
A Novel Homozygous Missense Variant of PIGT Related to Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 with Elevated of Serum ALP Level in a Thai Newborn Patient

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
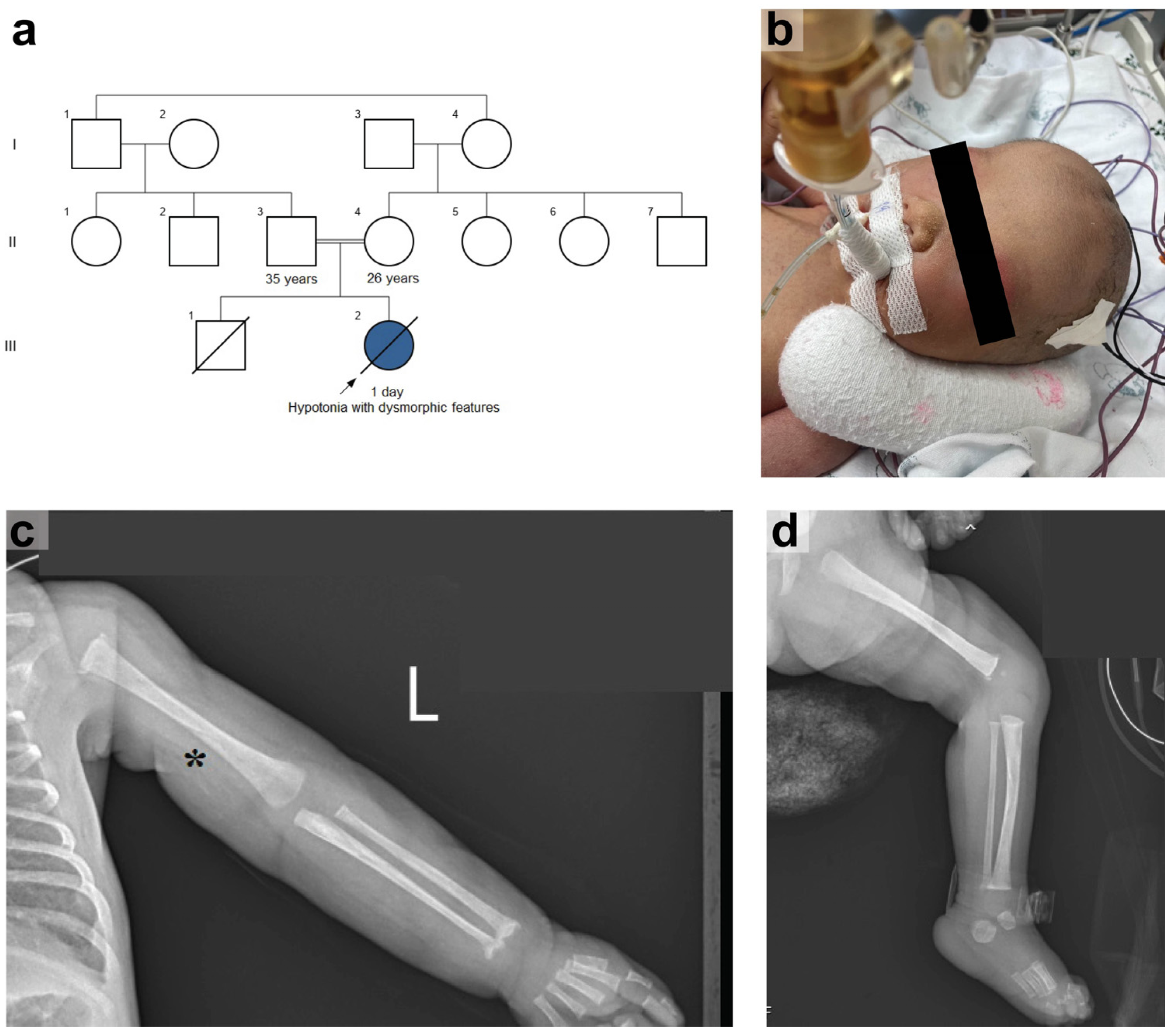
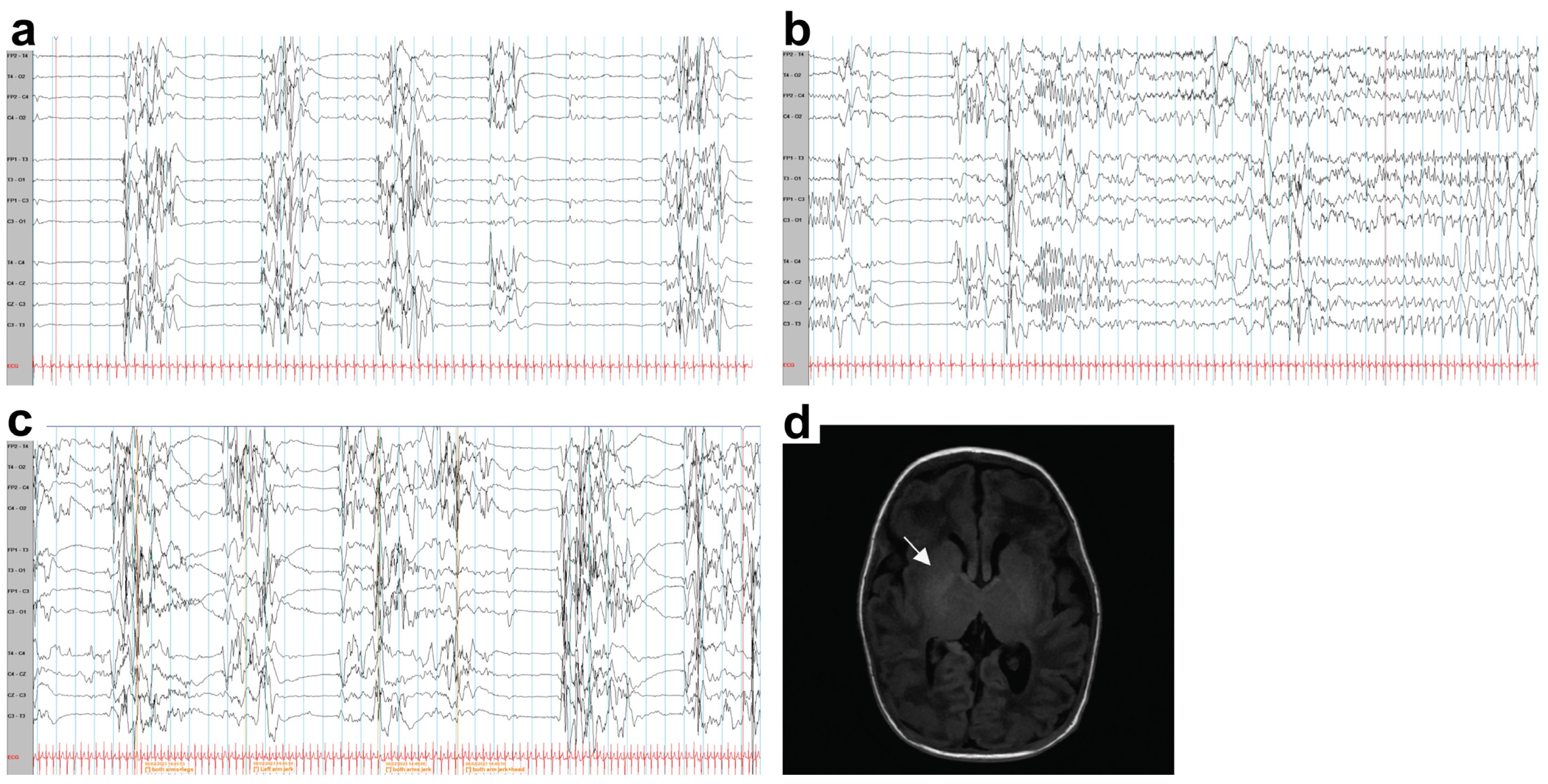
2.1. Case Report
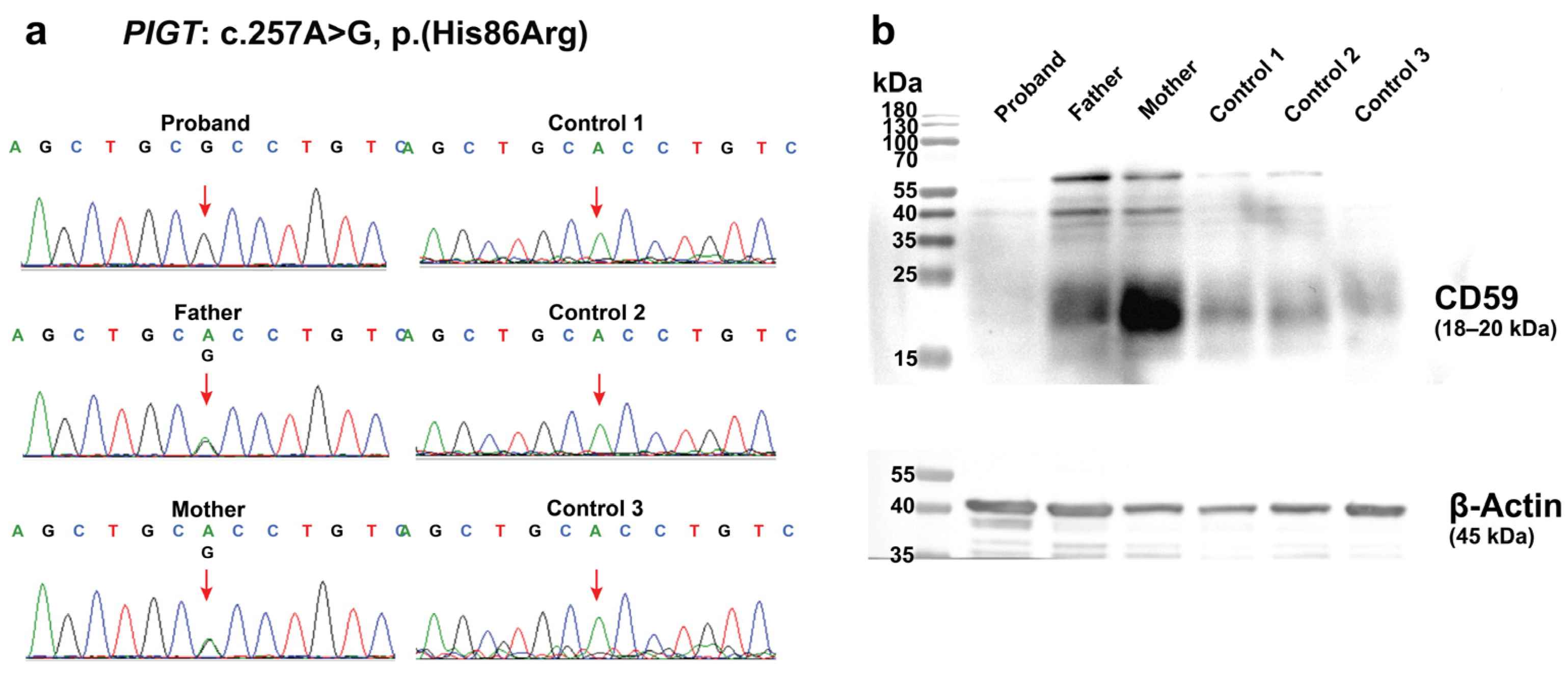
2.2. Germline Mutation Analysis
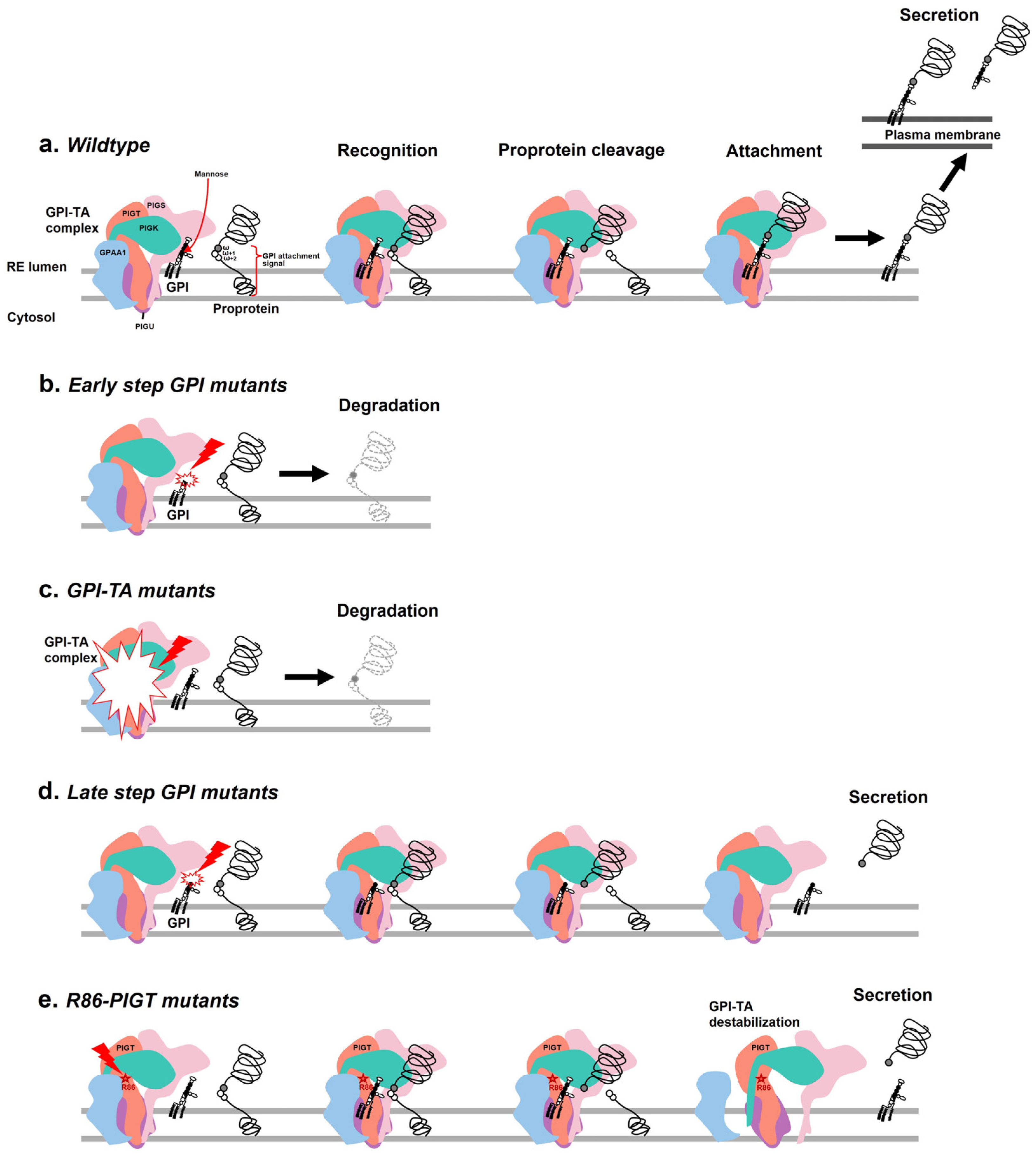
2.3. Effects of the c.257A>G on PIGT Function Demonstrated by GPI-AP Expression Analysis
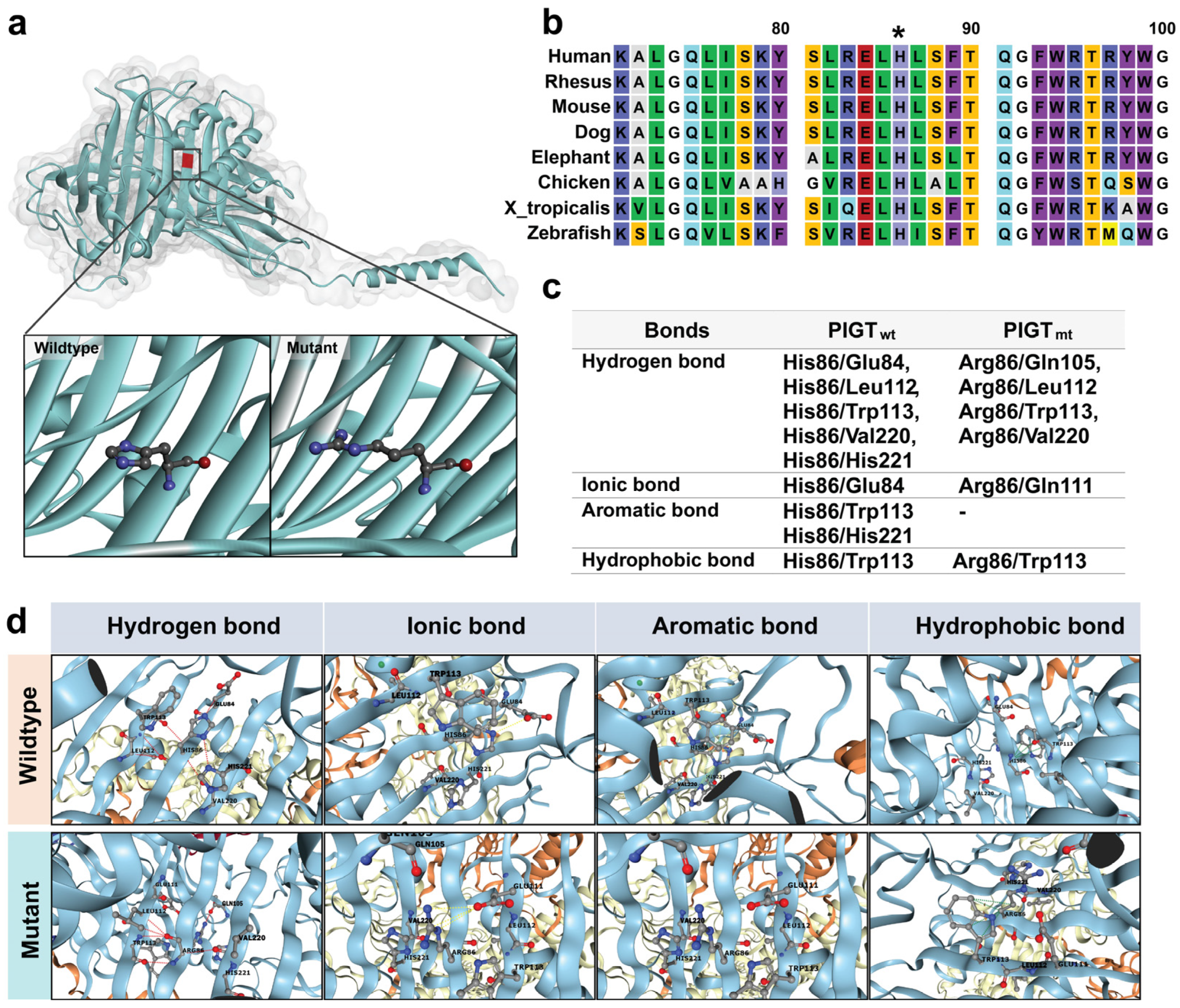
2.4. The Intramolecular Interactions Among Amino Acids at the Site of Alteration
2.5. The Prediction of Protein Stability
3. Discussion
4. Materials and Methods
4.1. Whole Exome Sequencing and Data Analysis
4.2. Variant Analysis
4.3. Sanger Sequencing
4.4. Western Blot Analysis for CD59 Expression
4.5. In-Silico Analysis of the PIGT Mutation on Protein Stability and Structural Dynamics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PIGT | Phosphatidylinositol glycan class T |
| GPI-TA | Glycosylphosphatidylinositol transamidase |
| GPI-AP | Glycosylphosphatidylinositol-anchored protein |
| MCAHS3 | Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 |
| WES | Whole exome sequencing |
| ALP | Alkaline phosphatase |
| ER | Endoplasmic reticulum |
| EEG | Electroencephalogram |
| ACMG/AMP | The American College of Medical Genetics and Genomics and the Association for Molecular Pathology |
| PBMC | Peripheral blood mononuclear cell |
| VCF | Variant calling file |
| HPO | Human phenotype ontology |
| GnomAD | The Genome Aggregation Database |
| T-REx | The Thai Reference Exome Database |
References
- Kinoshita, T.; Fujita, M. Biosynthesis of GPI-anchored proteins: Special emphasis on GPI lipid remodeling. J. Lipid Res. 2016, 57, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.; Knaus, A.; Pendziwiat, M.; Afenjar, A.; Barakat, T.S.; Bosch, F.; Callewaert, B.; Calvas, P.; Ceulemans, B.; Chassaing, N.; et al. Lessons learned from 40 novel PIGA patients and a review of the literature. Epilepsia 2020, 61, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.; Pendziwiat, M.; Obersztyn, E.; Goldenberg, P.; Zacher, P.; Döring, J.H.; Syrbe, S.; Begtrup, A.; Borovikov, A.; Sharkov, A.; et al. Deep-Phenotyping the Less Severe Spectrum of PIGT Deficiency and Linking the Gene to Myoclonic Atonic Seizures. Front. Genet. 2021, 12, 663643. [Google Scholar] [CrossRef]
- Kvarnung, M.; Nilsson, D.; Lindstrand, A.; Korenke, G.C.; Chiang, S.C.C.; Blennow, E.; Bergmann, M.; Stödberg, T.; Mäkitie, O.; Anderlid, B.-M.; et al. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J. Med. Genet. 2013, 50, 521–528. [Google Scholar] [CrossRef]
- Zhou, Y.; Pan, Q.; Pires, D.E.V.; Rodrigues, C.H.M.; Ascher, D.B. DDMut: Predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res. 2023, 51, W122–W128. [Google Scholar] [CrossRef]
- Sparks, S.E. Neonatal hypotonia. Clin. Perinatol. 2015, 42, 363–371, ix. [Google Scholar] [CrossRef]
- Nakashima, M.; Kashii, H.; Murakami, Y.; Kato, M.; Tsurusaki, Y.; Miyake, N.; Kubota, M.; Kinoshita, T.; Saitsu, H.; Matsumoto, N. Novel compound heterozygous PIGT mutations caused multiple congenital anomalies-hypotonia-seizures syndrome 3. Neurogenetics 2014, 15, 193–200. [Google Scholar] [CrossRef]
- Xu, Y.; Jia, G.; Li, T.; Zhou, Z.; Luo, Y.; Chao, Y.; Bao, J.; Su, Z.; Qu, Q.; Li, D. Molecular insights into biogenesis of glycosylphosphatidylinositol anchor proteins. Nat. Commun. 2022, 13, 2617. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Inoue, N.; Takeda, J. Defective glycosyl phosphatidylinositol anchor synthesis and paroxysmal nocturnal hemoglobinuria. Adv. Immunol. 1995, 60, 57–103. [Google Scholar] [CrossRef]
- Sidpra, J.; Sudhakar, S.; Biswas, A.; Massey, F.; Turchetti, V.; Lau, T.; Cook, E.; Alvi, J.R.; Elbendary, H.M.; Jewell, J.L.; et al. The clinical and genetic spectrum of inherited glycosylphosphatidylinositol deficiency disorders. Brain 2024, 147, 2775–2790. [Google Scholar] [CrossRef]
- Murakami, Y.; Kanzawa, N.; Saito, K.; Krawitz, P.M.; Mundlos, S.; Robinson, P.N.; Karadimitris, A.; Maeda, Y.; Kinoshita, T. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J. Biol. Chem. 2012, 287, 6318–6325. [Google Scholar] [CrossRef]
- Mabry, C.C.; Bautista, A.; Kirk, R.F.; Dubilier, L.D.; Braunstein, H.; Koepke, J.A. Familial hyperphosphatase with mental retardation, seizures, and neurologic deficits. J. Pediatr. 1970, 77, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Krawitz, P.M.; Schweiger, M.R.; Rödelsperger, C.; Marcelis, C.; Kölsch, U.; Meisel, C.; Stephani, F.; Kinoshita, T.; Murakami, Y.; Bauer, S.; et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 2010, 42, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Krawitz, P.M.; Murakami, Y.; Hecht, J.; Krüger, U.; Holder, S.E.; Mortier, G.R.; Chiaie, B.D.; De Baere, E.; Thompson, M.D.; Roscioli, T.; et al. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2012, 91, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Tawamie, H.; Murakami, Y.; Mang, Y.; Rehman, S.U.; Buchert, R.; Schaffer, S.; Muhammad, S.; Bak, M.; Nöthen, M.M.; et al. Hypomorphic mutations in PGAP2, encoding a GPI-anchor-remodeling protein, cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2013, 92, 575–583. [Google Scholar] [CrossRef]
- Howard, M.F.; Murakami, Y.; Pagnamenta, A.T.; Daumer-Haas, C.; Fischer, B.; Hecht, J.; Keays, D.A.; Knight, S.J.; Kölsch, U.; Krüger, U.; et al. Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 2014, 94, 278–287. [Google Scholar] [CrossRef]
- Chiyonobu, T.; Inoue, N.; Morimoto, M.; Kinoshita, T.; Murakami, Y. Glycosylphosphatidylinositol (GPI) anchor deficiency caused by mutations in PIGW is associated with West syndrome and hyperphosphatasia with mental retardation syndrome. J. Med. Genet. 2014, 51, 203–207. [Google Scholar] [CrossRef]
- Ranjan, A.; Alam, M.S.; Kumar, V.; Samanta, S.; Kumar, R.; Saifullah, K.M. Multiple congenital anomalies-hypotonia-seizures syndrome 3 secondary to phosphatidylinositol glycan class T mutation: A neonatal case report. Int. J. Contemp. Pediatr. 2023, 10, 394–397. [Google Scholar] [CrossRef]
- Hur, Y.J.; Lee, B.L.; Chung, W.Y.; Yu, S.; Jun, K.R.; Oh, S.H. Compound Heterozygous PIGT Mutations in Multiple Congenital Anomalies-Hypotonia-Seizures Syndrome: First Case in Korea and Characterization by Persistent Hypophosphatasia. Ann. Clin. Lab. Sci. 2021, 51, 422–425. [Google Scholar]
- Kohashi, K.; Ishiyama, A.; Yuasa, S.; Tanaka, T.; Miya, K.; Adachi, Y.; Sato, N.; Saitsu, H.; Ohba, C.; Matsumoto, N.; et al. Epileptic apnea in a patient with inherited glycosylphosphatidylinositol anchor deficiency and PIGT mutations. Brain Dev. 2018, 40, 53–57. [Google Scholar] [CrossRef]
- Yang, L.; Peng, J.; Yin, X.-M.; Pang, N.; Chen, C.; Wu, T.-H.; Zou, X.-M.; Yin, F. Homozygous PIGT Mutation Lead to Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3. Front. Genet. 2018, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- The DDD Study; Pagnamenta, A.T.; Murakami, Y.; Taylor, J.M.; Anzilotti, C.; Howard, M.F.; Miller, V.; Johnson, D.S.; Tadros, S.; Mansour, S.; et al. Analysis of exome data for 4293 trios suggests GPI-anchor biogenesis defects are a rare cause of developmental disorders. Eur. J. Hum. Genet. 2017, 25, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Skauli, N.; Wallace, S.; Chiang, S.C.C.; Barøy, T.; Holmgren, A.; Stray-Pedersen, A.; Bryceson, Y.T.; Strømme, P.; Frengen, E.; Misceo, D. Novel PIGT Variant in Two Brothers: Expansion of the Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 Phenotype. Genes 2016, 7, 108. [Google Scholar] [CrossRef]
- Lam, C.; Golas, G.A.; Davids, M.; Huizing, M.; Kane, M.S.; Krasnewich, D.M.; Malicdan, M.C.V.; Adams, D.R.; Markello, T.C.; Zein, W.M.; et al. Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors. Mol. Genet. Metab. 2015, 115, 128–140. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Cambridge, UK, 2010. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:13033997. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- BIOVIA DS. Discovery Studio Visualizer; v21. 1.0. 20298; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Our Study | Ranjan A. 2023 [18] | Hur Y. J. 2021 [19] | Kohashi K. 2018 [20] | Yang L. 2018 [21] | Pagnamenta A.T. 2017 [22] | Skauli N. 2016 [23] | Lam C. 2015 [24] | Nakashima M. 2014 [7] | Kvarnung M. 2013 [4] |
|---|---|---|---|---|---|---|---|---|---|---|
| Case number | 1 | 1 | 1 | 1 | 1 | 3 | 2 | 2 | 1 | 4 |
| Sex | Female | Male | Female | Male | Male | 1 Female/ 2 Male | Male | 1 Male 1 Female | Female | Female |
| Consanguinity | Yes | No | No | No | No | NA | Yes | No | No | Yes |
| Ethnicity | Thai | Indian | Korean | Japanese | Chinese | Caucasian/ Afghanistan | Somalian | NA | Japanese | Turkish |
| Dysmorphic features | + | + | + | + | + | +/NA | + | + | NA | + |
| Hypotonia | + | + | + | + | + | NA | + | + | + | + |
| Seizure | + | + | + | + | + | + | + | + | + | + |
| Seizure type | Myoclonic jerk | GTC, myoclonic, tonic | GTC | Myoclonic, Tonic, GTC, epileptic apnea | Myoclonic | GTC | Myoclonic, Tonic, GTC, complex partial seizures | Tonic, myoclonic | Myoclonic, Tonic, GTC | Myoclonic, GTC, head jerk, blinking, Absence |
| Brain atrophy | + | NA | + | + | + | + | + | + | + | +(3/4) |
| EEG | Multifocal epileptiform discharges from the right central parietal, left temporal, and left frontal areas | NA | Normal background rhythm, no epileptiform discharges | High-amplitude slow wave | Slow background wave | bilateral slow activity intermixed with sharp and spike waves /NA | Multiple spike-wave | Multifocal epileptiform, theta waves | High-amplitude slow wave | Multifocal epileptiform, theta waves |
| AED | LEV, PB | PB, FOS, LEV, CZP | VPA | LEV | LEV | NA/NA | NA | LEV, TPM, PB, CZP, MZ, BDZ, CLN, and ketogenic diet | CBZ, CLO, PLP, VPA, ZNS, PHE | NA |
| Developmental delay | Severe | NA | Severe | Severe | Severe | Severe/ Profound | Severe/ Moderate-severe | Profound | Profound | Severe |
| Skeletal | + | + | Normal | + | Normal | +/NA | Normal | + | + | + |
| Alkaline phosphatase level | High | Low | Low | Low | Normal | Normal/ Low, Normal | Normal | Normal | Low | Low |
| Variants in PIGT | Homozygous c.257A>G (p.His86Arg) | Homozygous c.709G>C (p.Glu237Gln) | c.250G>T (p.Glu84*) and c.1582G>A (p.Val528Met) | c.250 G>T (p.Glu84*) and c.1096 G>T (p.Gly366Trp) | Homozygous c.550G>A (p.Glu184Lys) | c.1582G4A (p.Val528Met) and c.1730dupC (p.Leu578fs*35) Homozygous c.709G>C (p.Glu237Gln) | Homozygous c.1079G>T (p.Gly360Val) | c.918dupC (p.Val307Argfs*13) and c.1342C>T (p.Arg448Trp) | c.250G>T (p.Glu84*) and c.1342C>T (p.Arg488Trp) | Homozygous c.547A>C (p.Thr183Pro) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klangjorhor, J.; Wiwattanadittakul, N.; Jaimalai, T.; Thongkumkoon, P.; Noisagul, P.; Khiaomai, R.; Sirikaew, N.; Moonsan, N.; Pasena, A.; Suksakit, P.; et al. A Novel Homozygous Missense Variant of PIGT Related to Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 with Elevated of Serum ALP Level in a Thai Newborn Patient. Int. J. Mol. Sci. 2025, 26, 2790. https://doi.org/10.3390/ijms26062790
Klangjorhor J, Wiwattanadittakul N, Jaimalai T, Thongkumkoon P, Noisagul P, Khiaomai R, Sirikaew N, Moonsan N, Pasena A, Suksakit P, et al. A Novel Homozygous Missense Variant of PIGT Related to Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 with Elevated of Serum ALP Level in a Thai Newborn Patient. International Journal of Molecular Sciences. 2025; 26(6):2790. https://doi.org/10.3390/ijms26062790
Chicago/Turabian StyleKlangjorhor, Jeerawan, Natrujee Wiwattanadittakul, Thanapak Jaimalai, Patcharawadee Thongkumkoon, Pitiporn Noisagul, Ratchadaporn Khiaomai, Nutnicha Sirikaew, Nonthanan Moonsan, Arnat Pasena, Pathacha Suksakit, and et al. 2025. "A Novel Homozygous Missense Variant of PIGT Related to Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 with Elevated of Serum ALP Level in a Thai Newborn Patient" International Journal of Molecular Sciences 26, no. 6: 2790. https://doi.org/10.3390/ijms26062790
APA StyleKlangjorhor, J., Wiwattanadittakul, N., Jaimalai, T., Thongkumkoon, P., Noisagul, P., Khiaomai, R., Sirikaew, N., Moonsan, N., Pasena, A., Suksakit, P., Teeyakasem, P., Chaiyawat, P., & Tengsujaritkul, M. (2025). A Novel Homozygous Missense Variant of PIGT Related to Multiple Congenital Anomalies-Hypotonia Seizures Syndrome 3 with Elevated of Serum ALP Level in a Thai Newborn Patient. International Journal of Molecular Sciences, 26(6), 2790. https://doi.org/10.3390/ijms26062790