
PAR2 Serves an Indispensable Role in Controlling PAR4 Oncogenicity: The β-Catenin–p53 Axis


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
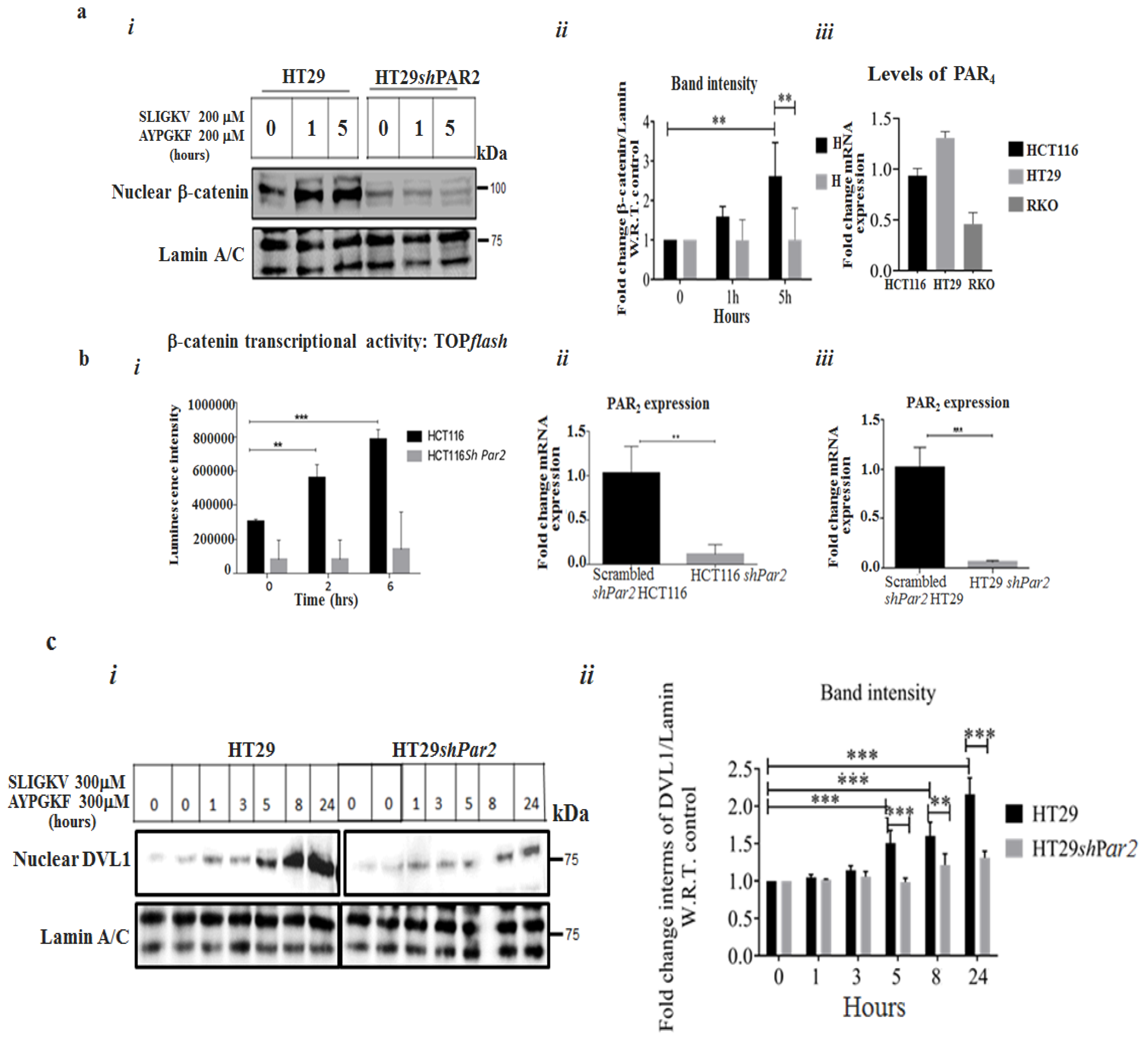
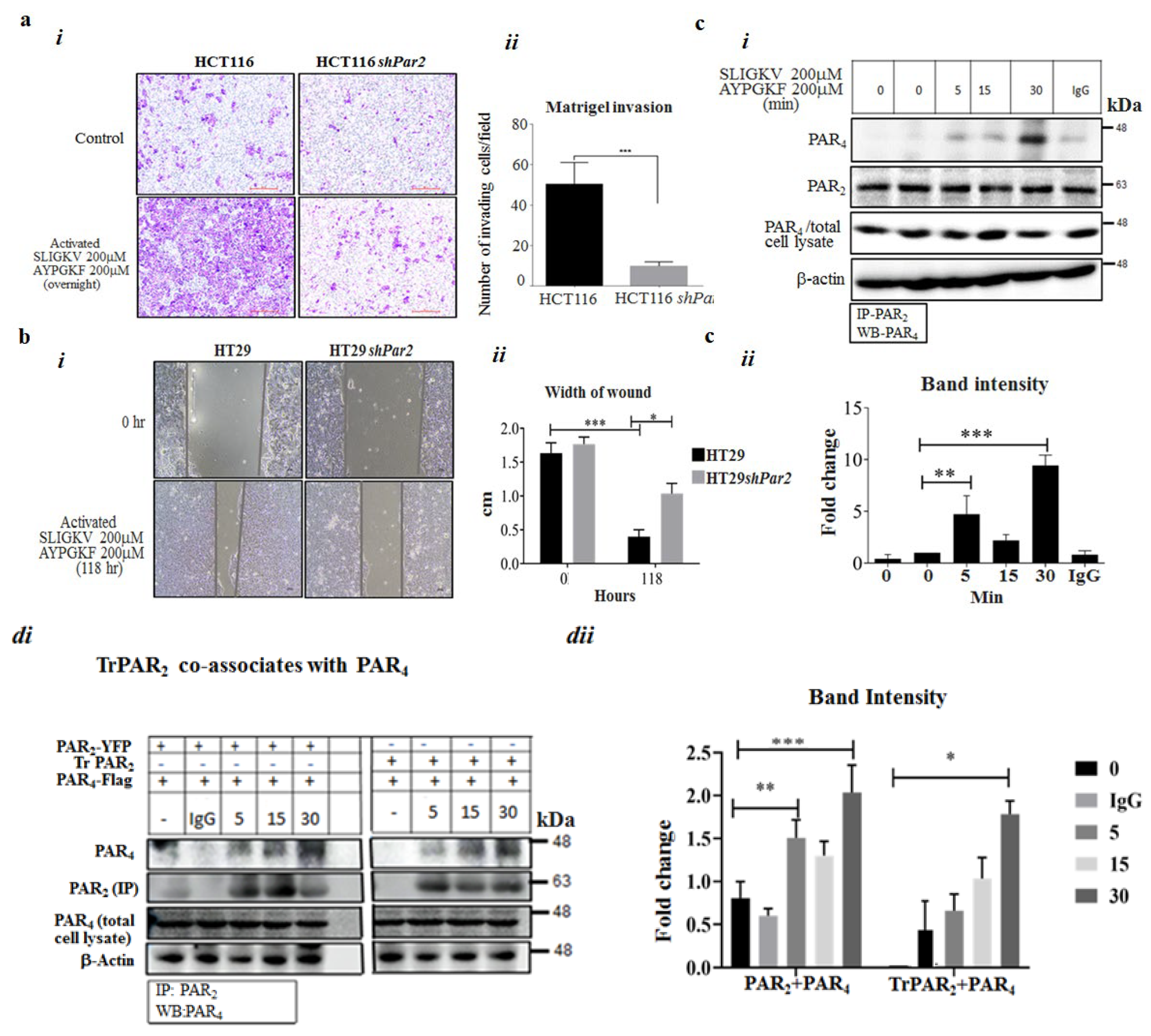
2.1. Knockdown of Par2/f2rl1 Inhibits Events in PAR2 and PAR4-Induced β-Catenin Stabilization and Cell Invasion
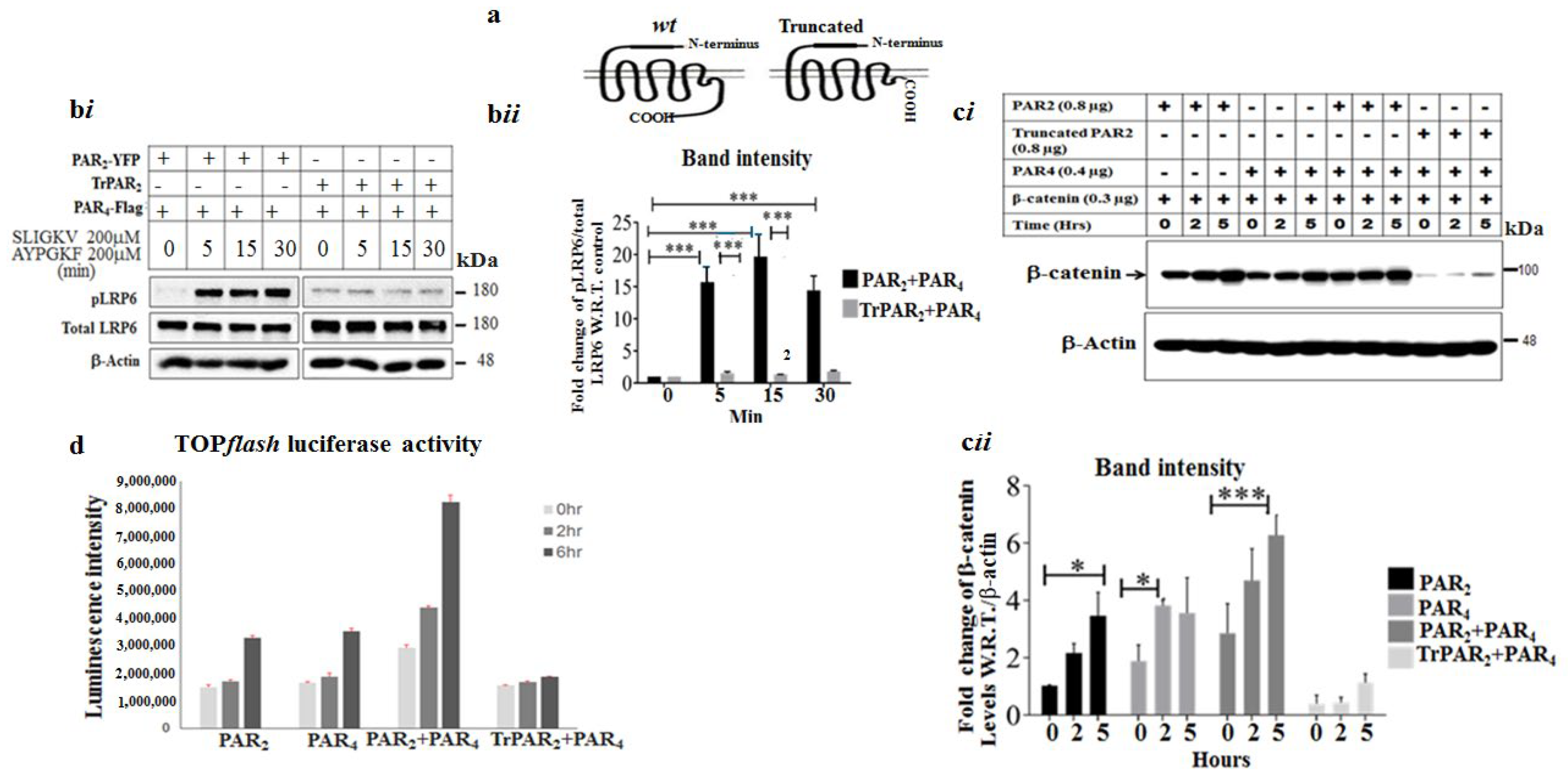
2.2. Co-Association of PAR2 and PAR4
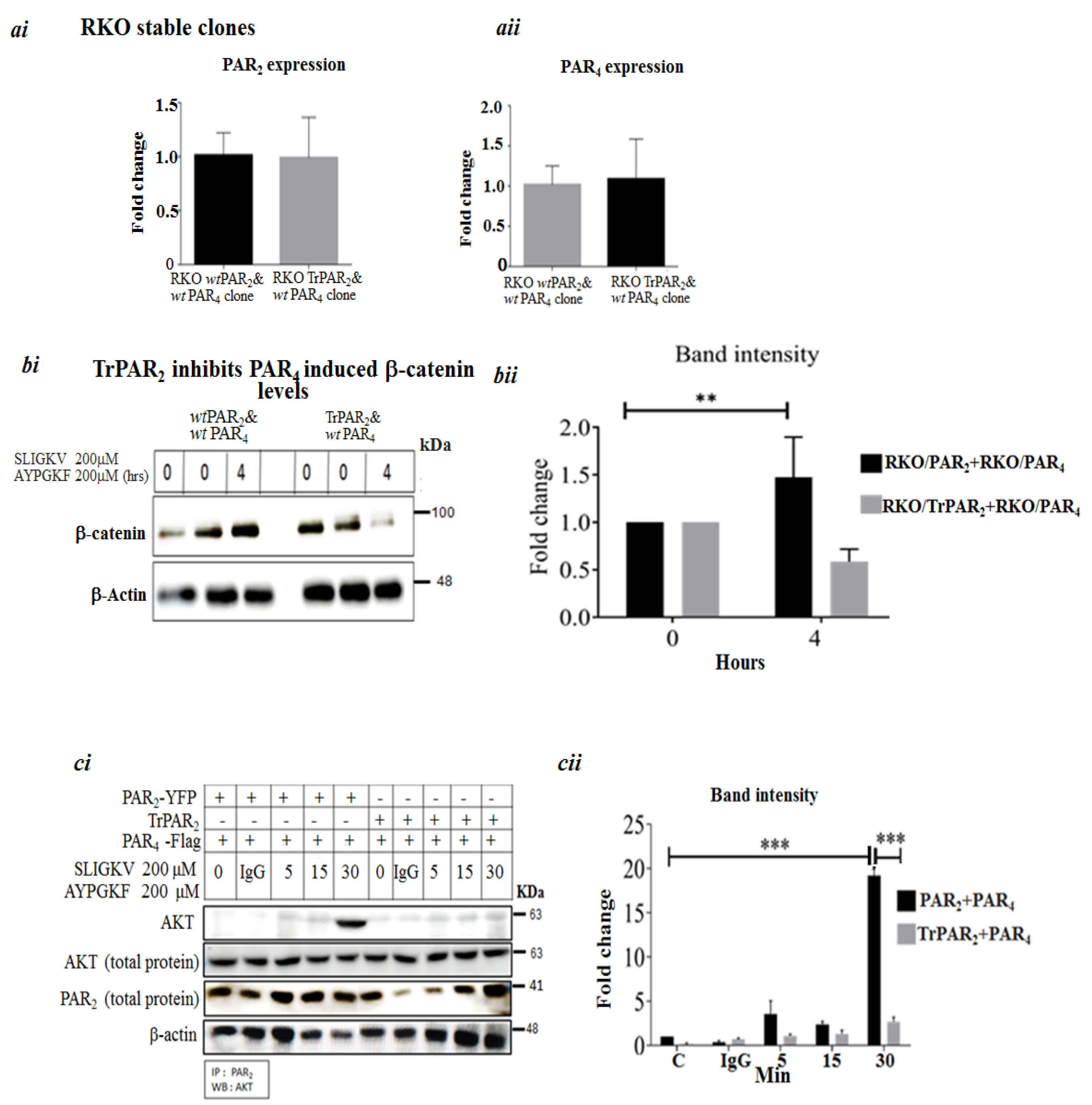
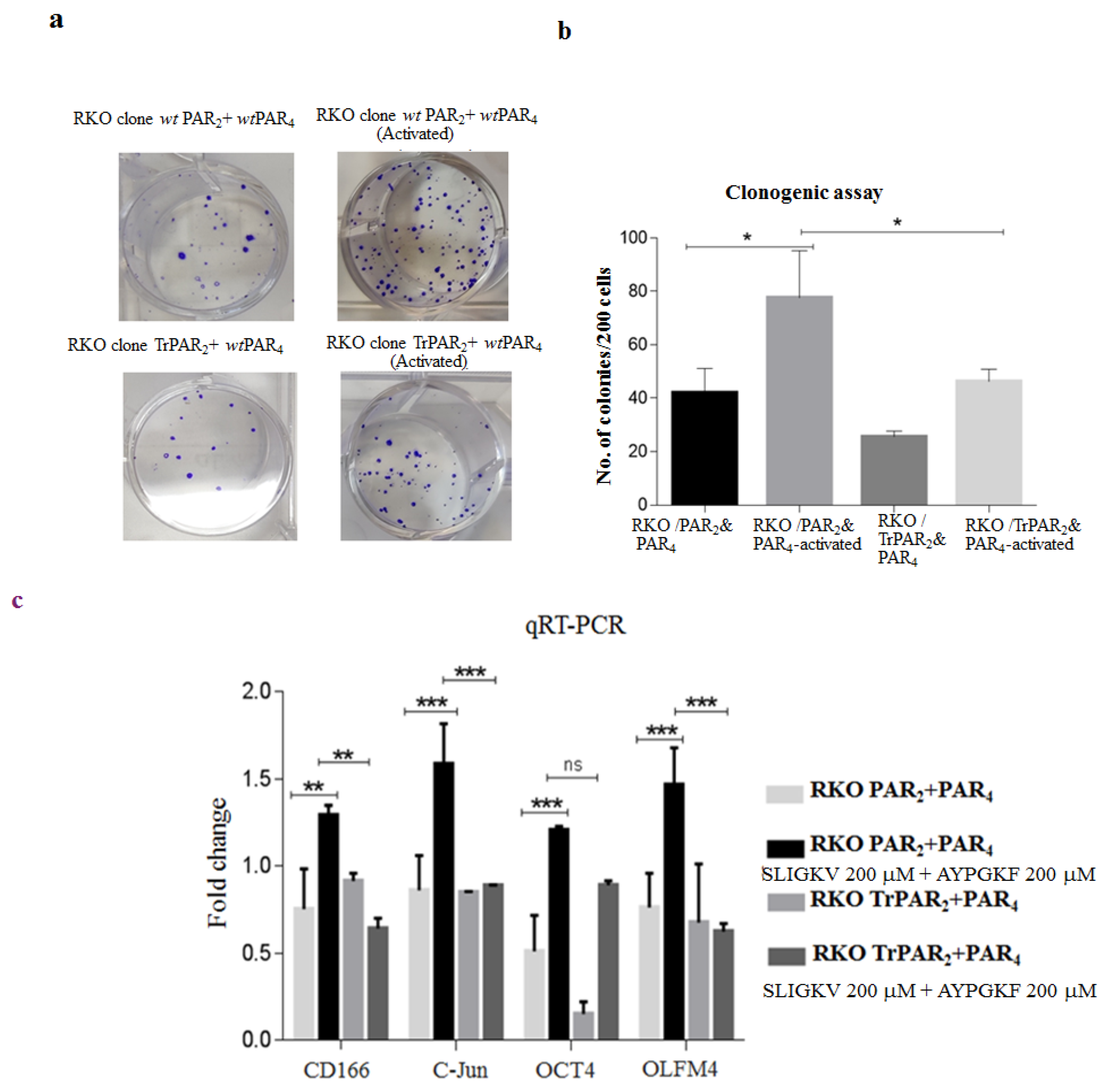
2.3. TrPAR2 Inhibits PAR2&4-Induced β-Catenin Stabilization, Transcriptional Activity, Colony Formation and Stem-Cell Marker Levels
2.4. TrPAR2 Inhibits PAR2 and PAR4-Induced Tumor Growth In Vivo
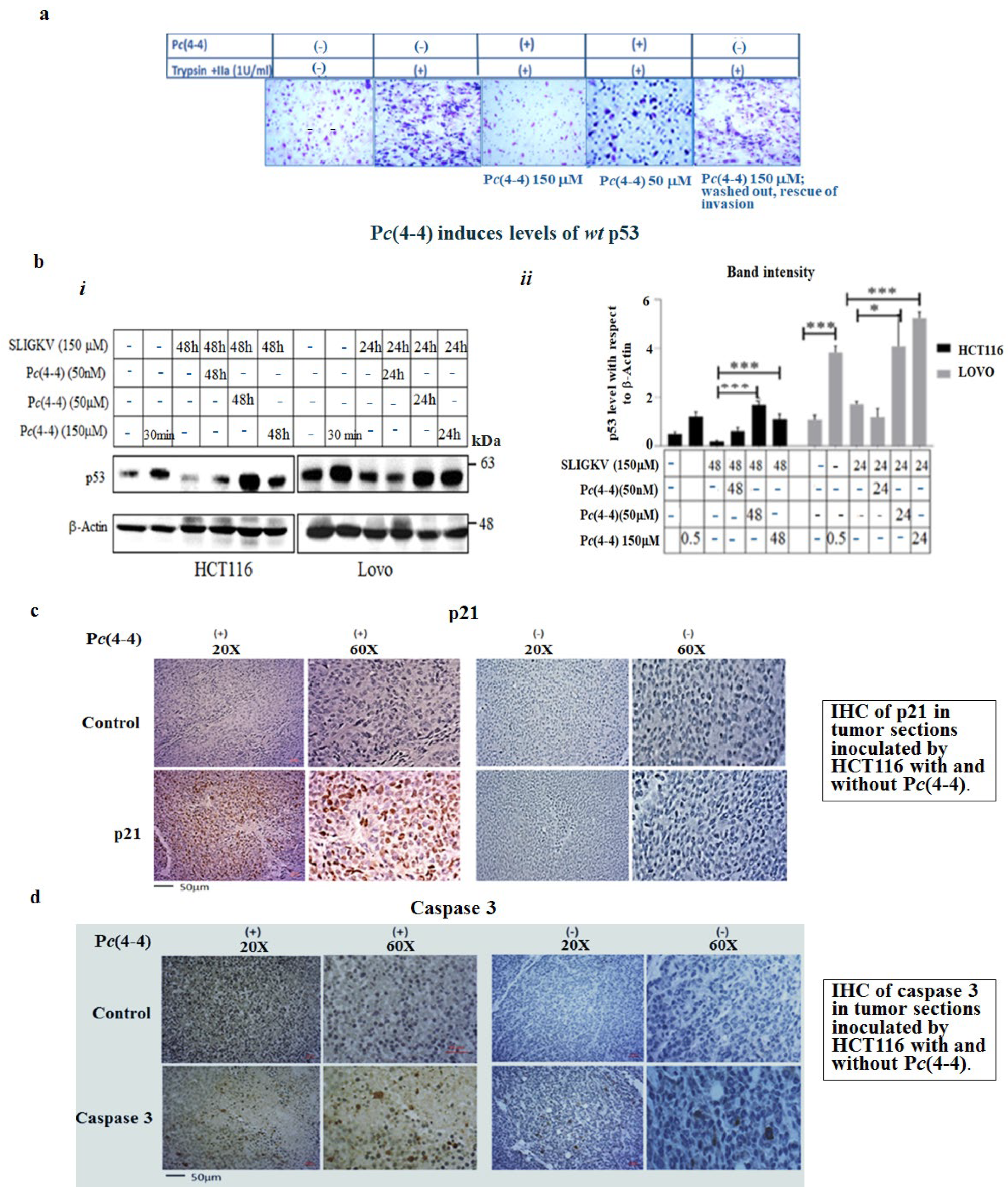
2.5. The Oncogenic Properties of PAR2 Are Significantly Inhibited in the Presence of Pc(4-4) via Induced wt p53 Levels
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Cell Culture
4.3. Matrigel Invasion
4.4. Scratch-Wound Healing
4.5. Clonogenic Assay
4.6. Plasmids and Reagents
4.7. Cell Transfections and PAR Activation
4.8. Small Hairpin (sh)-RNA Construct Preparation and Viral Particle Generation
4.9. Preparation of RKO Stable Clones Expressing wt PAR2/wtPAR4 and trPAR2/wtPAR4
4.10. Quantitative Real-Time (qRT) PCR and Reverse Transcriptase (RT) PCR
4.11. Cell Lysate Preparations, Immunoprecipitation (IP), and Western Blot
4.12. TOPflash Luciferase Reporter
4.13. Ectopic Tumor Xenograft Mouse Model
4.14. Immunohistochemistry (IHC)
4.15. Pc(4-4)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E. Harnessing the genome for characterization of G-protein coupled receptors in cancer pathogenesis. FEBS J. 2013, 280, 4729–4738. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A.C.A. Cancer statistics, 2024. Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Sonkin, D.; Thomas, A.; Teicher, B.A. Cancer treatments: Past, present, and future. Cancer Genet. 2024, 286, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.-K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; Van De Wetering, M.; Van Es, J.H.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.-X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Thrombin signaling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.J.; Katz, V.; Salah, Z.; Maoz, M.; Cohen, I.; Uziely, B.; Turm, H.; Grisaru-Granovsky, S.; Suzuki, H.; Bar-Shavit, R. Mammary gland tissue targeted overexpression of human protease-activated receptor 1 reveals a novel link to beta-catenin stabilization. Cancer Res. 2006, 66, 5224–5233. [Google Scholar] [CrossRef] [PubMed]
- Turm, H.; Maoz, M.; Katz, V.; Yin, Y.J.; Offermanns, S.; Bar-Shavit, R. Protease-activated receptor-1 (PAR1) acts via a novel Galpha13-dishevelled axis to stabilize beta-catenin levels. J. Biol. Chem. 2010, 285, 15137–15148. [Google Scholar] [CrossRef] [PubMed]
- Nag, J.K.; Kancharla, A.; Maoz, M.; Turm, H.; Agranovich, D.; Gupta, C.L.; Uziely, B.; Bar-Shavit, R. Low-density lipoprotein receptor-related protein 6 is a novel coreceptor of protease-activated receptor-2 in the dynamics of cancer-associated β-catenin stabilization. Oncotarget 2017, 8, 38650–38667. [Google Scholar] [CrossRef] [PubMed]
- Grisaru-Granovsky, S.; Kumar Nag, J.; Zakar, L.; Rudina, T.; Lal Gupta, C.; Maoz, M.; Kozlova, D.; Bar-Shavit, R. PAR1&2 driven placenta EVT invasion act via LRP5/6 as coreceptors. FASEB J. 2020, 34, 15701–15717. [Google Scholar] [PubMed]
- Nag, J.K.; Appasamy, P.; Sedley, S.; Malka, H.; Rudina, T.; Bar-Shavit, R. RNF43 induces the turnover of protease-activated receptor 2 in colon cancer. FASEB J. 2023, 37, e22675. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Saha, S.K.; Kim, K.; Kim, S.; Yang, G.-M.; Kim, B.; Kim, J.-H.; Cho, S.-G. G protein-coupled receptors in stem cell maintenance and somatic reprogramming to pluripotent or cancer stem cells. BMB Rep. 2015, 48, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Li, S.D.; Li, Z.G.; Zhu, Y.C.; Yi, X.J.; Li, S.M. The expression of protease-activated receptors in esophageal carcinoma cells: The relationship between changes in gene expression and cell proliferation, apoptosis in vitro and growing ability in vivo. Cancer Cell Int. 2018, 18, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Jiang, P.; Xiang, Y.; Zhang, Y.; Zhu, Z.; Zhang, C.; Lee, S.; Lee, W.; Zhang, Y. Increased expression of protease-activated receptor 4 and Trefoil factor 2 in human colorectal cancer. PLoS ONE 2015, 10, e0122678. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Gallagher, S.; Warpeha, K.; Hawthorne, S.J. Amplification of MMP-2 and MMP-9 production by prostate cancer cell lines via activation of protease activated receptors. Prostate 2004, 60, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Mußbach, F.; Henklein, P.; Westermann, M.; Settmacher, U.; Böhmer, F.D.; Kaufmann, R. Proteinase-activated receptor 1- and 4-promoted migration of Hep3B hepatocellular carcinoma cells depends on ROS formation and RTK transactivation. J. Cancer Res. Clin. Oncol. 2015, 141, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Elste, A.P.; Petersen, I. Expression of proteinase-activated receptor 1-4 (PAR 1-4) in human cancer. J. Mol. Histol. 2010, 41, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Nag, J.K.; Malka, H.; Sedley, S.; Appasamy, P.; Rudina, T.; Levi, T.; Hoffman, A.; Gilon, C.; Uziely, B.; Bar-Shavit, R. PH-Binding Motif in PAR4 Oncogene: From Molecular Mechanism to Drug Design. Mol. Cancer Ther. 2022, 21, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Hurevich, M.; Swed, A.; Joubran, S.; Cohen, S.; Freeman, N.S.; Britan-Rosich, E.; Briant-Longuet, L.; Bardy, M.; Devaux, C.; Kotler, M.; et al. Rational conversion of noncontinuous active region in proteins into a small orally bioavailable macrocyclic drug-like molecule: The HIV-1 CD4:Gp120 paradigm. Bioorg. Med. Chem. 2010, 18, 5754–5761. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.; Linde, Y.; Ovadia, O.; Safrai, E.; Shalev, D.E.; Swed, A.; Halbfinger, E.; Lapidot, T.; Winkler, I.; Gabinet, Y.; et al. Backbone cyclic peptidomimetic melanocortin-4 receptor agonist as a novel orally administrated drug lead for treating obesity. J. Med. Chem. 2008, 51, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Talhami, A.; Swed, A.; Hess, S.; Ovadia, O.; Greenberg, S.; Schumacher-Klinger, A.; Rosenthal, D.; Shalev, D.E.; Hurevich, M.; Lazarovici, P.; et al. Cyclizing painkillers: Development of backbone-cyclic TAPS analogs. Front. Chem. 2020, 8, 532577–532596. [Google Scholar] [CrossRef] [PubMed]
- Kancharla, A.; Maoz, M.; Jaber, M.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. PH motifs in PAR1&2 endow breast cancer growth. Nat. Commun. 2015, 6, 8853–8863. [Google Scholar] [PubMed]
- Even-Ram, S.; Uziely, B.; Cohen, P.; Grisaru-Granovsky, S.; Maoz, M.; Ginzburg, Y.; Reich, R.; Vlodavsky, I.; Bar-Shavit, R. Thrombin receptor overexpression in malignant and physiological invasion processes. Nat. Med. 1998, 4, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.; Maoz, M.; Kancharla, A.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. Protease-activated-receptor-2 affects protease-activated-receptor-1-driven breast cancer. Cell. Mol. Life Sci. 2014, 71, 2517–2533. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, L.M.; Austin, K.M.; Zhang, P.; Kasuda, S.; Koukos, G.; Sharifi, S.; Covic, L.; Kuliopulos, A. Protease-activated receptor-2 modulates protease-activated receptor-1-driven neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2012, 31, e100–e106. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, S.; Ramakrishnan, V.; Sundelin, J. The proteinase-activated receptor 2 is induced by inflammatory mediators in human endothelial cells. Comparison with the thrombin receptor. J. Biol. Chem. 1996, 271, 14910–14915. [Google Scholar] [CrossRef] [PubMed]
- Blackhart, B.D.; Emilsson, K.; Nguyen, D.; Teng, W.; Martelli, A.J.; Nystedt, S.; Sundelin, J.; Scarborough, R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996, 271, 16466–16471. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Sakai, E.; Echizen, K.; Yamada, Y.; Oshima, H.; Han, T.-S.; Ohki, R.; Fujii, S.; Ochiai, A.; Robine, S.; et al. Intestinal cancer progression by mutant p53 through the acquisition of invasiveness associated with complex glandular formation. Oncogene 2017, 36, 5885–5896. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; Van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Hou, C.; Zhang, Z.; Ji, S.; Zhong, C.; Li, J.; Zhang, Q.; Huang, J.C.; Cheng, J.C. Effects of concurrent TP53 mutations on the efficacy and prognosis of targeted therapy for advanced EGFR mutant lung adenocarcinoma. Cancer Genet. 2023, 278, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Farengo-Clark, D.; McKenna, D.B.; Safonov, A.; Good, M.; Le, A.; Kessler, L.; Shah, P.D.; Bradbury, A.R.; Domchek, S.M.; et al. Clinical management of TP53 mosaic variants found on germline genetic testing. Cancer Genet. 2024, 284, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Sadot, E.; Geiger, B.; Oren, M.; Ben-Ze’ev, A. Down-regulation of beta-catenin by activated p53. Mol. Cell. Biol. 2001, 36, 6768–6781. [Google Scholar] [CrossRef] [PubMed]
- Sedley, S.; Nag, J.K.; Rudina, T.; Bar-Shavit, R. PAR-Induced Harnessing of EZH2 to β-Catenin: Implications for Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 8758. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.Q.; Wang, J.Y.; Xi, Y.; Wu, Z.L.; Li, Y.P.; Li, L. Nuclear Dvl, c-Jun, b-catenin, and TCF form a complex leading to stabilization of b -catenin—TCF interaction. J. Cell Biol. 2008, 180, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; An, S.; Wang, D.; Ji, H.; Guo, X.; Wang, Z. Activation of PAR4 Upregulates p16 through Inhibition of DNMT1 and HDAC2 Expression via MAPK Signals in Esophageal Squamous Cell Carcinoma. Cells J. Immunol. Res. 2018, 2018, 4735752–4735762. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Yu, G.Y.; Zhang, Y.; Xiang, Y.; Hua, H.R.; Bian, L.; Wang, C.Y.; Lee, W.H.; Zhang, Y. Down-regulation of protease-activated receptor 4 in lung adenocarcinoma is associated with a more aggressive phenotype. Asian Pac. J. Cancer Prev. 2013, 14, 3793–3798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, G.; Jiang, P.; Xiang, Y.; Li, W.; Lee, W.; Zhang, Y. Decreased expression of protease-activated receptor 4 in human gastric cancer. Int. J. Biochem. Cell Biol. 2011, 43, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jiang, P.; Wang, W.; Feng, W.; Yu, G. The decreased expression of protease-activated receptor 4 in esophageal squamous carcinoma. Neoplasma 2014, 61, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Hou, W.; Yang, L.; Liu, R.; Gao, Y.; Kong, X.; Shi, Z.; Li, W.; Zheng, H.; Jiang, S.; et al. Increased expression of protease-activated receptor 2 and 4 within dorsal root ganglia in a rat model of bone cancer pain. J. Mol. Neurosci. 2015, 55, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Amano, A.; Lamont, R.J.; Murakami, Y. Involvement of protease-activated receptor 4 in over-expression of matrix metalloproteinase 9 induced by Porphyromonas gingivalis. Med. Microbiol. Immunol. 2015, 204, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Black, P.C.; Mize, G.J.; Karlin, P.; Greenberg, D.L.; Hawley, S.J.; True, L.D.; Vessella, R.L.; Takayama, T.K. Overexpression of protease-activated receptors-1,-2, and-4 (PAR-1, -2, and -4) in prostate cancer. Prostate 2007, 67, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Gratio, V.; Walker, F.; Lehy, T.; Laburthe, M.; Darmoul, D. Aberrant expression of proteinase-activated receptor 4 promotes colon cancer cell proliferation through a persistent signaling that involves Src and ErbB-2 kinase. Int. J. Cancer 2009, 124, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Otani, H.; Yagi, Y.; Kawai, K.; Araki, H.; Fukuhara, S.; Inagaki, C. Proteinase-activated receptor 4 stimulation-induced epithelial-mesenchymal transition in alveolar epithelial cells. Respir. Res. 2007, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, R.; Rahn, S.; Pollrich, K.; Hertel, J.; Dittmar, Y.; Hommann, M.; Henklein, P.; Biskup, C.; Westermann, M.; Hollenberg, M.D.; et al. Thrombin-mediated hepatocellular carcinoma cell migration: Cooperative action via proteinase-activated receptors 1 and 4. J. Cell Physiol. 2007, 211, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Cheng, J.; Gao, Y.; Zhang, Y.; Chen, X.; Xie, J. Protease-activated receptor 4: A critical participator in inflammatory response. Inflammation 2015, 38, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, P.; Zhang, C.; Lee, S.; Wang, W.; Zou, H. PAR4 overexpression promotes colorectal cancer cell proliferation and migration. Oncol. Lett. 2018, 16, 5745–5752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schöttker, B.; Ordóñez-Mena, J.; Holleczek, B.; Yang, R.; Burwinkel, B.; Butterbach, K.; Brenner, H. F2RL3 methylation, lung cancer incidence and mortality. Int. J. Cancer 2015, 137, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Arachiche, A.; Mumaw, M.M.; de la Fuente, M.; Nieman, M.T. Protease-activated Receptor 1 (PAR1) and PAR4 Heterodimers Are Required for PAR1-enhanced Cleavage of PAR4 by α-Thrombin. J. Biol. Chem. 2013, 288, 32553–32562. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Jacques, S.L.; Badar, J.; Kaneider, N.C.; Derian, C.K.; Andrade-Gordon, P.; Covic, L.; Kuliopulos, A. Blocking the Protease-Activated Receptor 1-4 Heterodimer in Platelet-Mediated Thrombosis. Circulation 2006, 113, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Sveshnikova, A.N.; Balatskiy, A.V.; Demianova, A.S.; Shepelyuk, T.O.; Shakhidzhanov, S.S.; Balatskaya, M.N.; Pichugin, A.V.; Ataullakhanov, F.I.; Panteleev, M.A. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J. Thromb. Haemost. 2016, 14, 2045–2057. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Park, E.; Kan, K.; Landgraf, R. Functional isolation of activated and unilaterally phosphorylated heterodimers of ERBB2 and ERBB3 as scaffolds in ligand-dependent signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 13237–13242. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Brown, B.P.; Meiler, J.; Marín, A.; Jayanthan, H.S.; Ye, D.; Lin, C.C.; Akamatsu, H.; Lee, K.M.; Chatterjee, S.; et al. Co-occurring gain-of-function mutations in HER2 and HER3 modulate HER2/HER3 activation, oncogenesis, and HER2 inhibitor sensitivity. Cancer Cell 2021, 39, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 341–395. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, C.; Lv, B.; Jiang, Y.; Wang, L. Proteinase-activated receptor-2 enhances Bcl2-like protein-12 expression in lung cancer cells to suppress p53 expression. Arch. Med. Sci. 2019, 15, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Ishizawa, J.; Andreeff, M. Pharmacological activation of wild-type p53 in the therapy of leukemia. Exp. Hematol. 2016, 44, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer. 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wenng, J.; Huang, C.L.-H.; Jackson, A.P. Is the voltage-gated sodium channel β3 subunit (SCN3B) a biomarker for glioma? Funct. Integr. Genom. 2024, 24, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Oren, M. p53 and apoptosis. Semin. Cancer Biol. 1998, 8, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. 1999, 17, 2941–2953. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase 3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159–e202201172. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Ekedahl, J.; Lewensohn, R.; Marchetti, P.; Formstecher, P.; Zhivotovsky, B. Defective caspase-3 relocalization in non-small cell lung carcinoma. Oncogene 2001, 20, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.A.; Hopcroft, L.E.M.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.K.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Pribluda, A.; Goldstein, R.E.; Morgenstern, Y.; Brachya, G.; Cojocaru, G.; Snir-Alkalay, I.; Burstain, I.; Haffner-Kraus, R.; Jung, S.; et al. CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 2011, 470, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Kuo, C.J.; Ito, T.; Su, Y.Y.; Jiang, S.T.; Chiu, M.H.; Lin, Y.H.; Nist, A.; Mernberger, M.; Stiewe, T.; et al. CK1α ablation in keratinocytes induces p53-dependent, sunburn-protective skin hyperpigmentation. Proc. Natl. Acad. Sci. USA 2017, 114, E8035–E8044. [Google Scholar] [CrossRef] [PubMed]
- Minzel, W.; Venkatachalam, A.; Fink, A.; Hung, E.; Brachya, G.; Burstain, I.; Shaham, M.; Rivlin, A.; Omer, I.; Zinger, A.; et al. Small Molecules Co-targeting CKIα and the Transcriptional Kinases CDK7/9 Control AML in Preclinical Models. Cell 2018, 175, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.G.; Wu, X.; Guan, J.L. Wound-Healing Assay. Methods Mol. Biol. 2005, 294, 23–29. [Google Scholar] [PubMed]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Appasamy, P.; Nag, J.K.; Malka, H.; Bar-Shavit, R. PAR2 Serves an Indispensable Role in Controlling PAR4 Oncogenicity: The β-Catenin–p53 Axis. Int. J. Mol. Sci. 2025, 26, 2780. https://doi.org/10.3390/ijms26062780
Appasamy P, Nag JK, Malka H, Bar-Shavit R. PAR2 Serves an Indispensable Role in Controlling PAR4 Oncogenicity: The β-Catenin–p53 Axis. International Journal of Molecular Sciences. 2025; 26(6):2780. https://doi.org/10.3390/ijms26062780
Chicago/Turabian StyleAppasamy, Priyanga, Jeetendra Kumar Nag, Hodaya Malka, and Rachel Bar-Shavit. 2025. "PAR2 Serves an Indispensable Role in Controlling PAR4 Oncogenicity: The β-Catenin–p53 Axis" International Journal of Molecular Sciences 26, no. 6: 2780. https://doi.org/10.3390/ijms26062780
APA StyleAppasamy, P., Nag, J. K., Malka, H., & Bar-Shavit, R. (2025). PAR2 Serves an Indispensable Role in Controlling PAR4 Oncogenicity: The β-Catenin–p53 Axis. International Journal of Molecular Sciences, 26(6), 2780. https://doi.org/10.3390/ijms26062780