative Med. Cell. Longev. 2019, 2019, 8409329. [Google Scholar]
- Kim, J.H.; Kim, K.M.; Jung, M.H.; Jung, J.H.; Kang, K.M.; Jeong, B.K.; Kim, J.P.; Park, J.J.; Woo, S.H. Protective effects of alpha lipoic acid on radiation-induced salivary gland injury in rats. Oncotarget 2016, 7, 29143. [Google Scholar] [PubMed]
- Eshima, H.; Siripoksup, P.; Mahmassani, Z.S.; Johnson, J.M.; Ferrara, P.J.; Verkerke, A.R.; Salcedo, A.; Drummond, M.J.; Funai, K. Neutralizing mitochondrial ROS does not rescue muscle atrophy induced by hindlimb unloading in female mice. J. Appl. Physiol. 2020, 129, 124–132. [Google Scholar]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A double-edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K. Potential adverse effects of resveratrol: A literature review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef]
- Weiss-Sadan, T.; Ge, M.; Hayashi, M.; Gohar, M.; Yao, C.-H.; de Groot, A.; Harry, S.; Carlin, A.; Fischer, H.; Shi, L. NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer. Cell Metab. 2023, 35, 487–503. e487. [Google Scholar]
- Li, J.; Arest, S.; Olszowy, B.; Gordon, J.; Barrero, C.A.; Perez-Leal, O. CRISPR/Cas9-based screening of FDA-approved drugs for NRF2 activation: A novel approach to discover therapeutics for non-alcoholic fatty liver disease. Antioxidants 2023, 12, 1363. [Google Scholar] [CrossRef]
- Kny, M.; Fielitz, J. Hidden agenda-the involvement of endoplasmic reticulum stress and unfolded protein response in inflammation-induced muscle wasting. Front. Immunol. 2022, 13, 878755. [Google Scholar]
- Ji, Y.; Jiang, Q.; Chen, B.; Chen, X.; Li, A.; Shen, D.; Shen, Y.; Liu, H.; Qian, X.; Yao, X. Endoplasmic reticulum stress and unfolded protein response: Roles in skeletal muscle atrophy. Biochem. Pharmacol. 2025, 116799. [Google Scholar]
- Wu, J.; Kaufman, R. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar]
- Li, F.; Guan, Z.; Gao, Y.; Bai, Y.; Zhan, X.; Ji, X.; Xu, J.; Zhou, H.; Rao, Z. ER stress promotes mitochondrial calcium overload and activates the ROS/NLRP3 axis to mediate fatty liver ischemic injury. Hepatol. Commun. 2024, 8, e0399. [Google Scholar]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [PubMed]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar]
- Martinvalet, D. The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 2018, 9, 336. [Google Scholar] [PubMed]
- Li, H.; Wen, W.; Luo, J. Targeting endoplasmic reticulum stress as an effective treatment for alcoholic pancreatitis. Biomedicines 2022, 10, 108. [Google Scholar] [CrossRef]
- Geng, T.; Li, P.; Okutsu, M.; Yin, X.; Kwek, J.; Zhang, M.; Yan, Z. PGC-1α plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am. J. Physiol. -Cell Physiol. 2010, 298, C572–C579. [Google Scholar] [CrossRef]
- El Assar, M.; Álvarez-Bustos, A.; Sosa, P.; Angulo, J.; Rodríguez-Mañas, L. Effect of physical activity/exercise on oxidative stress and inflammation in muscle and vascular aging. Int. J. Mol. Sci. 2022, 23, 8713. [Google Scholar] [CrossRef] [PubMed]
- Nash, D.; Hughes, M.G.; Butcher, L.; Aicheler, R.; Smith, P.; Cullen, T.; Webb, R. IL-6 signaling in acute exercise and chronic training: Potential consequences for health and athletic performance. Scand. J. Med. Sci. Sports 2023, 33, 4–19. [Google Scholar]
- Yamanashi, K.; Kinugawa, S.; Fukushima, A.; Kakutani, N.; Takada, S.; Obata, Y.; Nakano, I.; Yokota, T.; Kitaura, Y.; Shimomura, Y. Branched-chain amino acid supplementation ameliorates angiotensin II-induced skeletal muscle atrophy. Life Sci. 2020, 250, 117593. [Google Scholar]
- Fenercioglu, A.K. The anti-inflammatory roles of vitamin D for improving human health. Curr. Issues Mol. Biol. 2024, 46, 13514–13525. [Google Scholar] [CrossRef]
- Habedank, D.; Meyer, F.J.; Hetzer, R.; Anker, S.D.; Ewert, R. Relation of respiratory muscle strength, cachexia and survival in severe chronic heart failure. J. Cachexia Sarcopenia Muscle 2013, 4, 277–285. [Google Scholar] [CrossRef]
- Senf, S.M.; Dodd, S.L.; McClung, J.M.; Judge, A.R. Hsp70 overexpression inhibits NF-κB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008, 22, 3836–3845. [Google Scholar] [CrossRef] [PubMed]
- Macario, A.J.; de Macario, E.C. Sick chaperones, cellular stress, and disease. N. Engl. J. Med. 2005, 353, 1489–1501. [Google Scholar] [CrossRef]
- Becker, J.; Craig, E.A. Heat-shock proteins as molecular chaperones. Eur. J. Biochem. 1994, 219, 11–23. [Google Scholar] [PubMed]
- Mayer, M.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar]
- Park, C.-J.; Seo, Y.-S. Heat shock proteins: A review of the molecular chaperones for plant immunity. Plant Pathol. J. 2015, 31, 323. [Google Scholar] [CrossRef] [PubMed]
- Dybdahl, B.; Wahba, A.; Lien, E.; Flo, T.H.; Waage, A.; Qureshi, N.; Sellevold, O.F.; Espevik, T.; Sundan, A. Inflammatory response after open heart surgery: Release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation 2002, 105, 685–690. [Google Scholar]
- Elkenani, M.; Barakat, A.Z.; Held, T.; Rodrigues, D.M.; Mobarak, S.; Swarnka, S.; Adham, I.M.; Mohamed, B.A. Heat shock protein A4 ablation leads to skeletal muscle myopathy associated with dysregulated autophagy and induced apoptosis. J. Transl. Med. 2022, 20, 1–10. [Google Scholar]
- Giraldo, E.; Martin-Cordero, L.; Garcia, J.J.; Gerhmann, M.; Multhoff, G.; Ortega, E. Exercise-induced extracellular 72 kDa heat shock protein (Hsp72) stimulates neutrophil phagocytic and fungicidal capacities via TLR-2. Eur. J. Appl. Physiol. 2010, 108, 217–225. [Google Scholar]
- Weber, M.; Basu, S.; González, B.; Greslehner, G.P.; Singer, S.; Haskova, D.; Hasek, J.; Breitenbach, M.; Gourlay, C.; Cullen, P.J. Actin cytoskeleton regulation by the yeast NADPH oxidase Yno1p impacts processes controlled by MAPK pathways. Antioxidants 2021, 10, 322. [Google Scholar] [CrossRef]
- Bryantsev, A.L.; Loktionova, S.A.; Ilyinskaya, O.P.; Tararak, E.M.; Kampinga, H.H.; Kabakov, A.E. Distribution, phosphorylation, and activities of Hsp25 in heat-stressed H9c2 myoblasts: A functional link to cytoprotection. Cell Stress Chaperones 2002, 7, 146. [Google Scholar] [CrossRef]
- Lawler, J.M.; Kwak, H.-B.; Kim, J.-H.; Lee, Y.; Hord, J.M.; Martinez, D.A. Biphasic stress response in the soleus during reloading after hind limb unloading. Med. Sci. Sports Exerc. 2012, 44, 600–609. [Google Scholar] [PubMed]
- Naito, H.; Powers, S.K.; Demirel, H.A.; Sugiura, T.; Dodd, S.L.; Aoki, J. Heat stress attenuates skeletal muscle atrophy in hindlimb-unweighted rats. J. Appl. Physiol. 2000, 88, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Vitadello, M.; Gherardini, J.; Gorza, L. The stress protein/chaperone Grp94 counteracts muscle disuse atrophy by stabilizing subsarcolemmal neuronal nitric oxide synthase. Antioxid. Redox Signal. 2014, 20, 2479–2496. [Google Scholar] [CrossRef] [PubMed]
- McArdle, A.; Dillmann, W.H.; Mestril, R.; Faulkner, J.A.; Jackson, M.J. Overexpression of HSP70 in mouse skeletal muscle protects against muscle damage and age-related muscle dysfunction. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 355–357. [Google Scholar]
- Nascimento, T.L.; Mestril, R.; Miyabara, E.H. Overexpression of HSP70 attenuates sarcopenia by suppressing the expression of miR-133b. JCSM Rapid Commun. 2020, 3, 70–76. [Google Scholar] [CrossRef]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamal, K.Y.; Trombetta-Lima, M. Mechanotransduction and Skeletal Muscle Atrophy: The Interplay Between Focal Adhesions and Oxidative Stress. Int. J. Mol. Sci. 2025, 26, 2802. https://doi.org/10.3390/ijms26062802
Kamal KY, Trombetta-Lima M. Mechanotransduction and Skeletal Muscle Atrophy: The Interplay Between Focal Adhesions and Oxidative Stress. International Journal of Molecular Sciences. 2025; 26(6):2802. https://doi.org/10.3390/ijms26062802
Chicago/Turabian StyleKamal, Khaled Y., and Marina Trombetta-Lima. 2025. "Mechanotransduction and Skeletal Muscle Atrophy: The Interplay Between Focal Adhesions and Oxidative Stress" International Journal of Molecular Sciences 26, no. 6: 2802. https://doi.org/10.3390/ijms26062802
APA StyleKamal, K. Y., & Trombetta-Lima, M. (2025). Mechanotransduction and Skeletal Muscle Atrophy: The Interplay Between Focal Adhesions and Oxidative Stress. International Journal of Molecular Sciences, 26(6), 2802. https://doi.org/10.3390/ijms26062802