
Gemcitabine–Doxorubicin Combination Polymer-Drug Conjugate Prepared by SPAAC Click Chemistry: In Vitro Characterization

Abstract
1. Introduction
2. Results and Discussion
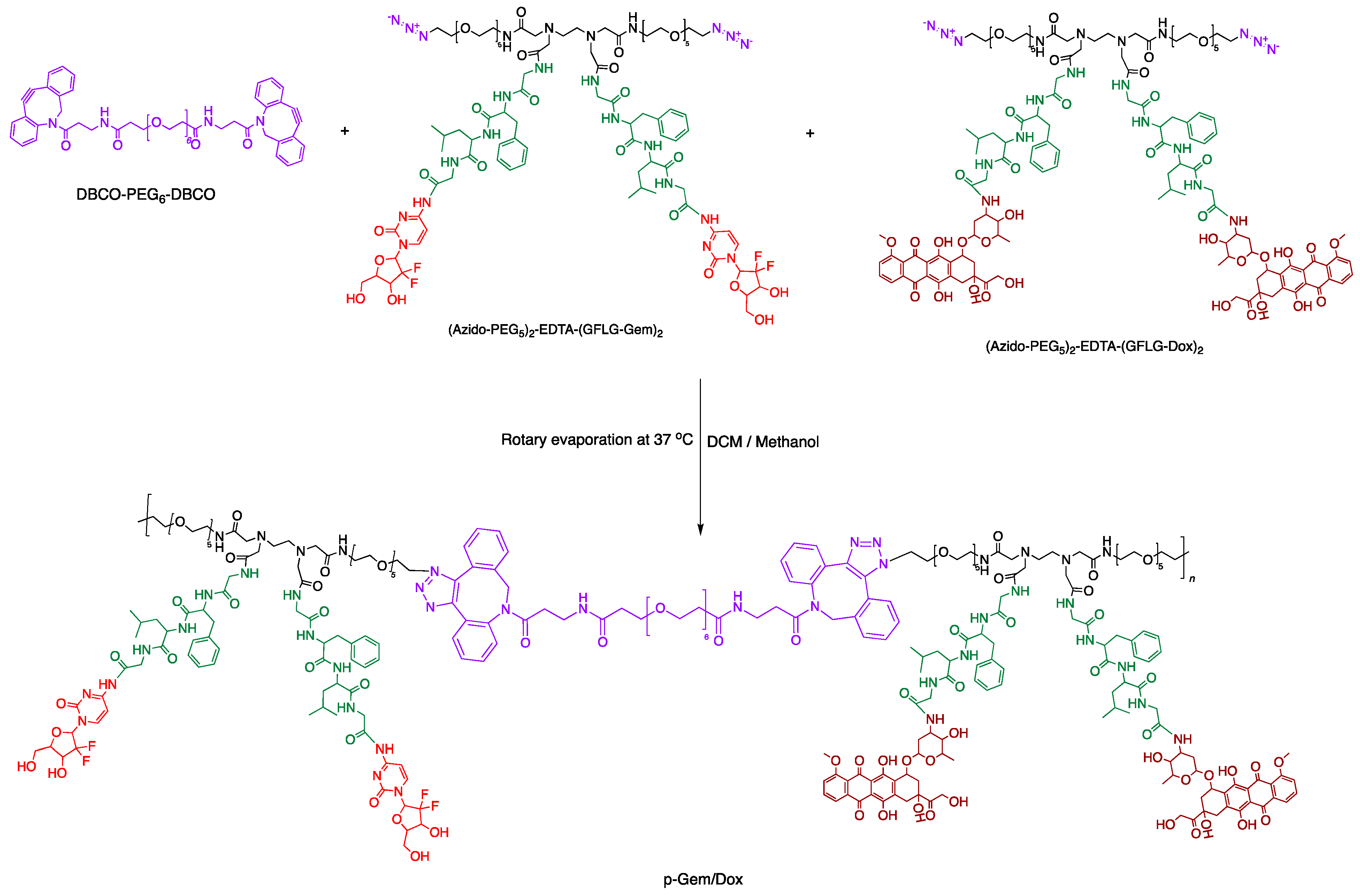
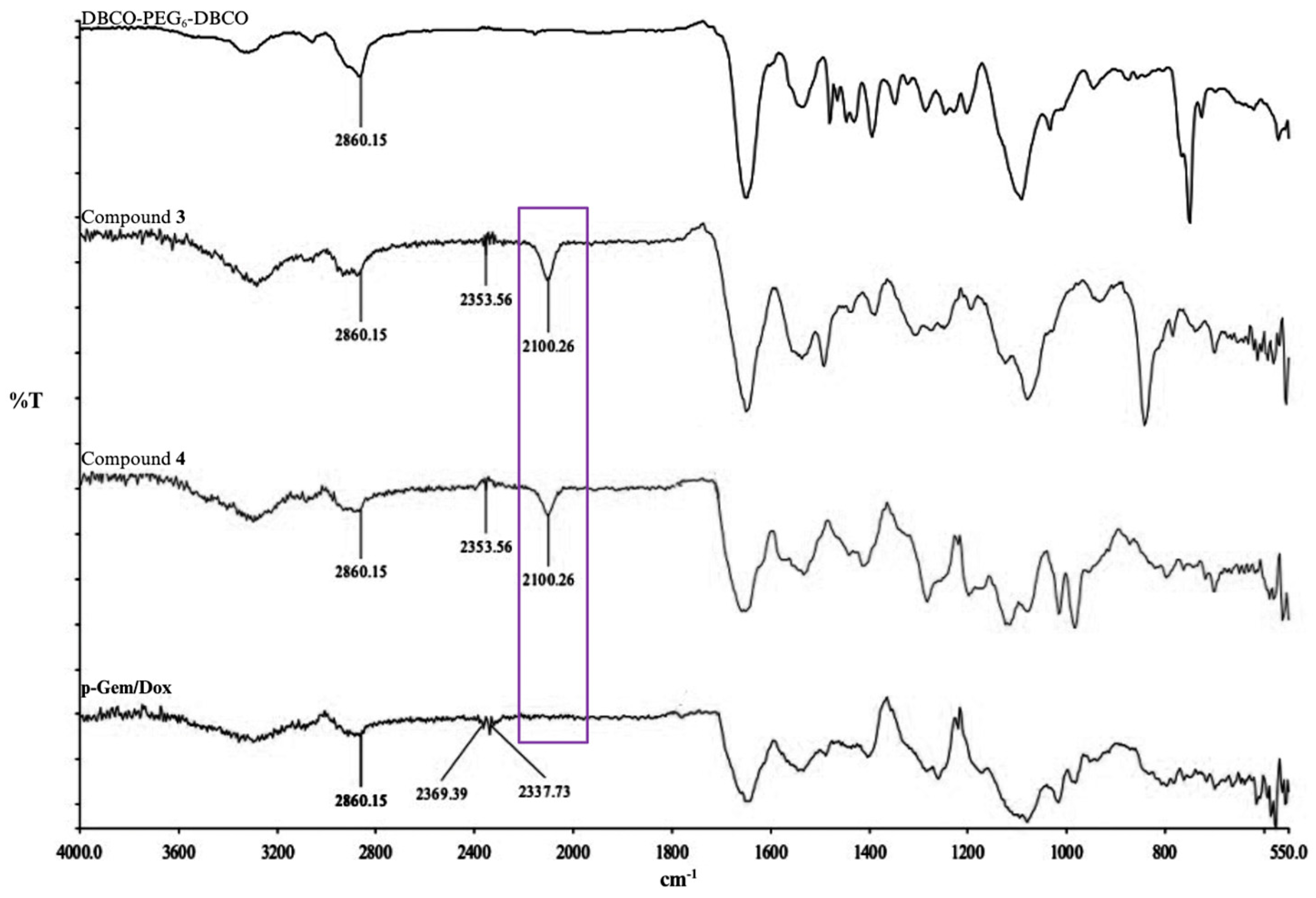
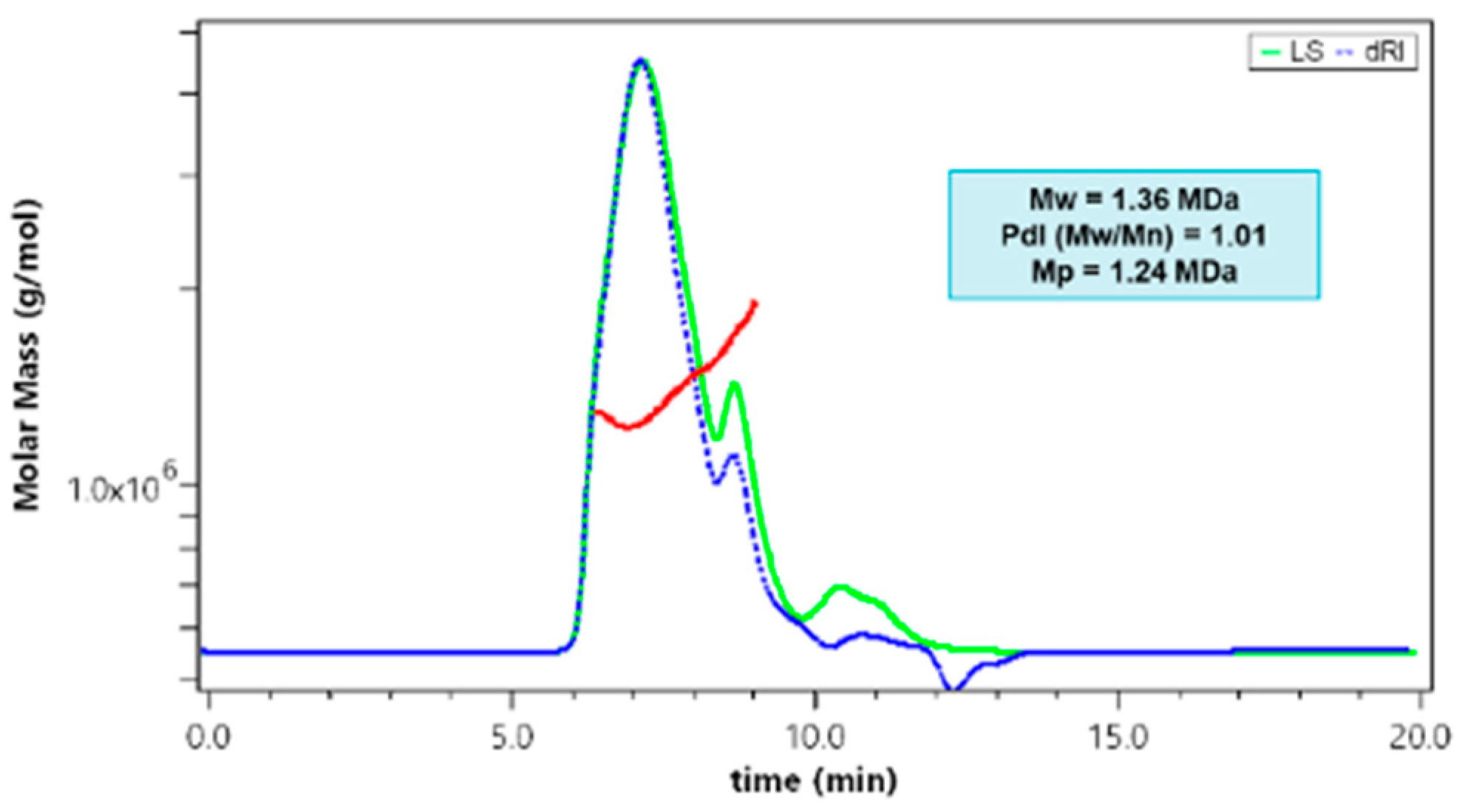
2.1. Synthesis and Characterization of P-Gem/Dox
2.2. In Vitro Cathepsin B-Catalyzed Cleavage of P-Gem/Dox
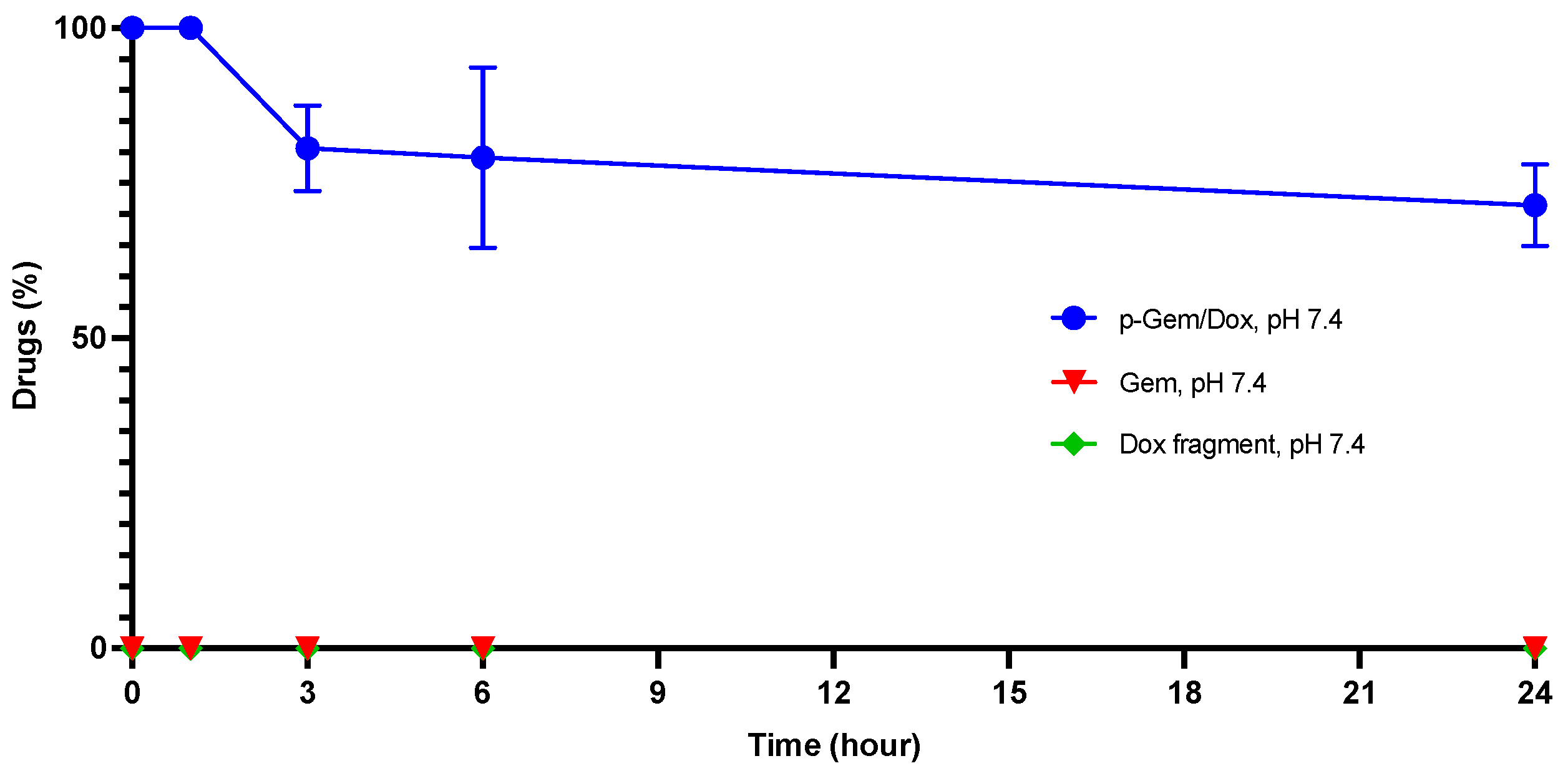
2.3. Hydrolytic Stability of P-Gem/Dox
2.4. In Vitro Cytotoxicity Evaluation
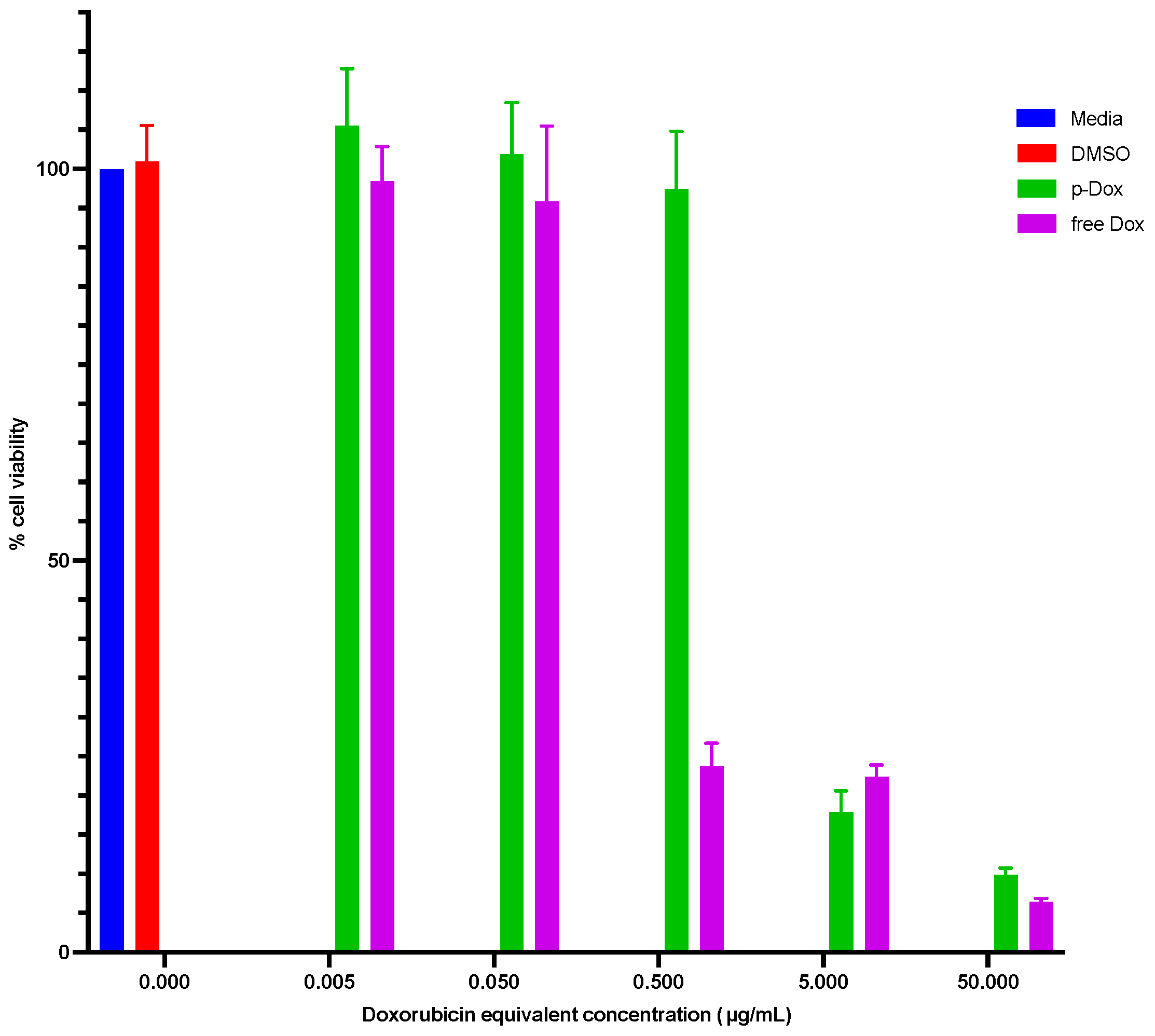
2.4.1. Cytotoxicity Evaluation of p-Dox
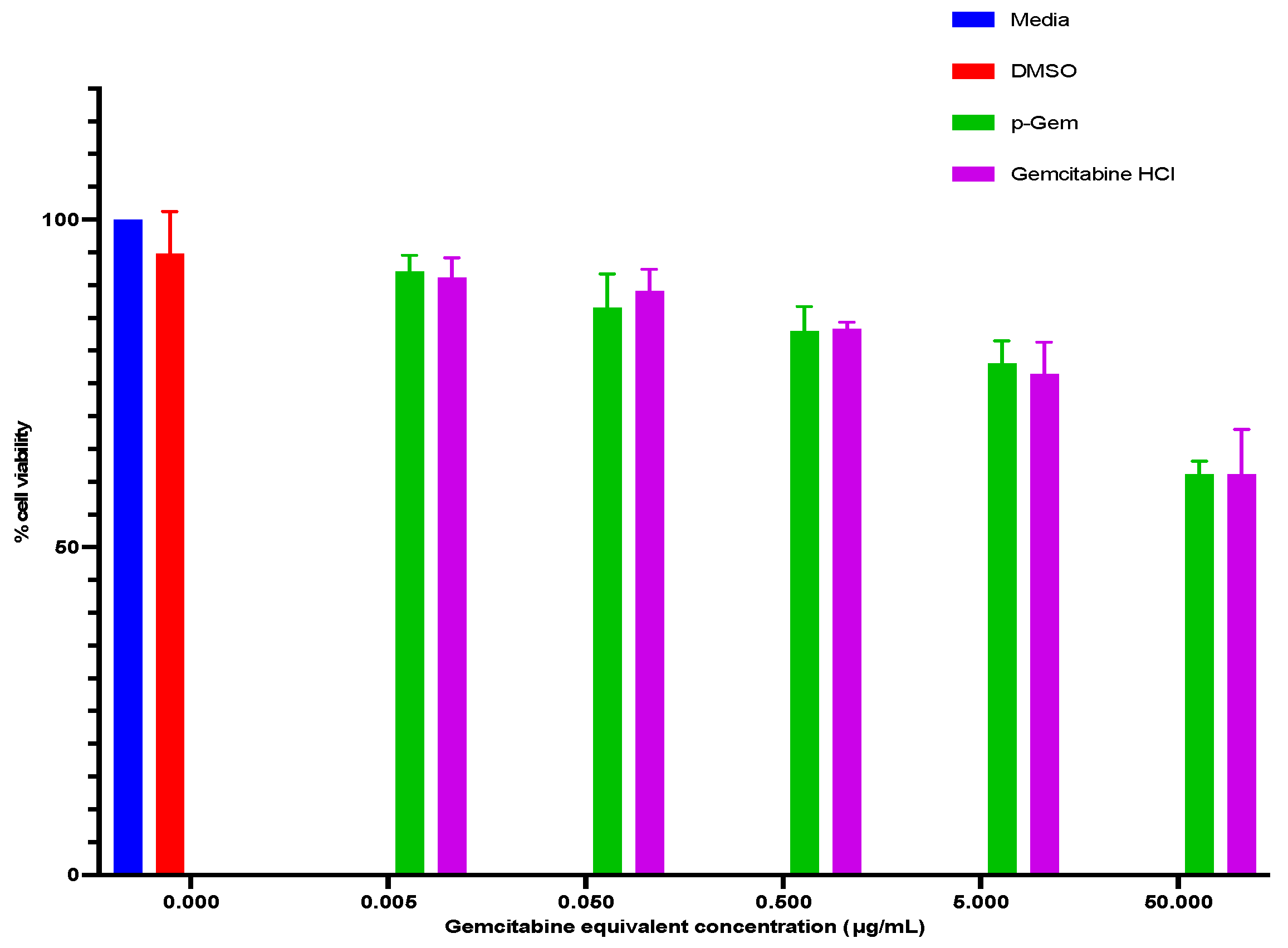
2.4.2. Cytotoxicity Evaluation of p-Gem
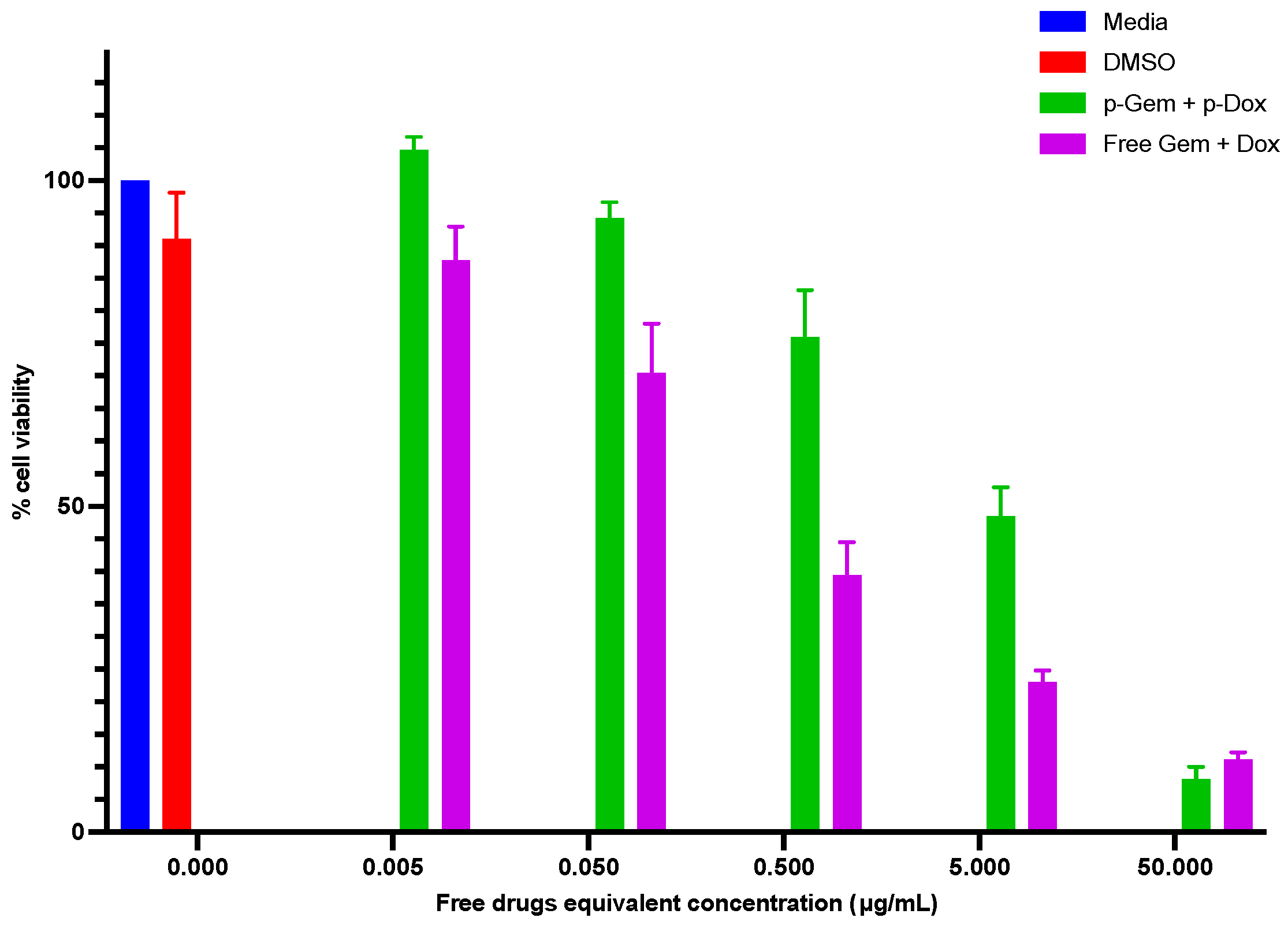
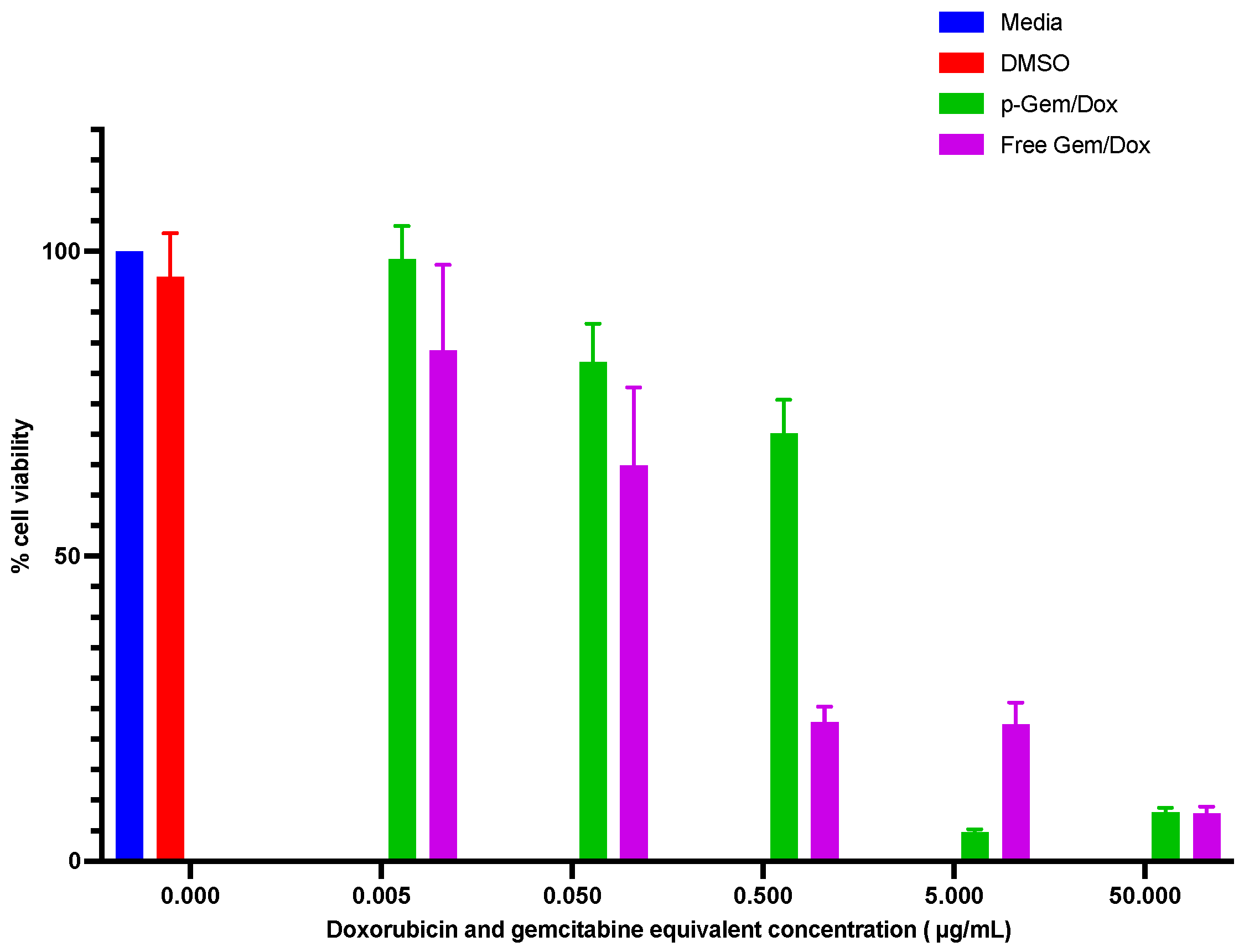
2.4.3. Cytotoxicity Evaluation of the Combination PDCs
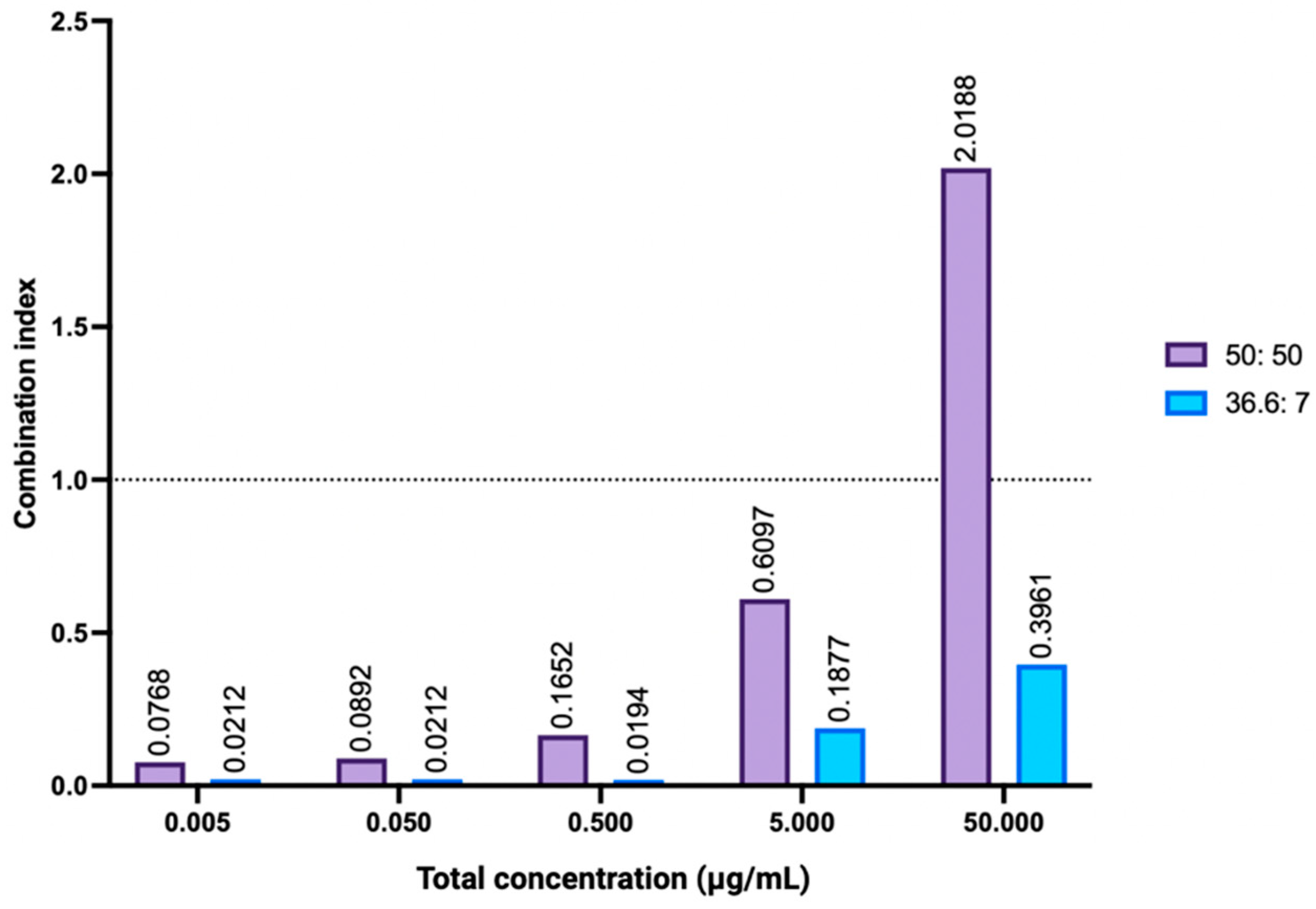
2.4.4. Combination Index Analysis
3. Materials and Method
3.1. Materials
3.2. Synthesis of Gemcitabine–Doxorubicin Combination PDC (P-Gem/Dox)
3.3. Molecular Weight Characterization
3.4. Drug Content Evaluation
3.5. In Vitro Cathepsin B-Catalyzed Cleavage
3.6. In Vitro Hydrolytic Stability
3.7. In Vitro Cytotoxicity Evaluation in OVCAR-3 Cell Line
3.7.1. Cell Culture
3.7.2. Cytotoxicity Evaluation of the Single Agents
3.7.3. The Cytotoxicity Evaluation of the Combination Agents and Combination Index Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian cancer: An integrated review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.; Bolina, A.; Mahajan, I.; Raza, S.A.; Clarke, M.; Pal, A.; Boussios, S. Hereditary Ovarian Cancer: Towards a Cost-Effective Prevention Strategy. Int. J. Environ. Res. Public Health 2022, 19, 12057. [Google Scholar] [CrossRef]
- Kwolek, D.G.; Gerstberger, S.; Tait, S.; Qiu, J.M. Ovarian, Uterine, and Vulvovaginal Cancers: Screening, Treatment Overview, and Prognosis. Med. Clin. 2023, 107, 329–355. [Google Scholar]
- Levine, M.; Naumann, R.W. Ovarian Cancer Screening and Early Detection. In Advances in Diagnosis and Management of Ovarian Cancer; Springer International Publishing: Cham, Switzerland, 2022; pp. 9–25. [Google Scholar]
- Sambasivan, S. Epithelial ovarian cancer. Cancer Treatment and Research Communications. Cancer. Treat Res. Commun. 2022, 33, 100629. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-grade serous ovarian cancer: Basic sciences, clinical and therapeutic standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.M.; Cristea, M.; DeRosa, M.; Eisenhauer, E.L.; et al. Ovarian Cancer, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 191–226. [Google Scholar] [CrossRef]
- Larson, N.; Yang, J.; Ray, A.; Cheney, D.L.; Ghandehari, H.; Kopeček, J. Biodegradable multiblock poly (N-2-hydroxypropyl) methacrylamide gemcitabine and paclitaxel conjugates for ovarian cancer cell combination treatment. Int. J. Pharm. 2013, 454, 435–443. [Google Scholar] [CrossRef]
- Cappella, P.; Tomasoni, D.; Faretta, M.; Lupi, M.; Montalenti, F.; Viale, F.; Banzato, F.; D’Incalci, M.; Ubezio, P. Cell cycle effects of gemcitabine. Int. J. Cancer 2001, 93, 401–408. [Google Scholar] [CrossRef]
- Montano, R.; Thompson, R.; Chung, I.; Hou, H.; Khan, N.; Eastman, A. Sensitization of human cancer cells to gemcitabine by the Chk1 inhibitor MK-8776: Cell cycle perturbation and impact of administration schedule in vitro and in vivo. BMC Cancer 2013, 13, 604. [Google Scholar] [CrossRef]
- Yang, F.; Kemp, C.J.; Henikoff, S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2015, 773, 9–15. [Google Scholar] [CrossRef]
- Ferrandina, G.; Paris, I.; Ludovisi, M.; D’Agostino, G.; Testa, A.; Lorusso, D.; Zanghi, M.; Pisconti, S.; Pezzella, G.; Adamo, V.; et al. Gemcitabine and liposomal doxorubicin in the salvage treatment of ovarian cancer: Updated results and long-term survival. Gynecol. Oncol. 2005, 98, 267–273. [Google Scholar] [CrossRef]
- Petru, E.; Angleitner-Boubenizek, L.; Reinthaller, A.; Deibl, M.; Zeimet, A.; Volgger, B.; Stempfl, A.; Marth, C. Combined PEG liposomal doxorubicin and gemcitabine are active and have acceptable toxicity in patients with platinum-refractory and resistant ovarian cancer after previous platinum–taxane therapy: A phase II Austrian AGO study. Gynecol. Oncol. 2006, 102, 226–229. [Google Scholar] [CrossRef]
- Gajera, K.; Patel, A. An Overview of FDA Approved Liposome Formulations for Cancer Therapy. J. Adv. Med. Pharm. Sci. 2022, 24, 1–7. [Google Scholar] [CrossRef]
- Moreno-Aspitia, A.; Perez, E.A. Nanoparticle albumin-bound paclitaxel (ABI-007): A newer taxane alternative in breast cancer. Future Oncol. 2005, 1, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Micha, J.P.; Goldstein, B.H.; Birk, C.L.; Rettenmaier, M.A.; Brown, J.V., III. Abraxane in the treatment of ovarian cancer: The absence of hypersensitivity reactions. Gynecol. Oncol. 2006, 100, 437–438. [Google Scholar] [CrossRef]
- Qi, R.; Wang, Y.; Bruno, P.M.; Xiao, H.; Yu, Y.; Li, T.; Ghoroghchian, P.P. Nanoparticle conjugates of a highly potent toxin enhance safety and circumvent platinum resistance in ovarian cancer. Nat. Commun. 2017, 8, 2166. [Google Scholar] [CrossRef]
- Olajubutu, O.; Ogundipe, O.D.; Adebayo, A.; Adesina, S.K. Drug delivery strategies for the treatment of pancreatic cancer. Pharmaceutics 2023, 15, 1318. [Google Scholar] [CrossRef] [PubMed]
- Ogundipe, O.D.; Olajubutu, O.; Adesina, S.K. Targeted drug conjugate systems for ovarian cancer chemotherapy. Biomed. Pharmacother. 2023, 165, 115151. [Google Scholar] [CrossRef]
- Javia, A.; Vanza, J.; Bardoliwala, D.; Ghosh, S.; Misra, A.; Patel, M.; Thakkar, H. Polymer-drug conjugates: Design principles, emerging synthetic strategies and clinical overview. Int. J. Pharm. 2022, 623, 121863. [Google Scholar] [CrossRef]
- Bazak, R.; Houri, M.; El Achy, S.; Hussein, W.; Refaat, T. Passive targeting of nanoparticles to cancer: A comprehensive review of the literature. Mol. Clin. Oncol. 2014, 2, 904–908. [Google Scholar] [CrossRef]
- Wang, Y.; Xia, H.; Chen, B.; Wang, Y. Rethinking nanoparticulate polymer–drug conjugates for cancer theranostics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 15, e1828. [Google Scholar] [CrossRef] [PubMed]
- Ejigah, V.; Owoseni, O.; Bataille-Backer, P.; Ogundipe, O.D.; Fisusi, F.A.; Adesina, S.K. Approaches to improve macromolecule and nanoparticle accumulation in the tumor microenvironment by the enhanced permeability and retention effect. Polymers 2022, 14, 2601. [Google Scholar] [CrossRef] [PubMed]
- Gbadegesin, O.D.; Adesina, S.K. Synthesis and Characterization of Polymer-Drug Conjugates by Strain-Promoted Azide-Alkyne Cycloaddition-Mediated Polymerization. J. Appl. Polym. Sci. 2025, e56928. [Google Scholar] [CrossRef]
- McNelles, S.A.; Pantaleo, J.L.; Meichsner, E.; Adronov, A. Strain-promoted azide-alkyne cycloaddition-mediated step-growth polymerization. Macromolecules 2019, 52, 7183–7187. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Chan, C.H.; Chen, J.T.; Farrell, W.S.; Fellows, C.M.; Keddie, D.J.; Luscombe, C.K.; Vargas, L.S. Reconsidering terms for mechanisms of polymer growth: The “step-growth” and “chain-growth” dilemma. Polym. Chem. 2022, 13, 2262–2270. [Google Scholar] [CrossRef]
- Lammers, T.; Subr, V.; Ulbrich, K.; Peschke, P.; Huber, P.E.; Hennink, W.E.; Storm, G. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials 2009, 30, 3466–3475. [Google Scholar] [CrossRef]
- Vogus, D.R.; Evans, M.A.; Pusuluri, A.; Barajas, A.; Zhang, M.; Krishnan, V.; Mitragotri, S. A hyaluronic acid conjugate engineered to synergistically and sequentially deliver gemcitabine and doxorubicin to treat triple negative breast cancer. J. Control. Release 2017, 267, 191–202. [Google Scholar] [CrossRef]
- Dvořák, M.; Kopečková, P.; Kopeček, J. High-molecular weight HPMA copolymer–adriamycin conjugates. J. Control. Release 1999, 60, 321–332. [Google Scholar] [CrossRef]
- Pola, R.; Janoušková, O.; Etrych, T. The pH-dependent and enzymatic release of cytarabine from hydrophilic polymer conjugates. Physiol. Res. 2016, 65, S225–S232. [Google Scholar] [CrossRef]
- Pechar, M.; Pola, R.; Studenovský, M.; Bláhová, M.; Grosmanová, E.; Dydowiczová, A.; Etrych, T. Polymer nanomedicines with enzymatically triggered activation: A comparative study of in vitro and in vivo anti-cancer efficacy related to the spacer structure. Nanomed. Nanotechnol. Biol. Med. 2022, 46, 102597. [Google Scholar] [CrossRef]
- Yamada, Y. Dimerization of doxorubicin causes its precipitation. ACS Omega 2020, 5, 33235–33241. [Google Scholar] [CrossRef]
- Hong, A.; Lee, H.H.; Heo, C.E.; Cho, Y.; Kim, S.; Kang, D.; Kim, H.I. Distinct Fragmentation Pathways of Anticancer Drugs Induced by Charge-Carrying Cations in the Gas Phase. J. Am. Soc. Mass Spectrom. 2016, 28, 628–637. [Google Scholar] [CrossRef]
- Subr, V.; Strohalm, J.; Ulbrich, K.; Duncan, R.; Hume, I. Polymers containing enzymatically degradable bonds, XII. Effect of spacer structure on the rate of release of daunomycin and adriamycin from poly [N-(2-hydroxypropyl)-methacrylamide] copolymer drag carriers in vitro and antitumour activity measured in vivo. J. Control. Release 1992, 18, 123–132. [Google Scholar] [CrossRef]
- Lammers, T.; Peschke, P.; Kühnlein, R.; Subr, V.; Ulbrich, K.; Huber, P.; Hennink, W.; Storm, G. Effect of Intratumoral Injection on the Biodistribution, the Therapeutic Potential of HPMA Copolymer-Based Drug Delivery Systems. Neoplasia 2006, 8, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cheetham, A.G.; Lock, L.L.; Cui, H. Cellular Uptake and Cytotoxicity of Drug Peptide Conjugates Regulated by Conjugation Site. Bioconjugate Chem. 2013, 24, 604–613. [Google Scholar] [CrossRef]
- Mahesh, S.; Tang, K.C.; Raj, M. Amide bond activation of biological molecules. Molecules 2018, 23, 2615. [Google Scholar] [CrossRef]
- Poreba, M.; Groborz, K.; Vizovisek, M.; Maruggi, M.; Turk, D.; Turk, B.; Salvesen, G.S. Fluorescent probes towards selective cathepsin B detection and visualization in cancer cells and patient samples. Chem. Sci. 2019, 10, 8461–8477. [Google Scholar] [CrossRef]
- Bourgeois, D.L.; Kabarowski, K.A.; Porubsky, V.L.; Kreeger, P.K. High-grade serous ovarian cancer cell lines exhibit heterogeneous responses to growth factor stimulation. Cancer. Cell Int. 2015, 15, 112. [Google Scholar] [CrossRef]
- Kamiloglu, S.; Sari, G.; Ozdal, T.; Capanoglu, E. Guidelines for cell viability assays. Food Front. 2020, 1, 332–349. [Google Scholar] [CrossRef]
- National Cancer Institute. Drugs Approved for Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. 2022. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/ovarian (accessed on 16 February 2024).
- Christie, R.J.; Grainger, D.W. Design strategies to improve soluble macromolecular delivery constructs. Adv. Drug Deliv. Rev. 2003, 55, 421–437. [Google Scholar] [CrossRef]
- Kostková, H.; Schindler, L.; Kotrchová, L.; Kovář, M.; Šírová, M.; Kostka, L.; Etrych, T. Star polymer-drug conjugates with pH-controlled drug release and carrier degradation. J. Nanomater. 2017, 1, 8675435. [Google Scholar] [CrossRef]
- Hovorka, O.; Št’astný, M.; Etrych, T.; Šubr, V.; Strohalm, J.; Ulbrich, K.; Říhová, B. Differences in the intracellular fate of free and polymer-bound doxorubicin. J. Control. Release 2002, 80, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Malugin, A.; Kopečková, P.; Kopeček, J. Liberation of doxorubicin from HPMA copolymer conjugate is essential for the induction of cell cycle arrest and nuclear fragmentation in ovarian carcinoma cells. J. Control. Release 2007, 124, 6–10. [Google Scholar] [CrossRef]
- Zhang, C.; Pan, D.; Luo, K.; Li, N.; Guo, C.; Zheng, X.; Gu, Z. Dendrimer–doxorubicin conjugate as enzyme-sensitive and polymeric nanoscale drug delivery vehicle for ovarian cancer therapy. Polym. Chem. 2014, 5, 5227–5235. [Google Scholar] [CrossRef]
- Zhang, C.; Pan, D.; Li, J.; Hu, J.; Bains, A.; Guys, N.; Gu, Z. Enzyme-responsive peptide dendrimer-gemcitabine conjugate as a controlled-release drug delivery vehicle with enhanced antitumor efficacy. Acta Biomater. 2017, 55, 153–162. [Google Scholar] [CrossRef]
- Hawryłkiewicz, A.; Ptaszyńska, N. Gemcitabine peptide-based conjugates and their application in targeted tumor therapy. Molecules 2021, 26, 364. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDCs | Mw (kDa) | Mn (kDa) | PdI | Mp (kDa) | % Weight Doxorubicin | % Weight Gemcitabine |
|---|---|---|---|---|---|---|
| p-Gem/Dox | 1360 | 1346.53 | 1.01 | 1240 | 7.00 ± 2.00 | 36.60 ± 0.24 |
| p-Gem ø | 40.18 | 29.33 | 1.37 | 40.37 | Not applicable | 29.20 ± 0.30 |
| p-Dox ø | 1800 | 1782.18 | 1.01 | 1920 | 10.30 ± 0.53 | Not applicable |
| Drug Treatment | IC50 (µg/mL) |
|---|---|
| Single | |
| p-Dox | 1.79 ± 0.17 |
| Doxorubicin HCl (DOX) | 0.17 ± 0.03 |
| p-Gem | >50 |
| Gemcitabine HCl (GEM) | >50 |
| Combination | |
| p-Gem + p-Dox admixture (50:50) | 3.87 ± 1.22 |
| GEM + DOX admixture (50:50) | 0.24 ± 0.12 |
| p-Gem/Dox | 0.99 ± 0.06 |
| GEM + DOX admixture (36.6:7) | 0.11 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gbadegesin, O.D.; Adesina, S.K. Gemcitabine–Doxorubicin Combination Polymer-Drug Conjugate Prepared by SPAAC Click Chemistry: In Vitro Characterization. Int. J. Mol. Sci. 2025, 26, 2798. https://doi.org/10.3390/ijms26062798
Gbadegesin OD, Adesina SK. Gemcitabine–Doxorubicin Combination Polymer-Drug Conjugate Prepared by SPAAC Click Chemistry: In Vitro Characterization. International Journal of Molecular Sciences. 2025; 26(6):2798. https://doi.org/10.3390/ijms26062798
Chicago/Turabian StyleGbadegesin, Omotola D., and Simeon K. Adesina. 2025. "Gemcitabine–Doxorubicin Combination Polymer-Drug Conjugate Prepared by SPAAC Click Chemistry: In Vitro Characterization" International Journal of Molecular Sciences 26, no. 6: 2798. https://doi.org/10.3390/ijms26062798
APA StyleGbadegesin, O. D., & Adesina, S. K. (2025). Gemcitabine–Doxorubicin Combination Polymer-Drug Conjugate Prepared by SPAAC Click Chemistry: In Vitro Characterization. International Journal of Molecular Sciences, 26(6), 2798. https://doi.org/10.3390/ijms26062798