

Pulmonary Myeloid Cells in Mild Cases of COVID-19 Upregulate the Intracellular Fc Receptor TRIM21 and Transcribe Proteasome-Associated Molecules
 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
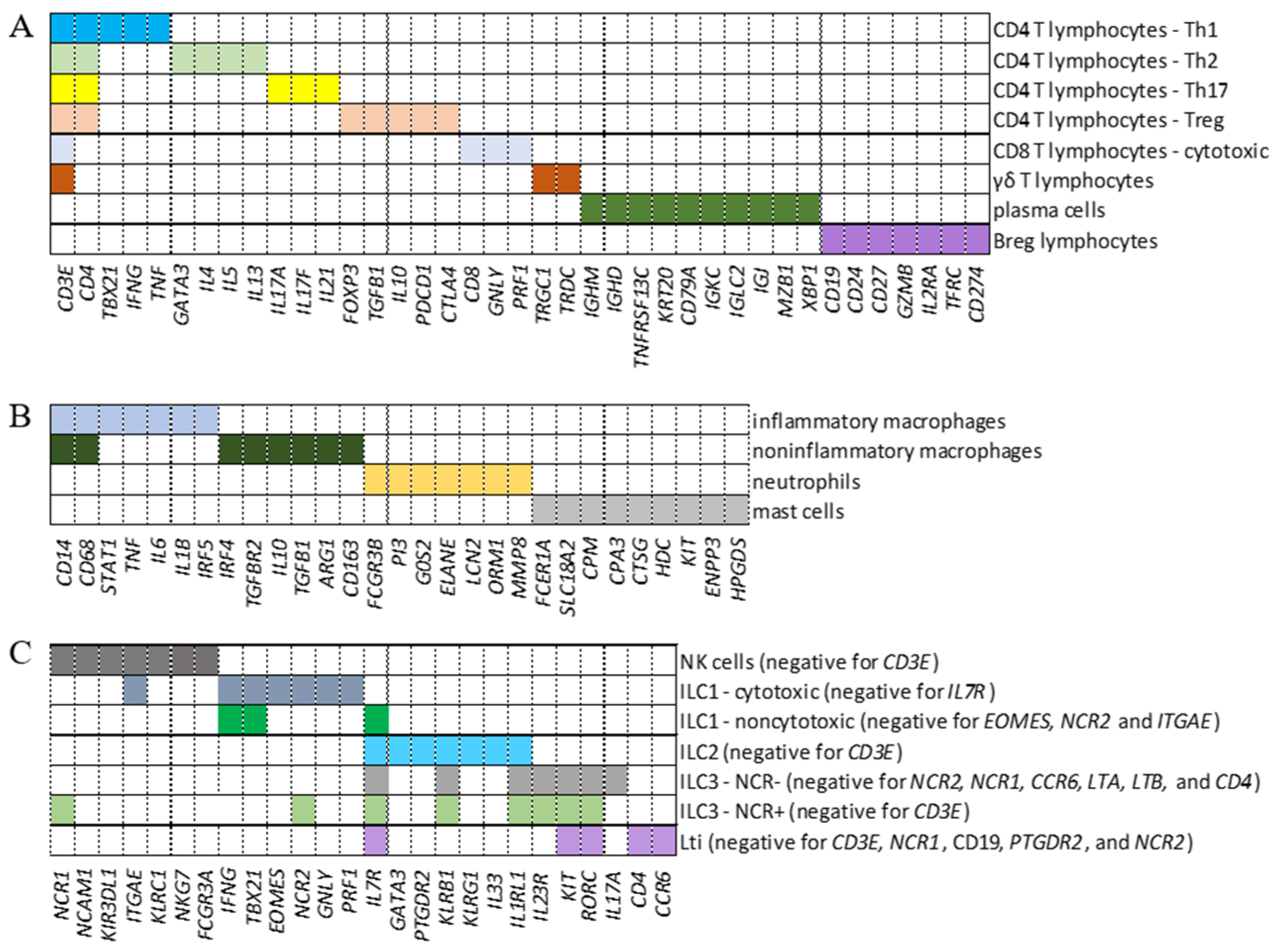
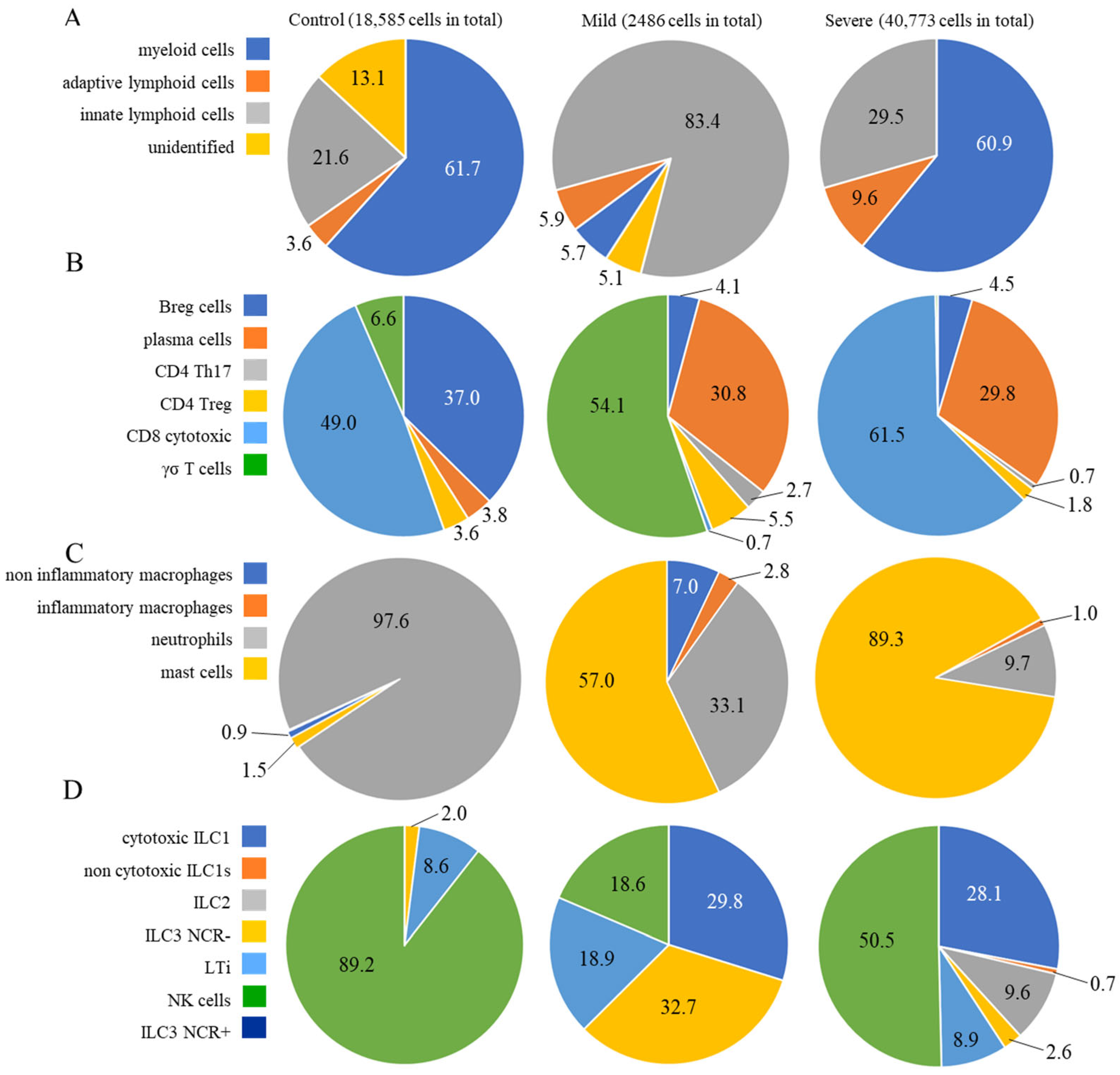
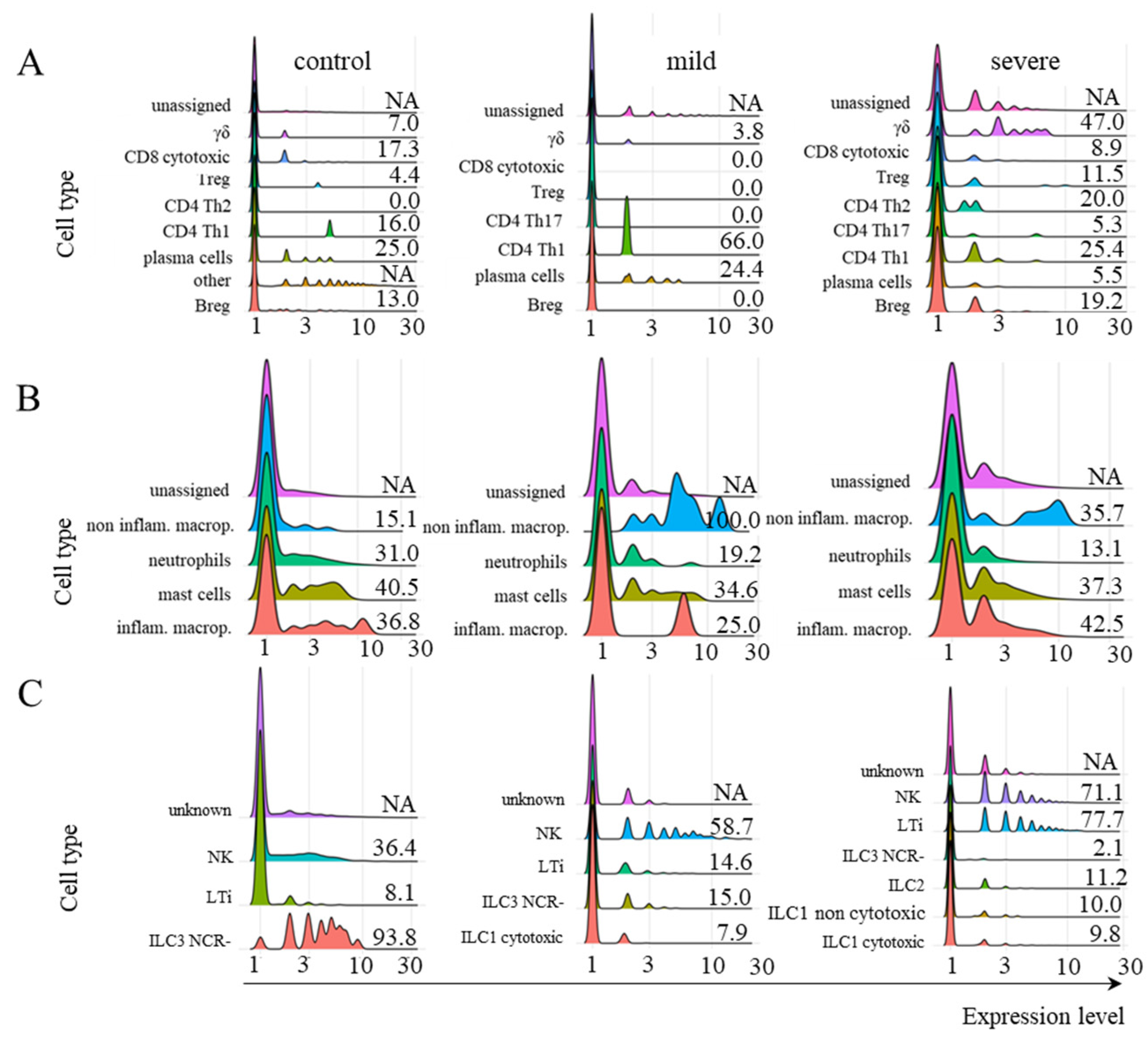
2.1. Identification of Cell Populations
2.2. FCGRT and TRIM21 Transcription
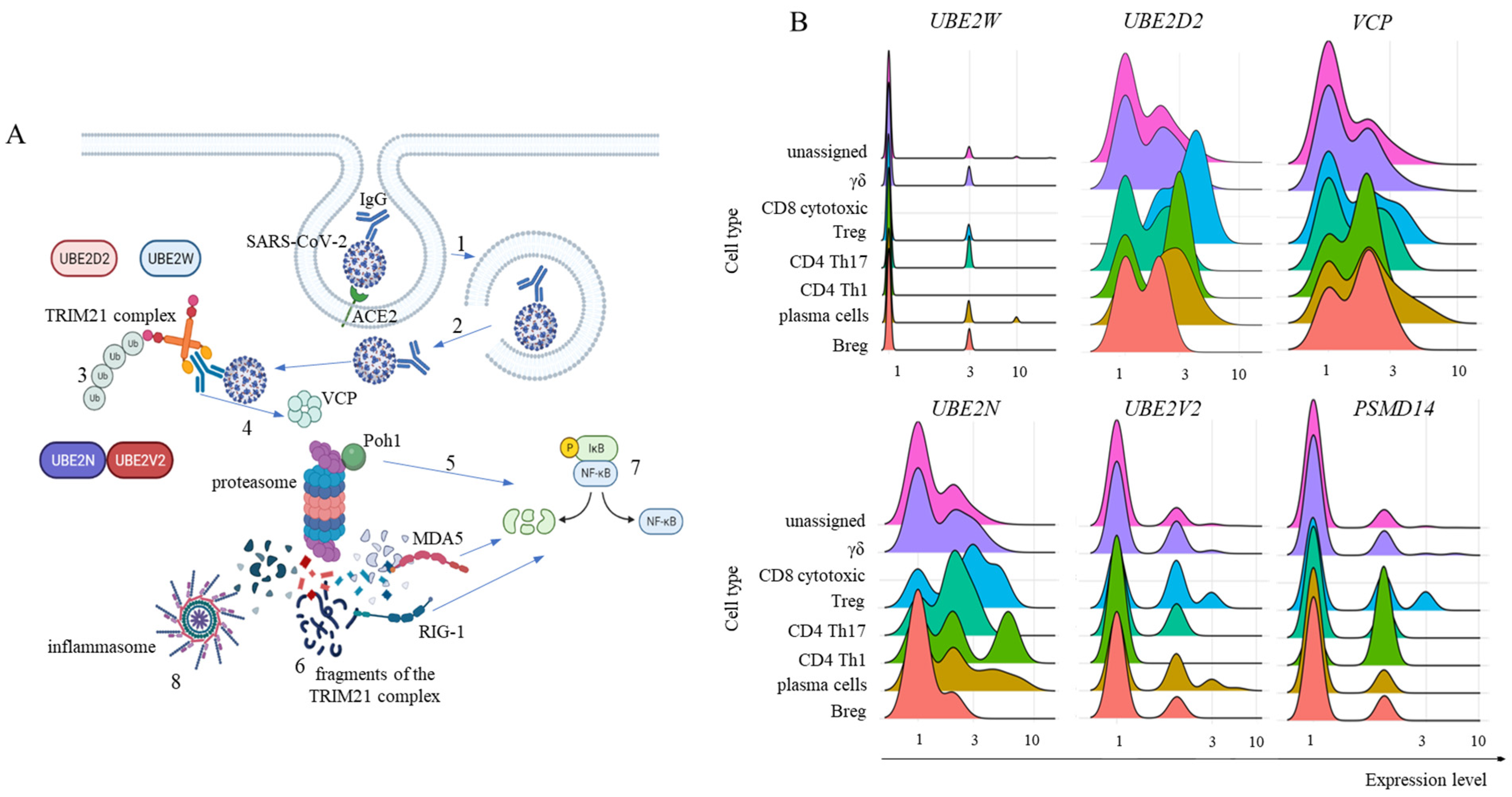
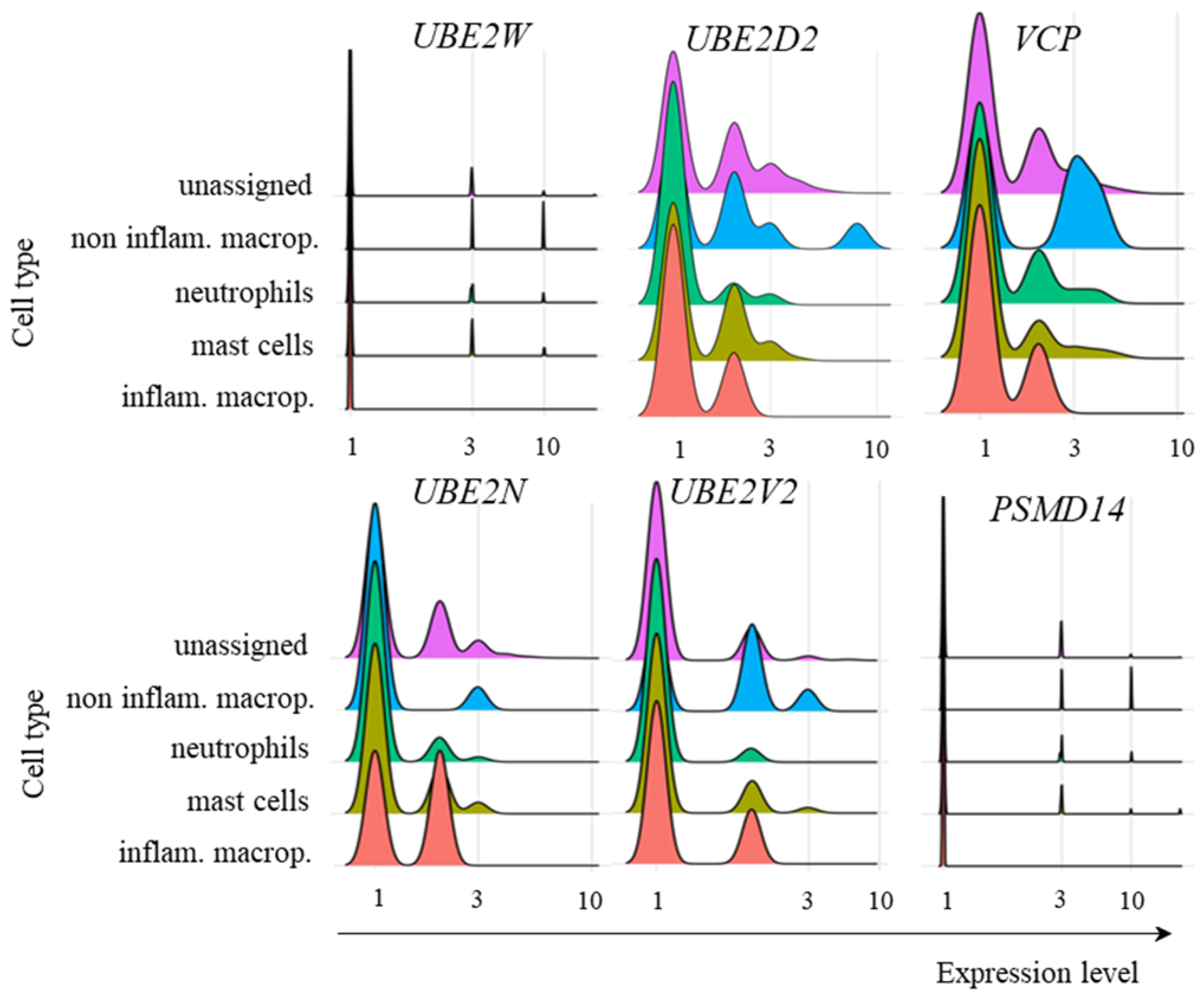
2.3. Transcription of TRIM21 Signaling Pathway Components
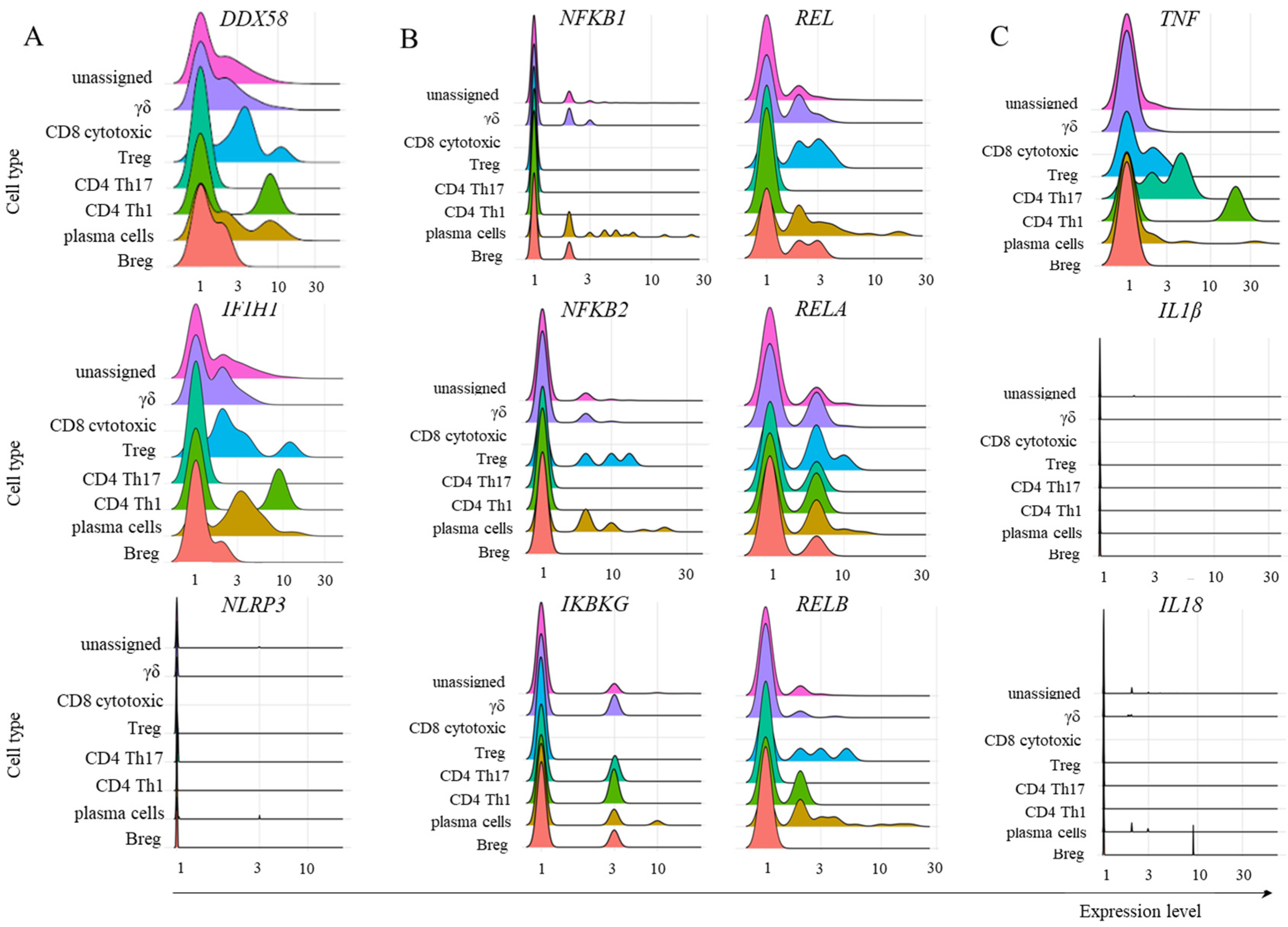
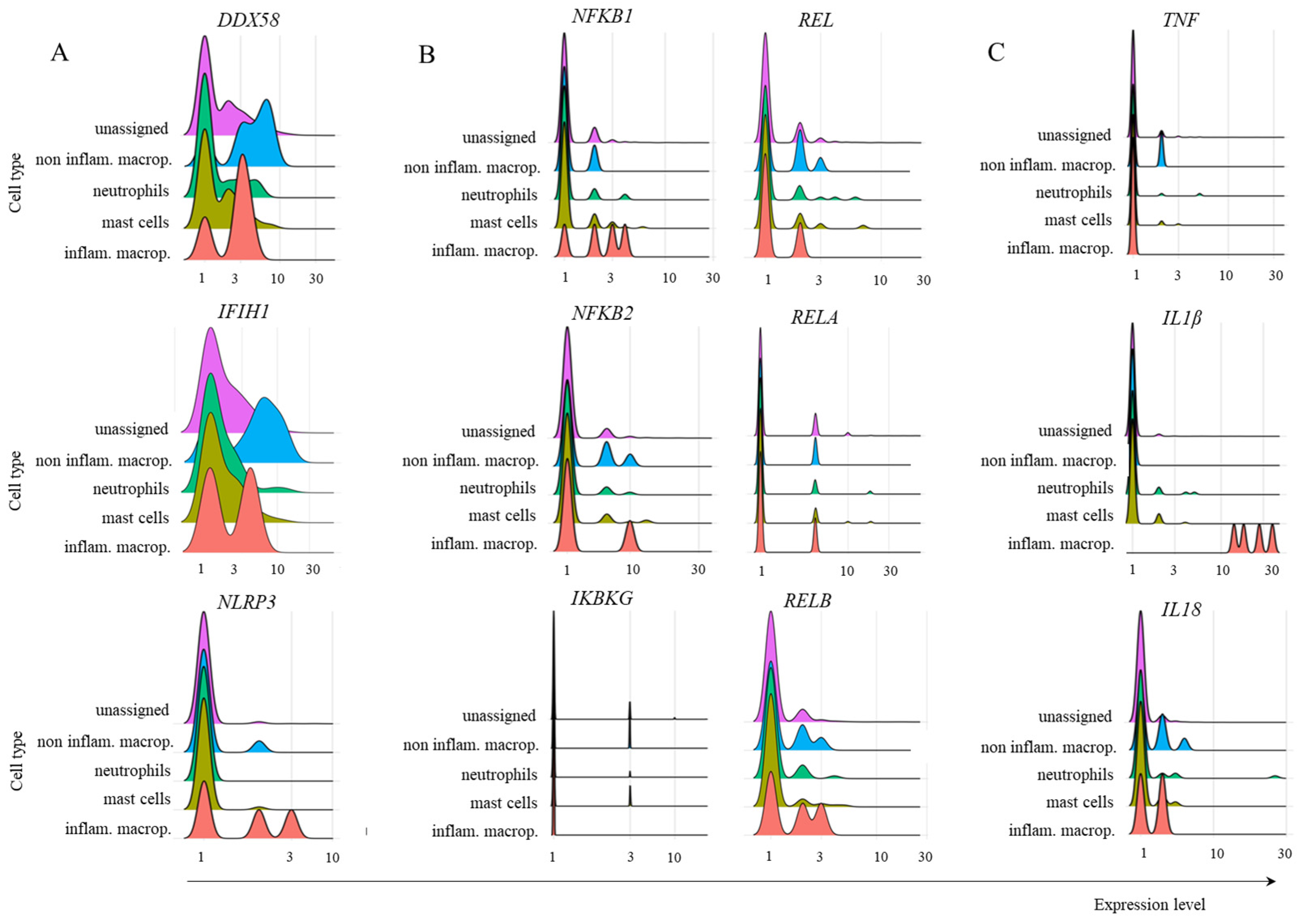
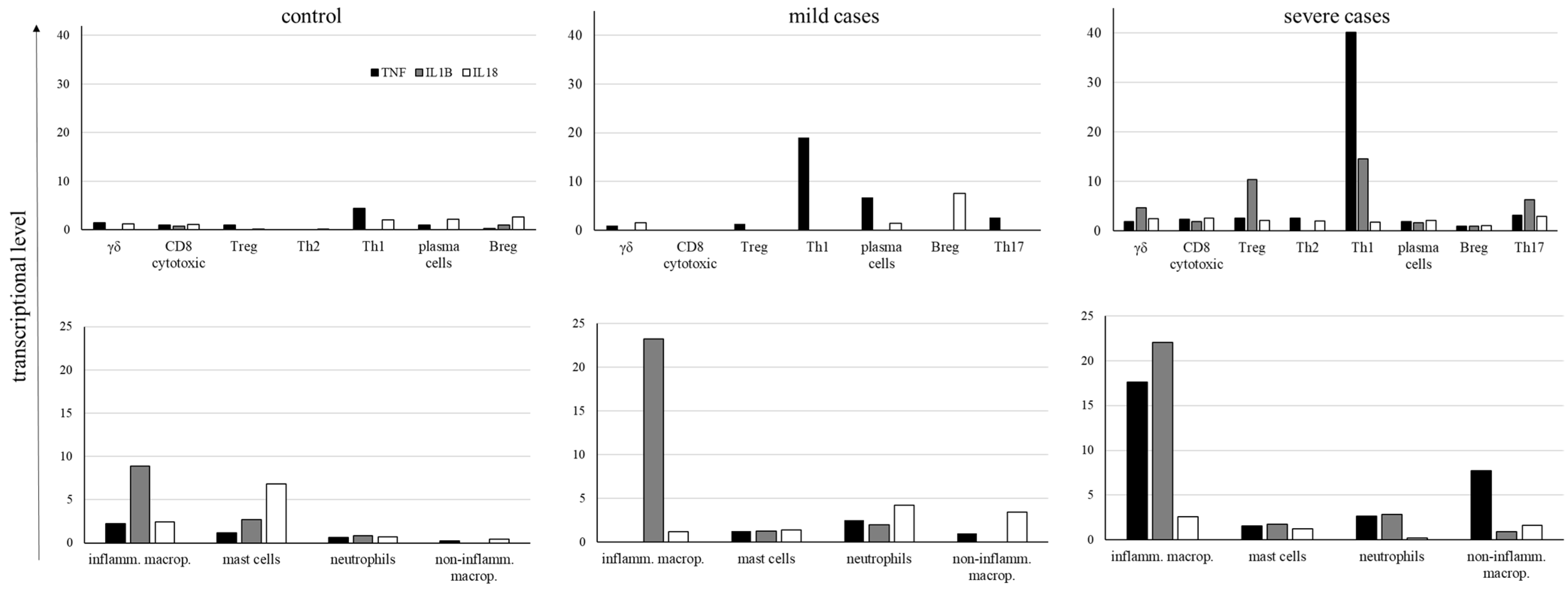
2.4. Inflammatory Cytokine Transcription
3. Discussion
4. Materials and Methods
- -
- The FindVariableGenes function detected the variable genes using the “vst” method and a number of features equal to 2000;
- -
- The datasets were integrated with Seurat’s FindIntegrationAnchors and IntegrataData functions by running a canonical correlation analysis (CCA) on each subset. We then performed dimensionality reduction using the PCA and UMAP algorithms;
- -
- In the final step of the clustering process, we calculated a shared nearest neighbor (SSN) graph between all cells through the FindClusters function, with a resolution parameter equal to 1.2. To identify markers for every cluster compared to all remaining cells, we used the FindAllMarkers function with default thresholds.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El Keshky, M.E.S.; Basyouni, S.S.; Al Sabban, A.M. Getting Through COVID-19: The Pandemic’s Impact on the Psychology of Sustainability, Quality of Life, and the Global Economy—A Systematic Review. Front. Psychol. 2020, 11, 585897. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Lu, L.; Ma, W. Immunopathological changes, complications, sequelae and immunological memory in COVID-19 patients. Heliyon 2022, 8, e09302. [Google Scholar] [CrossRef] [PubMed]
- Daeron, M. Fc receptors as adaptive immunoreceptors. Curr. Top Microbiol. Immunol. 2014, 382, 131–164. [Google Scholar]
- Lu, L.L.; Suscovich, T.J.; Fortune, S.M.; Alter, G. Beyond binding: Antibody effector functions in infectious diseases. Nat. Rev. Immunol. 2018, 18, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Daeron, M.; Jaeger, S.; Du Pasquier, L.; Vivier, E. Immunoreceptor tyrosine-based inhibition motifs: A quest in the past and future. Immunol. Rev. 2008, 224, 11–43. [Google Scholar] [CrossRef]
- Raghavan, M.; Bjorkman, P.J. Fc Receptors and their interactions with immunoglobulins. Annu. Rev. Cell Dev. Biol. 1996, 12, 181–220. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.; Pickkers, P.; Derde, L.; Leavis, H.; van Crevel, R.; Engel, J.J.; Wiersinga, W.J.; Vlaar, A.P.J.; Shankar-Hari, M.; et al. A guide to immunotherapy for COVID-19. Nat. Med. 2022, 28, 39–50. [Google Scholar] [CrossRef]
- Foss, S.; Bottermann, M.; Jonsson, A.; Sandlie, I.; James, L.C.; Andersen, J.T. TRIM21-From Intracellular Immunity to Therapy. Front. Immunol. 2019, 10, 2049. [Google Scholar] [CrossRef]
- Zeng, J.; Santos, A.F.; Mukadam, A.S.; Osswald, M.; Jacques, D.A.; Dickson, C.F.; McLaughlin, S.H.; Johnson, C.M.; Kiss, L.; Luptak, J.; et al. Target-induced clustering activates Trim-Away of pathogens and proteins. Nat. Struct. Mol. Biol. 2021, 28, 278–289. [Google Scholar] [CrossRef]
- McEwan, W.A.; Hauler, F.; Williams, C.R.; Bidgood, S.R.; Mallery, D.L.; Crowther, R.A.; James, L.C. Regulation of Virus Neutralization and the Persistent Fraction by TRIM21. J. Virol. 2012, 86, 8482–8491. [Google Scholar] [CrossRef] [PubMed]
- Bidgood, S.R.; Tam, J.C.H.; McEwan, W.A.; Mallery, D.L.; James, L.C. Translocalized IgA mediates neutralization and stimulates innate immunity inside infected cells. Proc. Natl. Acad. Sci. USA 2014, 111, 13463–13468. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Mallery, D.L.; Watkinson, R.E.; Dickson, C.F.; James, L.C. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc. Natl. Acad. Sci. USA 2015, 112, 10014–10019. [Google Scholar] [CrossRef]
- Mevissen, T.E.; Prasad, A.V.; Walter, J.C. TRIM21-dependent target protein ubiquitination mediates cell-free Trim-Away. Cell Rep. 2023, 42, 112125. [Google Scholar] [CrossRef]
- Rhodes, D.A.; Isenberg, D.A. TRIM21 and the Function of Antibodies inside Cells. Trends Immunol. 2017, 38, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Hauler, F.; Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; James, L.C. AAA ATPase p97/VCP is essential for TRIM21-mediated virus neutralization. Proc. Natl. Acad. Sci. USA 2012, 109, 19733–19738. [Google Scholar] [CrossRef]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef]
- Full, F.; Gack, M.U. Delicate coordination of TRIM21’s dual activity in virus neutralization and signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 9797–9798. [Google Scholar] [CrossRef]
- Mao, S.; Cai, X.; Niu, S.; Wei, J.; Jiang, N.; Deng, H.; Wang, W.; Zhang, J.; Shen, S.; Ma, Y.; et al. TRIM21 promotes ubiquitination of SARS-CoV-2 nucleocapsid protein to regulate innate immunity. J. Med. Virol. 2023, 95, e28719. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Wauters, E.; Van Mol, P.; Garg, A.D.; Jansen, S.; Van Herck, Y.; Vanderbeke, L.; Bassez, A.; Boeckx, B.; Malengier-Devlies, B.; Timmerman, A.; et al. Discriminating Mild from Critical COVID-19 by Innate and Adaptive Immune Single-cell Profiling of Bronchoalveolar Lavages. Cell Res. 2021, 31, 272–290. [Google Scholar] [CrossRef] [PubMed]
- Ben Mkaddem, S.; Benhamou, M.; Monteiro, R.C. Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front. Immunol. 2019, 10, 811. [Google Scholar] [CrossRef]
- Borger, J.G.; Lau, M.; Hibbs, M.L. The Influence of Innate Lymphoid Cells and Unconventional T Cells in Chronic Inflammatory Lung Disease. Front. Immunol. 2019, 10, 1597. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Cao, W.; Endrias, K.; Kuchipudi, S.V.; Mittal, S.K.; Sambhara, S. Innate lymphoid cells (ILC) in SARS-CoV-2 infection. Mol. Asp. Med. 2021, 80, 101008. [Google Scholar] [CrossRef]
- Gomes, A.M.C.; Farias, G.B.; Dias-Silva, M.; Laia, J.; Trombetta, A.C.; Godinho-Santos, A.; Rosmaninho, P.; Santos, D.F.; Conceicao, C.M.; Costa-Reis, R.; et al. SARS-CoV-2 pneumonia recovery is linked to expansion of innate lymphoid cells type 2 expressing CCR10. Eur. J. Immunol. 2021, 51, 3194–3201. [Google Scholar] [CrossRef]
- Silverstein, N.J.; Wang, Y.; Manickas-Hill, Z.; Carbone, C.; Dauphin, A.; Boribong, B.P. Innate lymphoid cells and COVID-19 severity in SARS-CoV-2 infection. Elife 2022, 11, e74681. [Google Scholar] [CrossRef] [PubMed]
- García, M.; Kokkinou, E.; García, A.C.; Parrot, T.; Medina, L.M.P.; Maleki, K.T.; Christ, W.; Varnaitė, R.; Filipovic, I.; Ljunggren, H.; et al. Innate lymphoid cell composition associates with COVID-19 disease severity. Clin. Transl. Immunol. 2020, 9, e1224. [Google Scholar] [CrossRef]
- Nagasawa, M.; Spits, H.; Ros, X.R. Innate Lymphoid Cells (ILCs): Cytokine Hubs Regulating Immunity and Tissue Homeostasis. Cold Spring Harb. Perspect. Biol. 2018, 10, a030304. [Google Scholar] [CrossRef]
- Walker, J.; Barlow, J.; McKenzie, A. Innate lymphoid cells—How did we miss them? Nat. Rev. Immunol. 2013, 13, 75–87. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Jung, M.K.; Lee, J.; Choi, S.J.; Choi, S.H.; Lee, H.W.; Lee, J.-J.; Kim, H.J.; Ahn, S.H.; Lee, D.H.; et al. Tumor Necrosis Factor-producing T-regulatory Cells Are Associated with Severe Liver Injury in Patients with Acute Hepatitis A. Gastroenterology 2018, 154, 1047–1060. [Google Scholar] [CrossRef]
- Weiner, L.M.; Murray, J.C.; Shuptrine, C.W. Antibody-Based Immunotherapy of Cancer. Cell 2012, 148, 1081–1084. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, X.; Yang, Y.; Hotez, P.J.; Du, L. Neutralizing antibodies for the treatment of COVID-19. Nat. Biomed. Eng. 2020, 4, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Landeck, L.; Sabat, R.; Ghoreschi, K.; Man, X.Y.; Fuhrmeister, K.; Gonzalez-Martinez, E.; Asadullah, K. Immunotherapy in psoriasis. Immunotherapy 2021, 13, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Ballow, M.; Haga, C.L. Why Do Some People Develop Serious COVID-19 Disease After Infection, While Others Only Exhibit Mild Symptoms? J. Allergy Clin. Immunol. Pract. 2021, 9, 1442–1448. [Google Scholar] [CrossRef]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef]
- Liu, L.; Wei, Q.; Lin, Q.; Fang, J.; Wang, H.; Kwok, H.; Tang, H.; Nishiura, K.; Peng, J.; Tan, Z.; et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4, e123158. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, F.; Yu, W.; He, T.; Yu, J.; Yi, C.E.; Ba, L.; Li, W.; Farzan, M.; Chen, Z.; et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J. Med. Virol. 2006, 78, 1–8. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Helmy, M.; Selvarajoo, K. Application of GeneCloudOmics: Transcriptomic Data Analytics for Synthetic Biology. In Computational Biology and Machine Learning for Metabolic Engineering and Synthetic Biology; Springer: New York, NY, USA, 2022; pp. 221–263. [Google Scholar]
- Higgins, J.J. An Introduction to Modern Nonparametric Statistics; Brooks/Cole: Pacific Grove, CA, USA, 2004; p. 366. [Google Scholar]
- Kothalawala, W.J.; Győrffy, B. Transcriptomic and Cellular Content Analysis of Colorectal Cancer by Combining Multiple Independent Cohorts. Clin. Transl. Gastroenterol. 2023, 14, e00517. [Google Scholar] [CrossRef] [PubMed]
- Yimthin, T.; Cliff, J.M.; Phunpang, R.; Ekchariyawat, P.; Kaewarpai, T.; Lee, J.-S.; Eckold, C.; Andrada, M.; Thiansukhon, E.; Tanwisaid, K.; et al. Blood transcriptomics to characterize key biological pathways and identify biomarkers for predicting mortality in melioidosis. Emerg. Microbes Infect. 2021, 10, 8–18. [Google Scholar] [CrossRef]
- Conforte, A.J.; Alves, L.; Coelho, F.C.; Carels, N.; Da Silva, F.A.B. Modeling Basins of Attraction for Breast Cancer Using Hopfield Networks. Front. Genet. 2020, 11, 314. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-Q.; Ma, W.-W.; Zeng, S.-X.; Zhang, C.-L.; Yan, J.; Sun, C.-C.; Li, X.; Wang, R.-M.; Li, Z.-Q. Transcriptome analysis of differential gene expression in the longissimus dorsi muscle from Debao and landrace pigs based on RNA-sequencing. Biosci. Rep. 2019, 39, BSR20192144. [Google Scholar] [CrossRef]
- Chowdhury, H.A.; Bhattacharyya, D.K.; Kalita, J.K. Differential expression analysis of RNA-seq reads: Overview, taxonomy, and tools. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 17, 566–586. [Google Scholar] [CrossRef]
- García-Ortega, L.F.; Martínez, O. How Many Genes Are Expressed in a Transcriptome? Estimation and Results for RNA-Seq. PLoS ONE 2015, 10, e0130262. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Weiss, Y.; Navon, Y.; Milo, I.; Azulay, N.; Keren, L.; Fuchs, S.; Ben-Zvi, D.; Noor, E.; Milo, R. The total mass, number, and distribution of immune cells in the human body. Proc. Natl. Acad. Sci. USA 2023, 120, e2308511120. [Google Scholar] [CrossRef]
- Li, C.K.-F.; Wu, H.; Yan, H.; Ma, S.; Wang, L.; Zhang, M.; Tang, X.; Temperton, N.J.; Weiss, R.A.; Brenchley, J.M.; et al. T Cell Responses to Whole SARS Coronavirus in Humans. J. Immunol. 2008, 181, 5490–5500. [Google Scholar] [CrossRef]
- Hotez, P.J.; Bottazzi, M.E.; Corry, D.B. The potential role of Th17 immune responses in coronavirus immunopathology and vaccine-induced immune enhancement. Microbes Infect. 2020, 22, 165–167. [Google Scholar] [CrossRef]
- Hsieh, W.C.; Lai, E.Y.; Liu, Y.T.; Wang, Y.F.; Tzeng, Y.S.; Cui, L.; Lai, Y.J.; Huang, H.C.; Huang, J.H.; Ni, H.C.; et al. NK cell receptor and ligand composition influences the clearance of SARS-CoV-2. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Qin, G.; Liu, S.; Yang, L.; Yu, W.; Zhang, Y. Myeloid cells in COVID-19 microenvironment. Signal Transduct. Target. Ther. 2021, 6, 372. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19-Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef]
- Montazersaheb, S.; Khatibi, S.M.H.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Sorbeni, F.G.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Scaramuzzo, G.; Nucera, F.; Asmundo, A.; Messina, R.; Mari, M.; Montanaro, F.; Johansen, M.D.; Monaco, F.; Fadda, G.; Tuccari, G.; et al. Cellular and molecular features of COVID-19 associated ARDS: Therapeutic relevance. J. Inflamm. 2023, 20, 11. [Google Scholar] [CrossRef]
- Shakiba, M.H.; Gemünd, I.; Beyer, M.; Bonaguro, L. Lung T cell response in COVID-19. Front. Immunol. 2023, 14, 1108716. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.; Hussell, T.; Shuwa, H.A. Regulatory B cells in respiratory health and diseases. Immunol. Rev. 2021, 299, 61–73. [Google Scholar] [CrossRef]
- Dhawan, M.; Rabaan, A.A.; Alwarthan, S.; Alhajri, M.; Halwani, M.A.; Alshengeti, A.; Najim, M.A.; Alwashmi, A.S.S.; Alshehri, A.A.; Alshamrani, S.A.; et al. Regulatory T Cells (Tregs) and COVID-19: Unveiling the Mechanisms, and Therapeutic Potentialities with a Special Focus on Long COVID. Vaccines 2023, 11, 699. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Silva, V.S.; Costa, M.O.C.; Castro, M.C.S.; Silva, H.S.; Walter, M.E.M.T.; Melo, A.C.M.A.; Ocaña, K.A.C.; dos Santos, M.T.; Nicolas, M.F.; Carvalho, A.C.C.; et al. CellHeap: A Workflow for Optimizing COVID-19 Single-Cell RNA-Seq Data Processing in the Santos Dumont Supercomputer. Adv. Bioinform. Comput. Biol. 2021, 13063, 41–52. [Google Scholar]
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol. Cell 2019, 73, 130–142.e5. [Google Scholar] [CrossRef]
- Young, M.D.; Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience 2020, 9, giaa151. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.W.; O’flanagan, C.; Chavez, E.A.; Lim, J.L.P.; Ceglia, N.; McPherson, A.; Wiens, M.; Walters, P.; Chan, T.; Hewitson, B.; et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods 2019, 16, 1007–1015. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 2022, 13, 1246. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henriques-Pons, A.; Castro, M.C.S.; Silva, V.S.; Costa, M.O.C.; Silva, H.S.I.L.; Walter, M.E.M.T.; Carvalho, A.C.C.; Melo, A.C.M.A.; Ocaña, K.; Santos, M.T.d.; et al. Pulmonary Myeloid Cells in Mild Cases of COVID-19 Upregulate the Intracellular Fc Receptor TRIM21 and Transcribe Proteasome-Associated Molecules. Int. J. Mol. Sci. 2025, 26, 2769. https://doi.org/10.3390/ijms26062769
Henriques-Pons A, Castro MCS, Silva VS, Costa MOC, Silva HSIL, Walter MEMT, Carvalho ACC, Melo ACMA, Ocaña K, Santos MTd, et al. Pulmonary Myeloid Cells in Mild Cases of COVID-19 Upregulate the Intracellular Fc Receptor TRIM21 and Transcribe Proteasome-Associated Molecules. International Journal of Molecular Sciences. 2025; 26(6):2769. https://doi.org/10.3390/ijms26062769
Chicago/Turabian StyleHenriques-Pons, Andrea, Maria Clicia S. Castro, Vanessa S. Silva, Maiana O. C. Costa, Helena S. I. L. Silva, Maria Emilia M. T. Walter, Anna Cristina C. Carvalho, Alba C. M. A. Melo, Kary Ocaña, Marcelo T. dos Santos, and et al. 2025. "Pulmonary Myeloid Cells in Mild Cases of COVID-19 Upregulate the Intracellular Fc Receptor TRIM21 and Transcribe Proteasome-Associated Molecules" International Journal of Molecular Sciences 26, no. 6: 2769. https://doi.org/10.3390/ijms26062769
APA StyleHenriques-Pons, A., Castro, M. C. S., Silva, V. S., Costa, M. O. C., Silva, H. S. I. L., Walter, M. E. M. T., Carvalho, A. C. C., Melo, A. C. M. A., Ocaña, K., Santos, M. T. d., Nicolas, M. F., & Silva, F. A. B. (2025). Pulmonary Myeloid Cells in Mild Cases of COVID-19 Upregulate the Intracellular Fc Receptor TRIM21 and Transcribe Proteasome-Associated Molecules. International Journal of Molecular Sciences, 26(6), 2769. https://doi.org/10.3390/ijms26062769