
The Crosstalk Between NETs and the Complement Cascade: An Overview in Nephrological Autoimmune Disease
 , , ,
, , ,  and
and
Abstract
1. Introduction
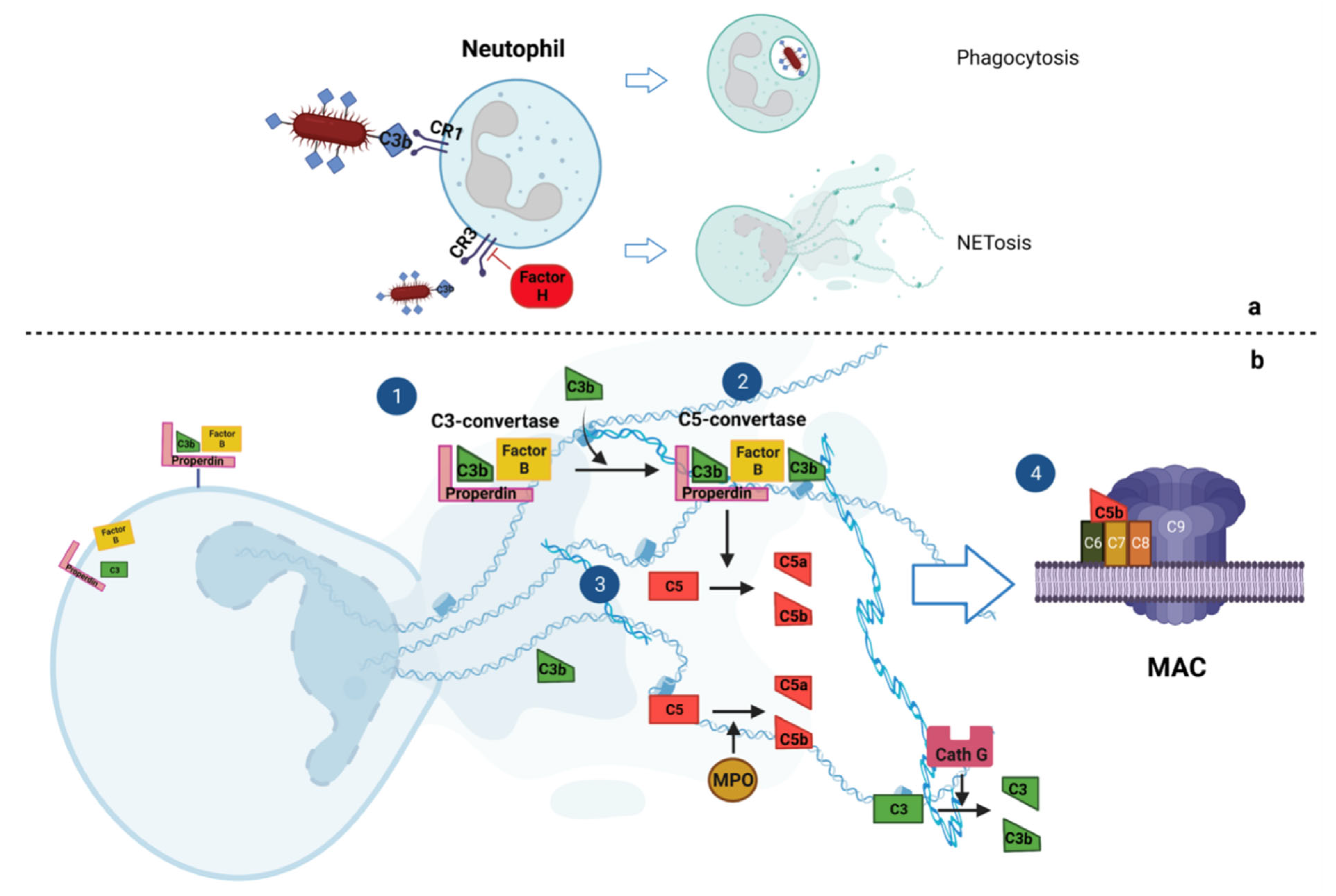
2. Interplay of NETosis and Complement Cascade
2.1. Complement Cascade Activates NETosis
2.2. NETs Activate Complement Cascade
3. NETosis and Complement Components in Renal Autoimmune Disease
3.1. Systemic Lupus Erythematosus
3.2. Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis
3.3. Antiphospholipid Syndrome
4. Possible Treatments
4.1. Modulation of NET Production
4.2. Modulation of NET Removal
4.3. Complement Component Inhibitors
4.4. Combined Therapies Modulating Complement Components and NETs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. Second of two parts. N. Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Wan, Y.; Shen, J.; Ouyang, J.; Dong, P.; Hong, Y.; Liang, L.; Liu, J. Bibliometric and visual analysis of neutrophil extracellular traps from 2004 to 2022. Front Immunol 2022, 13, 1025861. [Google Scholar] [CrossRef] [PubMed]
- Pinegin, B.; Vorobjeva, N.; Pinegin, V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun. Rev. 2015, 14, 633–640. [Google Scholar] [CrossRef]
- Vorobjeva, N.; Dagil, Y.; Pashenkov, M.; Pinegin, B.; Chernyak, B. Protein kinase C isoforms mediate the formation of neutrophil extracellular traps. Int. Immunopharmacol. 2023, 114, 109448. [Google Scholar] [CrossRef] [PubMed]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef]
- Karpati, E.; Kremlitzka, M.; Sandor, N.; Hajnal, D.; Schneider, A.E.; Jozsi, M. Complement Factor H Family Proteins Modulate Monocyte and Neutrophil Granulocyte Functions. Front. Immunol. 2021, 12, 660852. [Google Scholar] [CrossRef]
- Schneider, A.E.; Sandor, N.; Karpati, E.; Jozsi, M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol. Immunol. 2016, 72, 37–48. [Google Scholar] [CrossRef]
- Palmer, L.J.; Damgaard, C.; Holmstrup, P.; Nielsen, C.H. Influence of complement on neutrophil extracellular trap release induced by bacteria. J. Periodontal Res. 2016, 51, 70–76. [Google Scholar] [CrossRef]
- O’Flynn, J.; van der Pol, P.; Dixon, K.O.; Prohaszka, Z.; Daha, M.R.; van Kooten, C. Monomeric C-reactive protein inhibits renal cell-directed complement activation mediated by properdin. Am. J. Physiol. Renal Physiol. 2016, 310, F1308–F1316. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.O.; O’Flynn, J.; Klar-Mohamad, N.; Daha, M.R.; van Kooten, C. Properdin and factor H production by human dendritic cells modulates their T-cell stimulatory capacity and is regulated by IFN-gamma. Eur. J. Immunol. 2017, 47, 470–480. [Google Scholar] [CrossRef] [PubMed]
- O’Flynn, J.; Dixon, K.O.; Faber Krol, M.C.; Daha, M.R.; van Kooten, C. Myeloperoxidase directs properdin-mediated complement activation. J. Innate Immun. 2014, 6, 417–425. [Google Scholar] [CrossRef]
- Vogt, W. Complement activation by myeloperoxidase products released from stimulated human polymorphonuclear leukocytes. Immunobiology 1996, 195, 334–346. [Google Scholar] [CrossRef]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Maison, C.M.; Villiers, C.L.; Colomb, M.G. Proteolysis of C3 on U937 cell plasma membranes. Purification of cathepsin G. J. Immunol. 1991, 147, 921–926. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Zhao, M.H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tyden, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef]
- Yuen, J.; Pluthero, F.G.; Douda, D.N.; Riedl, M.; Cherry, A.; Ulanova, M.; Kahr, W.H.; Palaniyar, N.; Licht, C. NETosing Neutrophils Activate Complement Both on Their Own NETs and Bacteria via Alternative and Non-alternative Pathways. Front. Immunol. 2016, 7, 137. [Google Scholar] [CrossRef]
- Halder, L.D.; Abdelfatah, M.A.; Jo, E.A.; Jacobsen, I.D.; Westermann, M.; Beyersdorf, N.; Lorkowski, S.; Zipfel, P.F.; Skerka, C. Factor H Binds to Extracellular DNA Traps Released from Human Blood Monocytes in Response to Candida albicans. Front. Immunol. 2016, 7, 671. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Andersen, J.; Aringer, M.; Arnaud, L.; Bae, S.C.; Boletis, J.; Bruce, I.N.; Cervera, R.; Doria, A.; et al. EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann. Rheum. Dis. 2024, 83, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 2021, 100, S1–S276. [Google Scholar] [CrossRef]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 2019, 20, 1574–1583. [Google Scholar] [CrossRef]
- Herrada, A.A.; Escobedo, N.; Iruretagoyena, M.; Valenzuela, R.A.; Burgos, P.I.; Cuitino, L.; Llanos, C. Innate Immune Cells’ Contribution to Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 772. [Google Scholar] [CrossRef] [PubMed]
- Lodi, L.; Mastrolia, M.V.; Bello, F.; Rossi, G.M.; Angelotti, M.L.; Crow, Y.J.; Romagnani, P.; Vaglio, A. Type I interferon-related kidney disorders. Kidney Int. 2022, 101, 1142–1159. [Google Scholar] [CrossRef] [PubMed]
- Wigerblad, G.; Kaplan, M.J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 2023, 23, 274–288. [Google Scholar] [CrossRef]
- Alarcon, G.S.; McGwin, G., Jr.; Petri, M.; Reveille, J.D.; Ramsey-Goldman, R.; Kimberly, R.P.; PROFILE Study Group. Baseline characteristics of a multiethnic lupus cohort: PROFILE. Lupus 2002, 11, 95–101. [Google Scholar] [CrossRef]
- Tektonidou, M.G.; Dasgupta, A.; Ward, M.M. Risk of End-Stage Renal Disease in Patients With Lupus Nephritis, 1971–2015: A Systematic Review and Bayesian Meta-Analysis. Arthritis Rheumatol. 2016, 68, 1432–1441. [Google Scholar] [CrossRef]
- Contreras, G.; Mattiazzi, A.; Guerra, G.; Ortega, L.M.; Tozman, E.C.; Li, H.; Tamariz, L.; Carvalho, C.; Kupin, W.; Ladino, M.; et al. Recurrence of lupus nephritis after kidney transplantation. J. Am. Soc. Nephrol. 2010, 21, 1200–1207. [Google Scholar] [CrossRef]
- Bertelli, R.; Schena, F.; Antonini, F.; Reverberi, D.; Signa, S.; Pedemonte, N.; Consolaro, A.; Gattorno, M.; Negrini, S.; Pupo, F.; et al. Neutrophil Extracellular Traps in Systemic Lupus Erythematosus Stimulate IgG2 Production From B Lymphocytes. Front. Med. 2021, 8, 635436. [Google Scholar] [CrossRef]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 2016, 67, 19–28. [Google Scholar] [CrossRef]
- Bruschi, M.; Petretto, A.; Bertelli, R.; Galetti, M.; Bonanni, A.; Pratesi, F.; Migliorini, P.; Candiano, G.; Vaglio, A.; Ghiggeri, G.M. Post-translational modified proteins are biomarkers of autoimmune-processes: NETosis and the inflammatory-autoimmunity connection. Clin. Chim. Acta 2017, 464, 12–16. [Google Scholar] [CrossRef]
- Bruschi, M.; Petretto, A.; Santucci, L.; Vaglio, A.; Pratesi, F.; Migliorini, P.; Bertelli, R.; Lavarello, C.; Bartolucci, M.; Candiano, G.; et al. Neutrophil Extracellular Traps protein composition is specific for patients with Lupus nephritis and includes methyl-oxidized alphaenolase (methionine sulfoxide 93). Sci. Rep. 2019, 9, 7934. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Moroni, G.; Sinico, R.A.; Franceschini, F.; Fredi, M.; Vaglio, A.; Cavagna, L.; Petretto, A.; Pratesi, F.; Migliorini, P.; et al. Neutrophil Extracellular Traps in the Autoimmunity Context. Front. Med. 2021, 8, 614829. [Google Scholar] [CrossRef]
- Yu, Y.; Su, K. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J. Clin. Cell Immunol. 2013, 4, 139. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Bonanni, A.; Petretto, A.; Vaglio, A.; Pratesi, F.; Santucci, L.; Migliorini, P.; Bertelli, R.; Galetti, M.; Belletti, S.; et al. Neutrophil Extracellular Traps Profiles in Patients with Incident Systemic Lupus Erythematosus and Lupus Nephritis. J. Rheumatol. 2020, 47, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Su, J.; Yan, M.; Pan, J.; Zhang, X. Neutrophil extracellular traps in autoimmune diseases: Analysis of the knowledge map. Front. Immunol. 2023, 14, 1095421. [Google Scholar]
- Takagi, H.; Arimura, K.; Uto, T.; Fukaya, T.; Nakamura, T.; Choijookhuu, N.; Hishikawa, Y.; Sato, K. Plasmacytoid dendritic cells orchestrate TLR7-mediated innate and adaptive immunity for the initiation of autoimmune inflammation. Sci. Rep. 2016, 6, 24477. [Google Scholar] [CrossRef]
- Demkow, U. Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation. Int. J. Mol. Sci. 2023, 24, 4896. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Gudjonsson, J.E.; Kahlenberg, J.M.; Joseph McCune, W.; Bockenstedt, P.L.; Karp, D.R.; Knight, J.S. Anti-Neutrophil Extracellular Trap Antibodies and Impaired Neutrophil Extracellular Trap Degradation in Antiphospholipid Syndrome. Arthritis Rheumatol. 2020, 72, 2130–2135. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, T.; Nurbaeva, K. The Role of Neutrophil Extracellular Traps (NETs) in the Pathogenesis of Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Int. J. Mol. Sci. 2023, 24, 13581. [Google Scholar] [CrossRef] [PubMed]
- Mathapathi, S.; Chu, C.Q. Contribution of Impaired DNASE1L3 Activity to Anti-DNA Autoantibody Production in Systemic Lupus Erythematosus. Rheumatol. Immunol. Res. 2022, 3, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Almlof, J.C.; Nystedt, S.; Leonard, D.; Eloranta, M.L.; Grosso, G.; Sjowall, C.; Bengtsson, A.A.; Jonsen, A.; Gunnarsson, I.; Svenungsson, E.; et al. Whole-genome sequencing identifies complex contributions to genetic risk by variants in genes causing monogenic systemic lupus erythematosus. Hum. Genet. 2019, 138, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Yasutomo, K.; Horiuchi, T.; Kagami, S.; Tsukamoto, H.; Hashimura, C.; Urushihara, M.; Kuroda, Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001, 28, 313–314. [Google Scholar] [CrossRef]
- Kisla Ekinci, R.M.; Balci, S.; Ozcan, D.; Atmis, B.; Bisgin, A. Monogenic lupus due to DNASE1L3 deficiency in a pediatric patient with urticarial rash, hypocomplementemia, pulmonary hemorrhage, and immune-complex glomerulonephritis. Eur. J. Med. Genet. 2021, 64, 104262. [Google Scholar] [CrossRef]
- Hartl, J.; Serpas, L.; Wang, Y.; Rashidfarrokhi, A.; Perez, O.A.; Sally, B.; Sisirak, V.; Soni, C.; Khodadadi-Jamayran, A.; Tsirigos, A.; et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J. Exp. Med. 2021, 218, e20201138. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef]
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131. [Google Scholar] [CrossRef]
- Jarrot, P.A.; Kaplanski, G. Pathogenesis of ANCA-associated vasculitis: An update. Autoimmun. Rev. 2016, 15, 704–713. [Google Scholar] [CrossRef]
- Shiratori-Aso, S.; Nakazawa, D. The involvement of NETs in ANCA-associated vasculitis. Front. Immunol. 2023, 14, 1261151. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.U.; O’Sullivan, K.M. The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs. Int. J. Mol. Sci. 2022, 23, 3793. [Google Scholar] [CrossRef]
- Kambas, K.; Chrysanthopoulou, A.; Vassilopoulos, D.; Apostolidou, E.; Skendros, P.; Girod, A.; Arelaki, S.; Froudarakis, M.; Nakopoulou, L.; Giatromanolaki, A.; et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann. Rheum. Dis. 2014, 73, 1854–1863. [Google Scholar] [CrossRef]
- Nakazawa, D.; Tomaru, U.; Yamamoto, C.; Jodo, S.; Ishizu, A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front. Immunol. 2012, 3, 333. [Google Scholar] [CrossRef]
- Xia, Y.; He, J.; Zhang, H.; Wang, H.; Tetz, G.; Maguire, C.A.; Wang, Y.; Onuma, A.; Genkin, D.; Tetz, V.; et al. AAV-mediated gene transfer of DNase I in the liver of mice with colorectal cancer reduces liver metastasis and restores local innate and adaptive immune response. Mol. Oncol. 2020, 14, 2920–2935. [Google Scholar] [CrossRef]
- Patriarcheas, V.; Tsamos, G.; Vasdeki, D.; Kotteas, E.; Kollias, A.; Nikas, D.; Kaiafa, G.; Dimakakos, E. Antiphospholipid Syndrome: A Comprehensive Clinical Review. J. Clin. Med. 2025, 14, 733. [Google Scholar] [CrossRef]
- Oku, K.; Amengual, O.; Hisada, R.; Ohmura, K.; Nakagawa, I.; Watanabe, T.; Bohgaki, T.; Horita, T.; Yasuda, S.; Atsumi, T. Autoantibodies against a complement component 1 q subcomponent contribute to complement activation and recurrent thrombosis/pregnancy morbidity in anti-phospholipid syndrome. Rheumatology 2016, 55, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Lin, X.; Liang, H.; Liu, X.; Zhang, X.; Zhang, Q.; Zhou, F.; Yu, C.; Lei, L.; et al. Complement C5a induces the generation of neutrophil extracellular traps by inhibiting mitochondrial STAT3 to promote the development of arterial thrombosis. Thromb. J. 2022, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Kanthi, Y. Mechanisms of immunothrombosis and vasculopathy in antiphospholipid syndrome. Semin. Immunopathol. 2022, 44, 347–362. [Google Scholar] [CrossRef]
- Leung, H.H.L.; Perdomo, J.; Ahmadi, Z.; Yan, F.; McKenzie, S.E.; Chong, B.H. Inhibition of NADPH oxidase blocks NETosis and reduces thrombosis in heparin-induced thrombocytopenia. Blood Adv. 2021, 5, 5439–5451. [Google Scholar] [CrossRef]
- Bulnes, J.F.; Gonzalez, L.; Velasquez, L.; Orellana, M.P.; Venturelli, P.M.; Martinez, G. Role of inflammation and evidence for the use of colchicine in patients with acute coronary syndrome. Front. Cardiovasc. Med. 2024, 11, 1356023. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Biron, B.M.; Chung, C.S.; Chen, Y.; Wilson, Z.; Fallon, E.A.; Reichner, J.S.; Ayala, A. PAD4 Deficiency Leads to Decreased Organ Dysfunction and Improved Survival in a Dual Insult Model of Hemorrhagic Shock and Sepsis. J. Immunol. 2018, 200, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; You, Q.; Wu, Y.; Wu, J. Inhibition of PAD4-mediated NET formation by cl-amidine prevents diabetes development in nonobese diabetic mice. Eur. J. Pharmacol. 2022, 916, 174623. [Google Scholar] [CrossRef] [PubMed]
- Biron, B.M.; Chung, C.S.; O’Brien, X.M.; Chen, Y.; Reichner, J.S.; Ayala, A. Cl-Amidine Prevents Histone 3 Citrullination and Neutrophil Extracellular Trap Formation, and Improves Survival in a Murine Sepsis Model. J. Innate Immun. 2017, 9, 22–32. [Google Scholar] [CrossRef]
- Sondo, E.; Bertelli, R.; Pesce, E.; Ghiggeri, G.M.; Pedemonte, N. High-Content Screening Identifies Vanilloids as a Novel Class of Inhibitors of NET Formation. Front. Immunol. 2019, 10, 963. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kim, S.J.; Lei, Y.; Wang, S.; Wang, H.; Huang, H.; Zhang, H.; Tsung, A. Neutrophil extracellular traps in homeostasis and disease. Signal Transduct. Target. Ther. 2024, 9, 235. [Google Scholar] [CrossRef]
- Fan, X.; Shu, P.; Wang, Y.; Ji, N.; Zhang, D. Interactions between neutrophils and T-helper 17 cells. Front. Immunol. 2023, 14, 1279837. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Wang, Z.; Chang, X.; Wu, H.; Yan, Z.; Wu, J.; He, Z.; Kang, L.; Hu, W.; et al. Neutrophil-derived PAD4 induces citrullination of CKMT1 exacerbates mucosal inflammation in inflammatory bowel disease. Cell Mol. Immunol. 2024, 21, 620–633. [Google Scholar] [CrossRef]
- Zahm, J.M.; Debordeaux, C.; Maurer, C.; Hubert, D.; Dusser, D.; Bonnet, N.; Lazarus, R.A.; Puchelle, E. Improved activity of an actin-resistant DNase I variant on the cystic fibrosis airway secretions. Am. J. Respir. Crit. Care Med. 2001, 163, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R. Mining Clinical Case Reports to Identify New Lines of Investigation in Alzheimer’s Disease: The Curious Case of DNase I. J. Alzheimers Dis. Rep. 2019, 3, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.J. The curious case of protein splicing: Mechanistic insights suggested by protein semisynthesis. Protein Sci. 1993, 2, 697–705. [Google Scholar] [CrossRef]
- Manderson, A.P.; Carlucci, F.; Lachmann, P.J.; Lazarus, R.A.; Festenstein, R.J.; Cook, H.T.; Walport, M.J.; Botto, M. The in vivo expression of actin/salt-resistant hyperactive DNase I inhibits the development of anti-ssDNA and anti-histone autoantibodies in a murine model of systemic lupus erythematosus. Arthritis Res. Ther. 2006, 8, R68. [Google Scholar] [CrossRef]
- Davis, J.C., Jr.; Manzi, S.; Yarboro, C.; Rairie, J.; McInnes, I.; Averthelyi, D.; Sinicropi, D.; Hale, V.G.; Balow, J.; Austin, H.; et al. Recombinant human Dnase I (rhDNase) in patients with lupus nephritis. Lupus 1999, 8, 68–76. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, S.; Xia, Y.; Wang, H. The Therapeutic Strategies for SLE by Targeting Anti-dsDNA Antibodies. Clin. Rev. Allergy Immunol. 2022, 63, 152–165. [Google Scholar] [CrossRef]
- Marinho, A.; Delgado Alves, J.; Fortuna, J.; Faria, R.; Almeida, I.; Alves, G.; Araujo Correia, J.; Campar, A.; Brandao, M.; Crespo, J.; et al. Biological therapy in systemic lupus erythematosus, antiphospholipid syndrome, and Sjogren’s syndrome: Evidence- and practice-based guidance. Front. Immunol. 2023, 14, 1117699. [Google Scholar] [CrossRef] [PubMed]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef]
- Pickering, M.C.; Ismajli, M.; Condon, M.B.; McKenna, N.; Hall, A.E.; Lightstone, L.; Terence Cook, H.; Cairns, T.D. Eculizumab as rescue therapy in severe resistant lupus nephritis. Rheumatology 2015, 54, 2286–2288. [Google Scholar] [CrossRef]
- Ono, M.; Ohashi, N.; Namikawa, A.; Katahashi, N.; Ishigaki, S.; Tsuji, N.; Isobe, S.; Iwakura, T.; Sakao, Y.; Tsuji, T.; et al. A Rare Case of Lupus Nephritis Presenting as Thrombotic Microangiopathy with Diffuse Pseudotubulization Possibly Caused by Atypical Hemolytic Uremic Syndrome. Intern. Med. 2018, 57, 1617–1623. [Google Scholar] [CrossRef]
- Sciascia, S.; Radin, M.; Yazdany, J.; Tektonidou, M.; Cecchi, I.; Roccatello, D.; Dall’Era, M. Expanding the therapeutic options for renal involvement in lupus: Eculizumab, available evidence. Rheumatol. Int. 2017, 37, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.D.; Bannerman, F.; Beresford, M.W.; Oni, L. A systematic review of the role of eculizumab in systemic lupus erythematosus-associated thrombotic microangiopathy. BMC Nephrol. 2020, 21, 245. [Google Scholar] [CrossRef] [PubMed]
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 2756–2767. [Google Scholar] [CrossRef]
- Berdunov, V.; Ramirez de Arellano, A.; Li, T.; Vintderdag, H.; Baxter, G. Key considerations for modelling the long-term costs and benefits of treatments for ANCA-associated vasculitis. Clin. Exp. Rheumatol. 2024, 42, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Peffault de Latour, R.; Marano, L.; Frieri, C. Discovering C3 targeting therapies for paroxysmal nocturnal hemoglobinuria: Achievements and pitfalls. Semin. Immunol. 2022, 59, 101618. [Google Scholar] [CrossRef]
- D’Alessandro, M.; Conticini, E.; Bergantini, L.; Cameli, P.; Cantarini, L.; Frediani, B.; Bargagli, E. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis and Interstitial Lung Disease: A Scoping Review. Life 2022, 12, 317. [Google Scholar] [CrossRef]
- O’Sullivan, K.M.; Holdsworth, S.R. Neutrophil Extracellular Traps: A Potential Therapeutic Target in MPO-ANCA Associated Vasculitis? Front. Immunol. 2021, 12, 635188. [Google Scholar] [CrossRef]
- Herrmann, J.B.; Muenstermann, M.; Strobel, L.; Schubert-Unkmeir, A.; Woodruff, T.M.; Gray-Owen, S.D.; Klos, A.; Johswich, K.O. Complement C5a Receptor 1 Exacerbates the Pathophysiology of N. meningitidis Sepsis and Is a Potential Target for Disease Treatment. mBio 2018, 9, e01755-17. [Google Scholar] [CrossRef]
- Zhu, S.; Yu, Y.; Ren, Y.; Xu, L.; Wang, H.; Ling, X.; Jin, L.; Hu, Y.; Zhang, H.; Miao, C.; et al. The emerging roles of neutrophil extracellular traps in wound healing. Cell Death Dis. 2021, 12, 984. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Name | Class | Disease | Main Pathogenic Mechanism | Status |
|---|---|---|---|---|
| Modulation of NET production | ||||
| N-acetylcysteine | ROS inhibitor | Vasculitis, SLE APS | Reduces oxidative stress-induced NETosis | On the market |
| PAD4 inhibitors (Cl-amidine, GSK484) | Anti-inflammatory small molecule | Vasculitis, SLE APS | Prevents excessive NET release by neutrophils | Clinical trial Phase I-II |
| Colchicine, Hydroxychloroquine | Neutrophil inhibitor | SLE | Suppresses neutrophil activation and NET release | On the market |
| Capsaicin, Eugenol, Vanillin | Phytochemical | Vasculitis, SLE | Reduces NETosis by inhibiting ROS production and neutrophil activation | Pre-clinical studies |
| Modulation of NET removal | ||||
| Recombinant DNase I (Dornase alfa) | Enzyme | Vasculitis, SLE APS | Degrades extracellular DNA, promoting NET clearance | Clinical trial Phase I-II |
| Complement Inhibitors | ||||
| Eculizumab | Humanized monoclonal antibody | aHUS | Binds C5 to prevent generation of MAC | On the market |
| Ravalizumab | Humanized monoclonal antibody | aHUS | Binds C5 to prevent generation of MAC | On the market |
| CCX168 (Avacopan) | Anti-inflammatory small molecule | Vasculitis | Selective inhibitor of the complement C5a receptor | Clinical trial Phase III |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kajana, X.; Caridi, G.; Bruschi, M.; Spinelli, S.; Lugani, F.; Ghiggeri, G.M.; La Porta, E.; Mortari, G.; Verrina, E.E.; Angeletti, A.; et al. The Crosstalk Between NETs and the Complement Cascade: An Overview in Nephrological Autoimmune Disease. Int. J. Mol. Sci. 2025, 26, 2789. https://doi.org/10.3390/ijms26062789
Kajana X, Caridi G, Bruschi M, Spinelli S, Lugani F, Ghiggeri GM, La Porta E, Mortari G, Verrina EE, Angeletti A, et al. The Crosstalk Between NETs and the Complement Cascade: An Overview in Nephrological Autoimmune Disease. International Journal of Molecular Sciences. 2025; 26(6):2789. https://doi.org/10.3390/ijms26062789
Chicago/Turabian StyleKajana, Xhuliana, Gianluca Caridi, Maurizio Bruschi, Sonia Spinelli, Francesca Lugani, Gian Marco Ghiggeri, Edoardo La Porta, Gabriele Mortari, Enrico E. Verrina, Andrea Angeletti, and et al. 2025. "The Crosstalk Between NETs and the Complement Cascade: An Overview in Nephrological Autoimmune Disease" International Journal of Molecular Sciences 26, no. 6: 2789. https://doi.org/10.3390/ijms26062789
APA StyleKajana, X., Caridi, G., Bruschi, M., Spinelli, S., Lugani, F., Ghiggeri, G. M., La Porta, E., Mortari, G., Verrina, E. E., Angeletti, A., & Bigatti, C. (2025). The Crosstalk Between NETs and the Complement Cascade: An Overview in Nephrological Autoimmune Disease. International Journal of Molecular Sciences, 26(6), 2789. https://doi.org/10.3390/ijms26062789