
Understanding Neovascularization in Glioblastoma: Insights from the Current Literature
 , ,
, ,  , , , , , , ,
, , , , , , ,  , , ,
, , , 

Abstract
1. Introduction
2. GBM Vascularization: Morphological and Functional Aspects
3. Different Strategies of Neovascularization in GBM
3.1. Sprouting Angiogenesis
3.1.1. Hypoxia and Hypoxia-Inducible Factors
3.1.2. Pro-Angiogenic Factors
3.1.3. Proteinases
3.2. Vasculogenesis
3.3. Vessel Co-Option
3.4. Vascular Intussusception
3.5. Vasculogenic Mimicry
3.6. Trans-Differentiation of Cancer Stem-Like Cells
4. Treating GBM: Between Pitfalls and Emerging Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ah-Pine, F.; Khettab, M.; Bedoui, Y.; Slama, Y.; Daniel, M.; Doray, B.; Gasque, P. On the Origin and Development of Glioblastoma: Multifaceted Role of Perivascular Mesenchymal Stromal Cells. Acta Neuropathol. Commun. 2023, 11, 104. [Google Scholar] [CrossRef]
- Loras, A.; Gonzalez-Bonet, L.G.; Gutierrez-Arroyo, J.L.; Martinez-Cadenas, C.; Marques-Torrejon, M.A. Neural Stem Cells as Potential Glioblastoma Cells of Origin. Life 2023, 13, 905. [Google Scholar] [CrossRef] [PubMed]
- Youssef, G.; Miller, J.J. Lower Grade Gliomas. Curr. Neurol. Neurosci. Rep. 2020, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of Origin for Malignant Gliomas and Its Implication in Therapeutic Development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef]
- Weller, M.; Wen, P.Y.; Chang, S.M.; Dirven, L.; Lim, M.; Monje, M.; Reifenberger, G. Glioma. Nat. Rev. Dis. Primers 2024, 10, 33. [Google Scholar] [CrossRef]
- Garcia, M.R.; Feng, Y.; Vasudevaraja, V.; Galbraith, K.; Serrano, J.; Thomas, C.; Radmanesh, A.; Hidalgo, E.T.; Harter, D.H.; Allen, J.C.; et al. Clinical, Pathological, and Molecular Characteristics of Diffuse Spinal Cord Gliomas. J. Neuropathol. Exp. Neurol. 2022, 81, 865–872. [Google Scholar] [CrossRef]
- Aggarwal, P.; Luo, W.; Pehlivan, K.C.; Hoang, H.; Rajappa, P.; Cripe, T.P.; Cassady, K.A.; Lee, D.A.; Cairo, M.S. Pediatric versus Adult High Grade Glioma: Immunotherapeutic and Genomic Considerations. Front. Immunol. 2022, 13, 1038096. [Google Scholar] [CrossRef]
- Wang, G.-M.; Cioffi, G.; Patil, N.; Waite, K.A.; Lanese, R.; Ostrom, Q.T.; Kruchko, C.; Berens, M.E.; Connor, J.R.; Lathia, J.D.; et al. Importance of the Intersection of Age and Sex to Understand Variation in Incidence and Survival for Primary Malignant Gliomas. Neuro Oncol. 2022, 24, 302–310. [Google Scholar] [CrossRef]
- Mo, Z.; Xin, J.; Chai, R.; Woo, P.Y.; Chan, D.T.; Wang, J. Epidemiological Characteristics and Genetic Alterations in Adult Diffuse Glioma in East Asian Populations. Cancer Biol. Med. 2022, 19, 1440. [Google Scholar] [CrossRef]
- Persaud-Sharma, D.; Burns, J.; Trangle, J.; Castro, G.; Barengo, N.; Moulik, S.; Manuel Lozano, J. Demographic Variation in the Frequency of Gliomas in Florida. Medicina 2019, 55, 5. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Egan, K.M.; Nabors, L.B.; Gerke, T.; Thompson, R.C.; Olson, J.J.; LaRocca, R.; Chowdhary, S.; Eckel-Passow, J.E.; Armstrong, G.; et al. Glioma risk associated with extent of estimated European genetic ancestry in African-Americans and Hispanics. Int. J. Cancer 2019, 146, 739. [Google Scholar] [CrossRef] [PubMed]
- Frosina, G.; Casella, C.; Puppo, A.; Marani, E.; Campanella, D.; Boni, L.; Fontana, V. Epidemiology of Malignant Brain Tumors in Genova, Italy. 1993–2017. Sci. Rep. 2024, 14, 27300. [Google Scholar] [CrossRef]
- Mesti, T.; Ocvirk, J. Malignant Gliomas: Old and New Systemic Treatment Approaches. Radiol. Oncol. 2016, 50, 129. [Google Scholar] [CrossRef] [PubMed]
- Daswani, B.; Khan, Y. Insights into the Role of Estrogens and Androgens in Glial Tumorigenesis. J. Carcinog. 2021, 20, 10. [Google Scholar] [CrossRef]
- Rhun, E.L.; Weller, M. Sex-Specific Aspects of Epidemiology, Molecular Genetics and Outcome: Primary Brain Tumours. ESMO Open 2020, 5, e001034. [Google Scholar] [CrossRef]
- Stabellini, N.; Krebs, H.; Patil, N.; Waite, K.; Barnholtz-Sloan, J.S. Sex Differences in Time to Treat and Outcomes for Gliomas. Front. Oncol. 2021, 11, 630597. [Google Scholar] [CrossRef]
- Davis, M.E. Epidemiology and Overview of Gliomas. Semin. Oncol. Nurs. 2018, 34, 420–429. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Kinnersley, B.; Armstrong, G.; Rice, T.; Chen, Y.; Wiencke, J.K.; McCoy, L.S.; Hansen, H.M.; Amos, C.I.; Bernstein, J.L.; et al. Age-Specific Genome-Wide Association Study in Glioblastoma Identifies Increased Proportion of ‘Lower Grade Glioma’-like Features Associated with Younger Age. Int. J. Cancer 2018, 143, 2359–2366. [Google Scholar] [CrossRef]
- Krigers, A.; Demetz, M.; Thomé, C.; Freyschlag, C.F. Age Is Associated with Unfavorable Neuropathological and Radiological Features and Poor Outcome in Patients with WHO Grade 2 and 3 Gliomas. Sci. Rep. 2021, 11, 17380. [Google Scholar] [CrossRef]
- Choi, D.-J.; Armstrong, G.; Lozzi, B.; Vijayaraghavan, P.; Plon, S.E.; Wong, T.C.; Boerwinkle, E.; Muzny, D.M.; Chen, H.-C.; Gibbs, R.A.; et al. The Genomic Landscape of Familial Glioma. Sci. Adv. 2023, 9, eade2675. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Adel Fahmideh, M.; Cote, D.J.; Muskens, I.S.; Schraw, J.M.; Scheurer, M.E.; Bondy, M.L. Risk Factors for Childhood and Adult Primary Brain Tumors. Neuro-Oncology 2019, 21, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Kinnersley, B.; Houlston, R.S.; Bondy, M.L. Genome-Wide Association Studies in Glioma. Cancer Epidemiol. Biomark. Prev. 2018, 27, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Kinnersley, B.; Labussière, M.; Holroyd, A.; Di Stefano, A.-L.; Broderick, P.; Vijayakrishnan, J.; Mokhtari, K.; Delattre, J.-Y.; Gousias, K.; Schramm, J.; et al. Genome-Wide Association Study Identifies Multiple Susceptibility Loci for Glioma. Nat. Commun. 2015, 6, 8559. [Google Scholar] [CrossRef]
- Segura, P.P.; Quintela, N.V.; García, M.M.; Berrón, S.d.B.; Sarrió, R.G.; Gómez, J.G.; Castaño, A.G.; Martín, L.M.N.; Rubio, O.G.; Losada, E.P. SEOM-GEINO Clinical Guidelines for High-Grade Gliomas of Adulthood (2022). Clin. Transl. Oncol. 2023, 25, 2634. [Google Scholar] [CrossRef]
- Giannopoulou, A.-I.; Kanakoglou, D.S.; Piperi, C. Transcription Factors with Targeting Potential in Gliomas. Int. J. Mol. Sci. 2022, 23, 3720. [Google Scholar] [CrossRef]
- Toader, C.; Eva, L.; Costea, D.; Corlatescu, A.D.; Covache-Busuioc, R.-A.; Bratu, B.-G.; Glavan, L.A.; Costin, H.P.; Popa, A.A.; Ciurea, A.V. Low-Grade Gliomas: Histological Subtypes, Molecular Mechanisms, and Treatment Strategies. Brain Sci. 2023, 13, 1700. [Google Scholar] [CrossRef]
- Byun, Y.H.; Park, C.-K. Classification and Diagnosis of Adult Glioma: A Scoping Review. Brain NeuroRehabilitation 2022, 15, e23. [Google Scholar] [CrossRef]
- Nafe, R.; Porto, L.; Samp, P.-F.; You, S.-J.; Hattingen, E. Adult-Type and Pediatric-Type Diffuse Gliomas: What the Neuroradiologist Should Know. Clin. Neuroradiol. 2023, 33, 611–624. [Google Scholar] [CrossRef]
- Komori, T. Grading of Adult Diffuse Gliomas According to the 2021 WHO Classification of Tumors of the Central Nervous System. Lab. Investig. 2022, 102, 126–133. [Google Scholar] [CrossRef]
- Park, Y.W.; Vollmuth, P.; Foltyn-Dumitru, M.; Sahm, F.; Ahn, S.S.; Chang, J.H.; Kim, S.H. The 2021 WHO Classification for Gliomas and Implications on Imaging Diagnosis: Part 1—Key Points of the Fifth Edition and Summary of Imaging Findings on Adult-Type Diffuse Gliomas. J. Magn. Reson. Imaging 2023, 58, 677–689. [Google Scholar] [CrossRef]
- Fekete, B.; Werlenius, K.; Tisell, M.; Pivodic, A.; Smits, A.; Jakola, A.S.; Rydenhag, B. What Predicts Survival in Glioblastoma? A Population-Based Study of Changes in Clinical Management and Outcome. Front. Surg. 2023, 10, 1249366. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Song, K.; Wu, S.; Hameed, N.U.F.; Kudulaiti, N.; Xu, H.; Qin, Z.-Y.; Wu, J.-S. The Prognosis of Glioblastoma: A Large, Multifactorial Study. Br. J. Neurosurg. 2021, 35, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Marenco-Hillembrand, L.; Wijesekera, O.; Suarez-Meade, P.; Mampre, D.; Jackson, C.; Peterson, J.; Trifiletti, D.; Hammack, J.; Ortiz, K.; Lesser, E.; et al. Trends in Glioblastoma: Outcomes over Time and Type of Intervention: A Systematic Evidence Based Analysis. J. Neurooncol. 2020, 147, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, A.; Mohammed, R.; Zakieh, O. The Unique Invasiveness of Glioblastoma and Possible Drug Targets on Extracellular Matrix. Cancer Manag. Res. 2019, 11, 1843–1855. [Google Scholar] [CrossRef]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.-Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef]
- Bonosi, L.; Marrone, S.; Benigno, U.E.; Buscemi, F.; Musso, S.; Porzio, M.; Silven, M.P.; Torregrossa, F.; Grasso, G. Maximal Safe Resection in Glioblastoma Surgery: A Systematic Review of Advanced Intraoperative Image-Guided Techniques. Brain Sci. 2023, 13, 216. [Google Scholar] [CrossRef]
- Tomar, M.S.; Kumar, A.; Srivastava, C.; Shrivastava, A. Elucidating the Mechanisms of Temozolomide Resistance in Gliomas and the Strategies to Overcome the Resistance. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188616. [Google Scholar] [CrossRef]
- Brown, N.F.; Ottaviani, D.; Tazare, J.; Gregson, J.; Kitchen, N.; Brandner, S.; Fersht, N.; Mulholland, P. Survival Outcomes and Prognostic Factors in Glioblastoma. Cancers 2022, 14, 3161. [Google Scholar] [CrossRef]
- Mosteiro, A.; Pedrosa, L.; Ferrés, A.; Diao, D.; Sierra, À.; González, J.J. The Vascular Microenvironment in Glioblastoma: A Comprehensive Review. Biomedicines 2022, 10, 1285. [Google Scholar] [CrossRef]
- Tu, J.; Liang, H.; Li, C.; Huang, Y.; Wang, Z.; Chen, X.; Yuan, X. The Application and Research Progress of Anti-Angiogenesis Therapy in Tumor Immunotherapy. Front. Immunol. 2023, 14, 1198972. [Google Scholar] [CrossRef]
- Mukherjee, A.; Madamsetty, V.S.; Paul, M.K.; Mukherjee, S. Recent Advancements of Nanomedicine towards Antiangiogenic Therapy in Cancer. Int. J. Mol. Sci. 2020, 21, 455. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.D.; Aghi, M.K.; Taylor, J.W. Anti-Angiogenic Therapies in the Management of Glioblastoma. Chin. Clin. Oncol. 2021, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Bevacizumab (Avastin) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Sener, U.; Islam, M.; Webb, M.; Kizilbash, S.H. Antiangiogenic Exclusion Rules in Glioma Trials: Historical Perspectives and Guidance for Future Trial Design. Neuro-Oncol. Adv. 2024, 6, vdae039. [Google Scholar] [CrossRef]
- Qu, C.-Y.; Zheng, Y.; Zhou, M.; Zhang, Y.; Shen, F.; Cao, J.; Xu, L.-M. Value of Bevacizumab in Treatment of Colorectal Cancer: A Meta-Analysis. World J. Gastroenterol. 2015, 21, 5072–5080. [Google Scholar] [CrossRef]
- Cao, D.; Zheng, Y.; Xu, H.; Ge, W.; Xu, X. Bevacizumab Improves Survival in Metastatic Colorectal Cancer Patients with Primary Tumor Resection: A Meta-Analysis. Sci. Rep. 2019, 9, 20326. [Google Scholar] [CrossRef]
- Cutsem, E.V.; Rivera, F.; Berry, S.; Kretzschmar, A.; Michael, M.; DiBartolomeo, M.; Mazier, M.-A.; Canon, J.-L.; Georgoulias, V.; Peeters, M.; et al. Safety and Efficacy of First-Line Bevacizumab with FOLFOX, XELOX, FOLFIRI and Fluoropyrimidines in Metastatic Colorectal Cancer: The BEAT Study. Ann. Oncol. 2009, 20, 1842–1847. [Google Scholar] [CrossRef]
- Soria, J.-C.; Mauguen, A.; Reck, M.; Sandler, A.B.; Saijo, N.; Johnson, D.H.; Burcoveanu, D.; Fukuoka, M.; Besse, B.; Pignon, J.-P.; et al. Systematic Review and Meta-Analysis of Randomised, Phase II/III Trials Adding Bevacizumab to Platinum-Based Chemotherapy as First-Line Treatment in Patients with Advanced Non-Small-Cell Lung Cancer. Ann. Oncol. 2013, 24, 20–30. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.-M.; Wang, R. Effectiveness and Safety of Adding Bevacizumab to Platinum-Based Chemotherapy as First-Line Treatment for Advanced Non-Small-Cell Lung Cancer: A Meta-Analysis. Front. Med. 2021, 8, 616380. [Google Scholar] [CrossRef]
- Ascha, M.S.; Wang, J.F.; Kumthekar, P.; Sloan, A.E.; Kruchko, C.; Barnholtz-Sloan, J.S. Bevacizumab for the Treatment of Non-Small Cell Lung Cancer Patients with Synchronous Brain Metastases. Sci. Rep. 2019, 9, 17792. [Google Scholar] [CrossRef] [PubMed]
- Thompson Coon, J.S.; Liu, Z.; Hoyle, M.; Rogers, G.; Green, C.; Moxham, T.; Welch, K.; Stein, K. Sunitinib and Bevacizumab for First-Line Treatment of Metastatic Renal Cell Carcinoma: A Systematic Review and Indirect Comparison of Clinical Effectiveness. Br. J. Cancer 2009, 101, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.R.; Ged, Y.; Lee, C.-H.; Knezevic, A.; Molina, A.M.; Chen, Y.-B.; Chaim, J.; Coskey, D.T.; Murray, S.; Tickoo, S.K.; et al. Everolimus plus Bevacizumab Is Effective First-Line Treatment for Patients with Advanced Papillary Variant Renal Cell Carcinoma: Final Results from a Phase II Trial. Cancer 2020, 126, 5247–5255. [Google Scholar] [CrossRef]
- Zhang, A.B.; Mozaffari, K.; Aguirre, B.; Li, V.; Kubba, R.; Desai, N.C.; Wei, D.; Yang, I.; Wadehra, M. Exploring the Past, Present, and Future of Anti-Angiogenic Therapy in Glioblastoma. Cancers 2023, 15, 830. [Google Scholar] [CrossRef]
- Funakoshi, Y.; Takigawa, K.; Hata, N.; Kuga, D.; Hatae, R.; Sangatsuda, Y.; Fujioka, Y.; Otsuji, R.; Sako, A.; Yoshitake, T.; et al. Changes in the Relapse Pattern and Prognosis of Glioblastoma After Approval of First-Line Bevacizumab: A Single-Center Retrospective Study. World Neurosurg. 2022, 159, e479–e487. [Google Scholar] [CrossRef]
- Khan, A.B.; Lee, S.; Harmanci, A.S.; Patel, R.; Latha, K.; Yang, Y.; Marisetty, A.; Lee, H.-K.; Heimberger, A.B.; Fuller, G.N.; et al. CXCR4 expression is associated with proneural-to-mesenchymal transition in glioblastoma. Int. J. Cancer 2023, 152, 713–724. [Google Scholar] [CrossRef]
- Azam, Z.; To, S.T.; Tannous, B.A. Mesenchymal Transformation: The Rosetta Stone of Glioblastoma Pathogenesis and Therapy Resistance. Adv. Sci. 2020, 7, 2002015. [Google Scholar] [CrossRef]
- Luo, J.; Wang, Z.; Zhang, X.; Yu, H.; Chen, H.; Song, K.; Zhang, Y.; Schwartz, L.M.; Chen, H.; Liu, Y.; et al. Vascular Immune Evasion of Mesenchymal Glioblastoma Is Mediated by Interaction and Regulation of VE-Cadherin on PD-L1. Cancers 2023, 15, 4257. [Google Scholar] [CrossRef]
- Xu, C.; Hou, P.; Li, X.; Xiao, M.; Zhang, Z.; Li, Z.; Xu, J.; Liu, G.; Tan, Y.; Fang, C. Comprehensive Understanding of Glioblastoma Molecular Phenotypes: Classification, Characteristics, and Transition. Cancer Biol. Med. 2024, 21, 363–381. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Nan, G.-X. Oxidative Stress-Induced Angiogenesis. J. Clin. Neurosci. 2019, 63, 13–16. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The Role of Microenvironment in Tumor Angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.; Chapouly, C.; Andrique, L.; Bikfalvi, A.; Daubon, T. The Normal and Brain Tumor Vasculature: Morphological and Functional Characteristics and Therapeutic Targeting. Front. Physiol. 2021, 12, 622615. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, E.R.; Emblem, K.E.; Yen, Y.-F.; Dietrich, J.; Jordan, J.T.; Catana, C.; Wenchin, K.L.; Hooker, J.M.; Duda, D.G.; Rosen, B.R.; et al. Vascular Dysfunction Promotes Regional Hypoxia after Bevacizumab Therapy in Recurrent Glioblastoma Patients. Neurooncol. Adv. 2020, 2, vdaa157. [Google Scholar] [CrossRef]
- Brisson, L.; Henrique Geraldo, L.; Bikfalvi, A.; Mathivet, T. The Strange Microenvironment of Glioblastoma. Rev. Neurol. 2023, 179, 490–501. [Google Scholar] [CrossRef]
- Charalambous, C.; Chen, T.C.; Hofman, F.M. Characteristics of Tumor-Associated Endothelial Cells Derived from Glioblastoma Multiforme. Neurosurg. Focus. 2006, 20, E22. [Google Scholar] [CrossRef]
- Vartanian, A.; Singh, S.K.; Agnihotri, S.; Jalali, S.; Burrell, K.; Aldape, K.D.; Zadeh, G. GBM’s Multifaceted Landscape: Highlighting Regional and Microenvironmental Heterogeneity. Neuro Oncol. 2014, 16, 1167–1175. [Google Scholar] [CrossRef]
- Sounni, N.E.; Paye, A.; Host, L.; Noël, A. MT-MMPS as Regulators of Vessel Stability Associated with Angiogenesis. Front. Pharmacol. 2011, 1, 111. [Google Scholar] [CrossRef]
- Testa, E.; Palazzo, C.; Mastrantonio, R.; Viscomi, M.T. Dynamic Interactions between Tumor Cells and Brain Microvascular Endothelial Cells in Glioblastoma. Cancers 2022, 14, 3128. [Google Scholar] [CrossRef]
- Neubauer, K.; Zieger, B. Endothelial Cells and Coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Mathivet, T.; Bouleti, C.; Van Woensel, M.; Stanchi, F.; Verschuere, T.; Phng, L.; Dejaegher, J.; Balcer, M.; Matsumoto, K.; Georgieva, P.B.; et al. Dynamic Stroma Reorganization Drives Blood Vessel Dysmorphia during Glioma Growth. EMBO Mol. Med. 2017, 9, 1629–1645. [Google Scholar] [CrossRef] [PubMed]
- Barlow, K.D.; Sanders, A.M.; Soker, S.; Ergun, S.; Metheny-Barlow, L.J. Pericytes on the Tumor Vasculature: Jekyll or Hyde? Cancer Microenviron. 2013, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.C.; Aghi, M.K. Biology of Angiogenesis and Invasion in Glioma. Neurotherapeutics 2009, 6, 447–457. [Google Scholar] [CrossRef]
- Sanati, M.; Afshari, A.R.; Amini, J.; Mollazadeh, H.; Jamialahmadi, T.; Sahebkar, A. Targeting Angiogenesis in Gliomas: Potential Role of Phytochemicals. J. Funct. Foods 2022, 96, 105192. [Google Scholar] [CrossRef]
- Dudley, A.C.; Griffioen, A.W. Pathological Angiogenesis: Mechanisms and Therapeutic Strategies. Angiogenesis 2023, 26, 313–347. [Google Scholar] [CrossRef]
- Plate, K.H.; Scholz, A.; Dumont, D.J. Tumor Angiogenesis and Anti-Angiogenic Therapy in Malignant Gliomas Revisited. Acta Neuropathol. 2012, 124, 763–775. [Google Scholar] [CrossRef]
- Pezzella, F.; Ribatti, D. Vascular Co-option and Vasculogenic Mimicry Mediate Resistance to Antiangiogenic Strategies. Cancer Rep. 2020, 5, e1318. [Google Scholar] [CrossRef]
- Belotti, D.; Pinessi, D.; Taraboletti, G. Alternative Vascularization Mechanisms in Tumor Resistance to Therapy. Cancers 2021, 13, 1912. [Google Scholar] [CrossRef]
- Holst, C.B.; Pedersen, H.; Obara, E.A.A.; Vitting-Seerup, K.; Jensen, K.E.; Skjøth-Rasmussen, J.; Lund, E.L.; Poulsen, H.S.; Johansen, J.S.; Hamerlik, P. Perspective: Targeting VEGF-A and YKL-40 in Glioblastoma—Matter Matters. Cell Cycle 2021, 20, 702–715. [Google Scholar] [CrossRef]
- Cuypers, A.; Truong, A.-C.K.; Becker, L.M.; Saavedra-García, P.; Carmeliet, P. Tumor Vessel Co-Option: The Past & the Future. Front. Oncol. 2022, 12, 965277. [Google Scholar]
- Ribatti, D.; Annese, T.; Tamma, R. Vascular Co-Option in Resistance to Anti-Angiogenic Therapy. Front. Oncol. 2023, 13, 1323350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yao, Y.; Gao, H.; Hu, X. Mechanisms of Angiogenesis in Tumour. Front. Oncol. 2024, 14, 1359069. [Google Scholar] [CrossRef]
- Brat, D.J.; Van Meir, E.G. Glomeruloid Microvascular Proliferation Orchestrated by VPF/VEGF. Am. J. Pathol. 2001, 158, 789–796. [Google Scholar] [CrossRef]
- Ionescu, C.; Oprea, B.; Ciobanu, G.; Georgescu, M.; Bică, R.; Mateescu, G.-O.; Huseynova, F.; Barragan-Montero, V. The Angiogenic Balance and Its Implications in Cancer and Cardiovascular Diseases: An Overview. Medicina 2022, 58, 903. [Google Scholar] [CrossRef]
- Wong, A.L.; Haroon, Z.A.; Werner, S.; Dewhirst, M.W.; Greenberg, C.S.; Peters, K.G. Tie2 Expression and Phosphorylation in Angiogenic and Quiescent Adult Tissues. Circ. Res. 1997, 81, 567–574. [Google Scholar] [CrossRef]
- Al Sabti, H. Therapeutic Angiogenesis in Cardiovascular Disease. J. Cardiothorac. Surg. 2007, 2, 49. [Google Scholar] [CrossRef]
- Sund, M.; Nyberg, P.; Eikesdal, H.P. Endogenous Matrix-Derived Inhibitors of Angiogenesis. Pharmaceuticals 2010, 3, 3021–3039. [Google Scholar] [CrossRef]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef]
- Logsdon, E.A.; Finley, S.D.; Popel, A.S.; Gabhann, F.M. A Systems Biology View of Blood Vessel Growth and Remodelling. J. Cell. Mol. Med. 2014, 18, 1491–1508. [Google Scholar] [CrossRef]
- Senger, D.R.; Davis, G.E. Angiogenesis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005090. [Google Scholar] [CrossRef] [PubMed]
- Bulnes, S.; Bengoetxea, H.; Ortuzar, N.; Argandoña, E.G.; Garcia-Blanco, Á.; Rico-Barrio, I.; Lafuente, J.V. Angiogenic Signalling Pathways Altered in Gliomas: Selection Mechanisms for More Aggressive Neoplastic Subpopulations with Invasive Phenotype. J. Signal Transduct. 2012, 2012, 597915. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Qin, R.-Y. Mechanism and Its Regulation of Tumor-Induced Angiogenesis. World J. Gastroenterol. 2003, 9, 1144–1155. [Google Scholar] [CrossRef]
- Sebestyén, A.; Kopper, L.; Dankó, T.; Tímár, J. Hypoxia Signaling in Cancer: From Basics to Clinical Practice. Pathol. Oncol. Res. 2021, 27, 1609802. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, M.; Wang, L.; Jiao, B. HIFs, Angiogenesis, and Cancer. J. Cell Biochem. 2013, 114, 967–974. [Google Scholar] [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and Its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the Hypoxia-Inducible-Factor Pathway in Glioma Growth and Angiogenesis. Neuro-Oncology 2005, 7, 134–153. [Google Scholar] [CrossRef]
- Begagić, E.; Bečulić, H.; Džidić-Krivić, A.; Kadić Vukas, S.; Hadžić, S.; Mekić-Abazović, A.; Šegalo, S.; Papić, E.; Muchai Echengi, E.; Pugonja, R.; et al. Understanding the Significance of Hypoxia-Inducible Factors (HIFs) in Glioblastoma: A Systematic Review. Cancers 2024, 16, 2089. [Google Scholar] [CrossRef]
- Chédeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef]
- Calvo, P.M.; Hernández, R.G.; de la Cruz, R.R.; Pastor, A.M. Role of Vascular Endothelial Growth Factor as a Critical Neurotrophic Factor for the Survival and Physiology of Motoneurons. Neural Regen. Res. 2022, 18, 1691–1696. [Google Scholar] [CrossRef]
- Dakowicz, D.; Zajkowska, M.; Mroczko, B. Relationship between VEGF Family Members, Their Receptors and Cell Death in the Neoplastic Transformation of Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 3375. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Acharya, K.R. Tying the Knot: The Cystine Signature and Molecular-Recognition Processes of the Vascular Endothelial Growth Factor Family of Angiogenic Cytokines. FEBS J. 2011, 278, 4304–4322. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Stuttfeld, E.; Ballmer-Hofer, K. Structure and Function of VEGF Receptors. IUBMB Life 2009, 61, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Pia Ciotti, G.M.; Chiavari, M.; Ruffini, F.; Lacal, P.M.; Graziani, G.; Navarra, P. Vascular Endothelial Growth Factor Receptor 1 in Glioblastoma-associated Microglia/Macrophages. Oncol. Rep. 2020, 43, 2083–2092. [Google Scholar] [CrossRef]
- Azimi-Nezhad, M.; Stathopoulou, M.G.; Bonnefond, A.; Rancier, M.; Saleh, A.; Lamont, J.; Fitzgerald, P.; Ndiaye, N.C.; Visvikis-Siest, S. Associations of Vascular Endothelial Growth Factor (VEGF) with Adhesion and Inflammation Molecules in a Healthy Population. Cytokine 2013, 61, 602–607. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF Targets the Tumour Cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Maity, A.; Pore, N.; Lee, J.; Solomon, D.; O’Rourke, D.M. Epidermal Growth Factor Receptor Transcriptionally Up-Regulates Vascular Endothelial Growth Factor Expression in Human Glioblastoma Cells via a Pathway Involving Phosphatidylinositol 3′-Kinase and Distinct from That Induced by Hypoxia. Cancer Res. 2000, 60, 5879–5886. [Google Scholar]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular Endothelial Growth Factor Is a Potential Tumour Angiogenesis Factor in Human Gliomas in Vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef]
- Sonoda, Y.; Kanamori, M.; Deen, D.F.; Cheng, S.-Y.; Berger, M.S.; Pieper, R.O. Overexpression of Vascular Endothelial Growth Factor Isoforms Drives Oxygenation and Growth but Not Progression to Glioblastoma Multiforme in a Human Model of Gliomagenesis. Cancer Res. 2003, 63, 1962–1968. [Google Scholar] [PubMed]
- Flynn, J.R.; Wang, L.; Gillespie, D.L.; Stoddard, G.J.; Reid, J.K.; Owens, J.; Ellsworth, G.B.; Salzman, K.L.; Kinney, A.Y.; Jensen, R.L. Hypoxia-Regulated Protein Expression, Patient Characteristics, and Preoperative Imaging as Predictors of Survival in Adults with Glioblastoma Multiforme. Cancer 2008, 113, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Held-Feindt, J.; Buhl, R.; Mehdorn, H.M.; Mentlein, R. Expression of VEGF and Its Receptors in Different Brain Tumors. Neurol. Res. 2005, 27, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Ghalehbandi, S.; Yuzugulen, J.; Pranjol, M.Z.I.; Pourgholami, M.H. The Role of VEGF in Cancer-Induced Angiogenesis and Research Progress of Drugs Targeting VEGF. Eur. J. Pharmacol. 2023, 949, 175586. [Google Scholar] [CrossRef]
- Xu, C.; Wu, X.; Zhu, J. VEGF Promotes Proliferation of Human Glioblastoma Multiforme Stem-like Cells through VEGF Receptor 2. Sci. World J. 2013, 2013, 417413. [Google Scholar] [CrossRef]
- Martini, M.; de Pascalis, I.; D’Alessandris, Q.G.; Fiorentino, V.; Pierconti, F.; Marei, H.E.-S.; Ricci-Vitiani, L.; Pallini, R.; Larocca, L.M. VEGF-121 Plasma Level as Biomarker for Response to Anti-Angiogenetic Therapy in Recurrent Glioblastoma. BMC Cancer 2018, 18, 553. [Google Scholar] [CrossRef]
- Wallensten, J.; Mobarrez, F.; Åsberg, M.; Borg, K.; Beser, A.; Wilczek, A.; Nager, A. Isoforms of Soluble Vascular Endothelial Growth Factor in Stress-Related Mental Disorders: A Cross-Sectional Study. Sci. Rep. 2021, 11, 16693. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Okawa, M.; Takenouchi, K.; Bibek, A.; Yamada, S.; Inoue, K.; Higurashi, K.; Tabaru, A.; Tanoue, K.; Oyama, Y.; et al. VEGF-A165 Is the Predominant VEGF-A Isoform in Platelets, While VEGF-A121 Is Abundant in Serum and Plasma from Healthy Individuals. PLoS ONE 2023, 18, e0284131. [Google Scholar] [CrossRef]
- Vempati, P.; Popel, A.S.; Mac Gabhann, F. Extracellular Regulation of VEGF: Isoforms, Proteolysis, and Vascular Patterning. Cytokine Growth Factor Rev. 2014, 25, 1–19. [Google Scholar] [CrossRef]
- D’Alessandris, Q.G.; Martini, M.; Cenci, T.; Capo, G.; Ricci-Vitiani, L.; Larocca, L.M.; Pallini, R. VEGF Isoforms as Outcome Biomarker for Anti-Angiogenic Therapy in Recurrent Glioblastoma. Neurology 2015, 84, 1906–1908. [Google Scholar] [CrossRef]
- Lohela, M.; Bry, M.; Tammela, T.; Alitalo, K. VEGFs and Receptors Involved in Angiogenesis versus Lymphangiogenesis. Curr. Opin. Cell Biol. 2009, 21, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. Fibroblast Growth Factors. Genome Biol. 2001, 2, reviews3005.1–reviews3005.12. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF Family: Biology, Pathophysiology and Therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Khan, A.W.; Kim, M.S.; Choi, S. The Role of Fibroblast Growth Factor (FGF) Signaling in Tissue Repair and Regeneration. Cells 2021, 10, 3242. [Google Scholar] [CrossRef]
- Zhuang, L.; Falquet, L.; Trueb, B. Genome-Wide Comparison of FGFRL1 with Structurally Related Surface Receptors. Exp. Ther. Med. 2010, 1, 161–168. [Google Scholar] [CrossRef]
- Kostas, M.; Lampart, A.; Bober, J.; Wiedlocha, A.; Tomala, J.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. Translocation of Exogenous FGF1 and FGF2 Protects the Cell against Apoptosis Independently of Receptor Activation. J. Mol. Biol. 2018, 430, 4087–4101. [Google Scholar] [CrossRef]
- Giacomini, A.; Grillo, E.; Rezzola, S.; Ribatti, D.; Rusnati, M.; Ronca, R.; Presta, M. The FGF/FGFR System in the Physiopathology of the Prostate Gland. Physiol. Rev. 2021, 101, 569–610. [Google Scholar] [CrossRef]
- Liu, G.; Chen, T.; Ding, Z.; Wang, Y.; Wei, Y.; Wei, X. Inhibition of FGF-FGFR and VEGF-VEGFR Signalling in Cancer Treatment. Cell Prolif. 2021, 54, e13009. [Google Scholar] [CrossRef]
- Sahores, A.; Figueroa, V.; May, M.; Liguori, M.; Rubstein, A.; Fuentes, C.; Jacobsen, B.M.; Elía, A.; Rojas, P.; Sequeira, G.R.; et al. Increased High Molecular Weight FGF2 in Endocrine-Resistant Breast Cancer. Horm. Cancer 2018, 9, 338–348. [Google Scholar] [CrossRef]
- Donnem, T.; Al-Shibli, K.; Al-Saad, S.; Busund, L.-T.; Bremnes, R.M. Prognostic Impact of Fibroblast Growth Factor 2 in Non-Small Cell Lung Cancer: Coexpression with VEGFR-3 and PDGF-B Predicts Poor Survival. J. Thorac. Oncol. 2009, 4, 578–585. [Google Scholar] [CrossRef]
- Gnanapragasam, V.J.; Robinson, M.C.; Marsh, C.; Robson, C.N.; Hamdy, F.C.; Leung, H.Y. FGF8 Isoform b Expression in Human Prostate Cancer. Br. J. Cancer 2003, 88, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, P.; Gandini, O.; Gradilone, A.; Silvestri, I.; Giuliani, L.; Magnanti, M.; Gallucci, M.; Saccani, G.; Frati, L.; Agliano, A.M. Detection of Basic Fibroblast Growth Factor mRNA in Urinary Bladder Cancer: Correlation with Local Relapses. Int. J. Oncol. 1999, 14, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.L.S.; Sham, J.S.T.; Xie, D.; Fang, Y.; Wu, Y.-L.; Hu, L.; Deng, W.; Tsao, G.S.W.; Qiao, G.-B.; Cheung, A.L.M.; et al. Co-Overexpression of Fibroblast Growth Factor 3 and Epidermal Growth Factor Receptor Is Correlated with the Development of Nonsmall Cell Lung Carcinoma. Cancer 2006, 106, 146–155. [Google Scholar] [CrossRef]
- Suzuki, K.; Tokue, A.; Kamiakito, T.; Kuriki, K.; Saito, K.; Tanaka, A. Predominant Expression of Fibroblast Growth Factor (FGF) 8, FGF4, and FGF Receptor 1 in Nonseminomatous and Highly Proliferative Components of Testicular Germ Cell Tumors. Virchows Arch. 2001, 439, 616–621. [Google Scholar] [CrossRef]
- Sonvilla, G.; Allerstorfer, S.; Stättner, S.; Karner, J.; Klimpfinger, M.; Fischer, H.; Grasl-Kraupp, B.; Holzmann, K.; Berger, W.; Wrba, F.; et al. FGF18 in Colorectal Tumour Cells: Autocrine and Paracrine Effects. Carcinogenesis 2008, 29, 15–24. [Google Scholar] [CrossRef]
- Takahashi, J.A.; Fukumoto, M.; Igarashi, K.; Oda, Y.; Kikuchi, H.; Hatanaka, M. Correlation of Basic Fibroblast Growth Factor Expression Levels with the Degree of Malignancy and Vascularity in Human Gliomas. J. Neurosurg. 1992, 76, 792–798. [Google Scholar] [CrossRef]
- Takahashi, J.A.; Mori, H.; Fukumoto, M.; Igarashi, K.; Jaye, M.; Oda, Y.; Kikuchi, H.; Hatanaka, M. Gene Expression of Fibroblast Growth Factors in Human Gliomas and Meningiomas: Demonstration of Cellular Source of Basic Fibroblast Growth Factor mRNA and Peptide in Tumor Tissues. Proc. Natl. Acad. Sci. USA 1990, 87, 5710–5714. [Google Scholar] [CrossRef]
- Kaya, B.; Çiçek, O.; Erdi, F.; Findik, S.; Karatas, Y.; Esen, H.; Keskin, F.; Kalkan, E. Intratumoral Hemorrhage-Related Differences in the Expression of Vascular Endothelial Growth Factor, Basic Fibroblast Growth Factor and Thioredoxin Reductase 1 in Human Glioblastoma. Mol. Clin. Oncol. 2016, 5, 343–346. [Google Scholar] [CrossRef]
- Numasaki, M.; Ito, K. VEGF-A, HGF and bFGF Are Involved in IL-17A-Mediated Migration and Capillary-like Vessel Formation of Vascular Endothelial Cells. Iran. J. Immunol. 2021, 18, 103–110. [Google Scholar]
- Yoshida, A.; Anand-Apte, B.; Zetter, B.R. Differential Endothelial Migration and Proliferation to Basic Fibroblast Growth Factor and Vascular Endothelial Growth Factor. Growth Factors 1996, 13, 57–64. [Google Scholar] [CrossRef]
- Pepper, M.S.; Ferrara, N.; Orci, L.; Montesano, R. Potent Synergism between Vascular Endothelial Growth Factor and Basic Fibroblast Growth Factor in the Induction of Angiogenesis in Vitro. Biochem. Biophys. Res. Commun. 1992, 189, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Meyer, K.; Mundhenke, C.; Drew, S.A.; Friedl, A. Heparan Sulfate Proteoglycans as Regulators of Fibroblast Growth Factor-2 Signaling in Brain Endothelial Cells. Specific Role for Glypican-1 in Glioma Angiogenesis. J. Biol. Chem. 2003, 278, 16045–16053. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, P.; Carceller, F.; Angulo, J.; González-Corrochano, R.; Cuevas-Bourdier, A.; Giménez-Gallego, G. Antiglioma Effects of a New, Low Molecular Mass, Inhibitor of Fibroblast Growth Factor. Neurosci. Lett. 2011, 491, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast Growth Factors, Their Receptors and Signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef]
- Li, G.; Chen, Z.; Hu, Y.-D.; Wei, H.; Li, D.; Ji, H.; Wang, D.-L. Autocrine Factors Sustain Glioblastoma Stem Cell Self-Renewal. Oncol. Rep. 2009, 21, 419–424. [Google Scholar]
- Toyoda, K.; Tanaka, K.; Nakagawa, S.; Thuy, D.H.D.; Ujifuku, K.; Kamada, K.; Hayashi, K.; Matsuo, T.; Nagata, I.; Niwa, M. Initial Contact of Glioblastoma Cells with Existing Normal Brain Endothelial Cells Strengthen the Barrier Function via Fibroblast Growth Factor 2 Secretion: A New in Vitro Blood-Brain Barrier Model. Cell Mol. Neurobiol. 2013, 33, 489–501. [Google Scholar] [CrossRef]
- Zahra, F.T.; Sajib, M.S.; Mikelis, C.M. Role of bFGF in Acquired Resistance upon Anti-VEGF Therapy in Cancer. Cancers 2021, 13, 1422. [Google Scholar] [CrossRef]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a New Fibroblast Growth Factor Receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Harding, M.J.; Nechiporuk, A.V. Fgfr-Ras-MAPK Signaling Is Required for Apical Constriction via Apical Positioning of Rho-Associated Kinase during Mechanosensory Organ Formation. Development 2012, 139, 3130–3135. [Google Scholar] [CrossRef]
- Mohammadi, M.; Honegger, A.M.; Rotin, D.; Fischer, R.; Bellot, F.; Li, W.; Dionne, C.A.; Jaye, M.; Rubinstein, M.; Schlessinger, J. A Tyrosine-Phosphorylated Carboxy-Terminal Peptide of the Fibroblast Growth Factor Receptor (Flg) Is a Binding Site for the SH2 Domain of Phospholipase C-Gamma 1. Mol. Cell Biol. 1991, 11, 5068–5078. [Google Scholar]
- Reis-Filho, J.S.; Simpson, P.T.; Turner, N.C.; Lambros, M.B.; Jones, C.; Mackay, A.; Grigoriadis, A.; Sarrio, D.; Savage, K.; Dexter, T.; et al. FGFR1 Emerges as a Potential Therapeutic Target for Lobular Breast Carcinomas. Clin. Cancer Res. 2006, 12, 6652–6662. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.C.; Robertson, S.C.; Kanemitsu, M.Y.; Meyer, A.N.; Tynan, J.A.; Donoghue, D.J. Transformation and Stat Activation by Derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 2000, 19, 3309–3320. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Dono, A.; El Achi, H.; Bundrant, B.E.; Goli, P.S.; Zhu, P.; Ozkizilkaya, H.I.; Esquenazi, Y.; Ballester, L.Y. Infiltrating Gliomas with FGFR Alterations: Histologic Features, Genetic Alterations, and Potential Clinical Implications. Cancer Biomark. 2023, 36, 117–131. [Google Scholar] [CrossRef]
- Jimenez-Pascual, A.; Siebzehnrubl, F.A. Fibroblast Growth Factor Receptor Functions in Glioblastoma. Cells 2019, 8, 715. [Google Scholar] [CrossRef]
- Danielpour, D. Advances and Challenges in Targeting TGF-β Isoforms for Therapeutic Intervention of Cancer: A Mechanism-Based Perspective. Pharmaceuticals 2024, 17, 533. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β Receptors: In and beyond TGF-β Signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Peng, D.; Zhang, Y.; Shi, H. TGF-Beta Signal Transduction: Biology, Function and Therapy for Diseases. Mol. Biomed. 2022, 3, 45. [Google Scholar] [CrossRef]
- Oft, M.; Heider, K.-H.; Beug, H. TGFβ Signaling Is Necessary for Carcinoma Cell Invasiveness and Metastasis. Curr. Biol. 1998, 8, 1243–1252. [Google Scholar] [CrossRef]
- Davies, M.; Prime, S.S.; Eveson, J.W.; Price, N.; Ganapathy, A.; D’Mello, A.; Paterson, I.C. Transforming Growth Factor-β Enhances Invasion and Metastasis in Ras-Transfected Human Malignant Epidermal Keratinocytes. Int. J. Exp. Pathol. 2012, 93, 148–156. [Google Scholar] [CrossRef]
- Zhang, J.; Deng, Y.-T.; Liu, J.; Gan, L.; Jiang, Y. Role of Transforming Growth Factor-Β1 Pathway in Angiogenesis Induced by Chronic Stress in Colorectal Cancer. Cancer Biol. Ther. 2024, 25, 2366451. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, Z.; Wen, T. Multiplex Fluorescent Immunohistochemistry Quantitatively Analyses Microvascular Density (MVD) and the Roles of TGF-β Signalling in Orchestrating Angiogenesis in Colorectal Cancer. Transl. Cancer Res. 2019, 8, 429. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.J.; Shih, R.M.; Uy, B.R.; Shafer, A.; Chang, Z.L.; Comin-Anduix, B.; Guemes, M.; Galic, Z.; Phyu, S.; Okada, H.; et al. IL-13Rα2/TGF-β Bispecific CAR-T Cells Counter TGF-β-Mediated Immune Suppression and Potentiate Anti-Tumor Responses in Glioblastoma. Neuro-Oncology 2024, 26, 1850–1866. [Google Scholar] [CrossRef] [PubMed]
- Ghahremanifard, P.; Chanda, A.; Bonni, S.; Bose, P. TGF-β Mediated Immune Evasion in Cancer—Spotlight on Cancer-Associated Fibroblasts. Cancers 2020, 12, 3650. [Google Scholar] [CrossRef]
- Zhang, J.; Thorikay, M.; van der Zon, G.; van Dinther, M.; Ten Dijke, P. Studying TGF-β Signaling and TGF-β-Induced Epithelial-to-Mesenchymal Transition in Breast Cancer and Normal Cells. J. Vis. Exp. 2020, 27, e61830. [Google Scholar]
- Zhang, Q.; Yu, N.; Lee, C. Mysteries of TGF-β Paradox in Benign and Malignant Cells. Front. Oncol. 2014, 4, 94. [Google Scholar] [CrossRef]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocaña, A.; Peñuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFbeta-Smad Activity Confers Poor Prognosis in Glioma Patients and Promotes Cell Proliferation Depending on the Methylation of the PDGF-B Gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef]
- Pen, A.; Moreno, M.J.; Durocher, Y.; Deb-Rinker, P.; Stanimirovic, D.B. Glioblastoma-Secreted Factors Induce IGFBP7 and Angiogenesis by Modulating Smad-2-Dependent TGF-β Signaling. Oncogene 2008, 27, 6834–6844. [Google Scholar] [CrossRef]
- Hovis, G.; Chandra, N.; Kejriwal, N.; Hsieh, K.J.-Y.; Chu, A.; Yang, I.; Wadehra, M. Understanding the Role of Endothelial Cells in Glioblastoma: Mechanisms and Novel Treatments. Int. J. Mol. Sci. 2024, 25, 6118. [Google Scholar] [CrossRef]
- Krishnan, S.; Szabo, E.; Burghardt, I.; Frei, K.; Tabatabai, G.; Weller, M. Modulation of Cerebral Endothelial Cell Function by TGF-β in Glioblastoma: VEGF-Dependent Angiogenesis versus Endothelial Mesenchymal Transition. Oncotarget 2015, 6, 22480–22495. [Google Scholar] [CrossRef]
- Piek, E.; Westermark, U.; Kastemar, M.; Heldin, C.H.; van Zoelen, E.J.; Nistér, M.; Ten Dijke, P. Expression of Transforming-Growth-Factor (TGF)-Beta Receptors and Smad Proteins in Glioblastoma Cell Lines with Distinct Responses to TGF-Beta1. Int. J. Cancer 1999, 80, 756–763. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Westermark, B. Mechanism of Action and In Vivo Role of Platelet-Derived Growth Factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef] [PubMed]
- Michalevicz, R.; Katz, F.; Stroobant, P.; Janossy, G.; Tindle, R.W.; Hoffbrand, A.V. Platelet-Derived Growth Factor Stimulates Growth of Highly Enriched Multipotent Haemopoietic Progenitors. Br. J. Haematol. 1986, 63, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Deuel, T.F.; Senior, R.M.; Huang, J.S.; Griffin, G.L. Chemotaxis of Monocytes and Neutrophils to Platelet-Derived Growth Factor. J. Clin. Investig. 1982, 69, 1046–1049. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Chang, T.; Seppä, H.E.; Kleinman, H.K.; Martin, G.R. Platelet-Derived Growth Factor Is a Chemoattractant for Vascular Smooth Muscle Cells. J. Cell Physiol. 1982, 113, 261–266. [Google Scholar] [CrossRef]
- De Donatis, A.; Comito, G.; Buricchi, F.; Vinci, M.C.; Parenti, A.; Caselli, A.; Camici, G.; Manao, G.; Ramponi, G.; Cirri, P. Proliferation versus Migration in Platelet-Derived Growth Factor Signaling: The Key Role of Endocytosis. J. Biol. Chem. 2008, 283, 19948–19956. [Google Scholar] [CrossRef]
- Bartoschek, M.; Pietras, K. PDGF Family Function and Prognostic Value in Tumor Biology. Biochem. Biophys. Res. Commun. 2018, 503, 984–990. [Google Scholar] [CrossRef]
- Westermark, B.; Heldin, C.H.; Nistér, M. Platelet-Derived Growth Factor in Human Glioma. Glia 1995, 15, 257–263. [Google Scholar] [CrossRef]
- Mahadevan, D.; Yu, J.-C.; Saldanha, J.W.; Thanki, N.; McPhie, P.; Uren, A.; LaRochelle, W.J.; Heidaran, M.A. Structural Role of Extracellular Domain 1 of α-Platelet-Derived Growth Factor (PDGF) Receptor for PDGF-AA and PDGF-BB Binding (*). J. Biol. Chem. 1995, 270, 27595–27600. [Google Scholar] [CrossRef]
- Zhang, H.; Bajraszewski, N.; Wu, E.; Wang, H.; Moseman, A.P.; Dabora, S.L.; Griffin, J.D.; Kwiatkowski, D.J. PDGFRs Are Critical for PI3K/Akt Activation and Negatively Regulated by mTOR. J. Clin. Investig. 2007, 117, 730–738. [Google Scholar] [CrossRef]
- Zou, X.; Tang, X.-Y.; Qu, Z.-Y.; Sun, Z.-W.; Ji, C.-F.; Li, Y.-J.; Guo, S.-D. Targeting the PDGF/PDGFR Signaling Pathway for Cancer Therapy: A Review. Int. J. Biol. Macromol. 2022, 202, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Khan, F.; Upadhyay, T.K.; Seungjoon, M.; Park, M.N.; Kim, B. New Insights about the PDGF/PDGFR Signaling Pathway as a Promising Target to Develop Cancer Therapeutic Strategies. Biomed. Pharmacother. 2023, 161, 114491. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yuan, W.; Wu, L.; Tang, Q.; Xia, Q.; Ji, J.; Liu, Z.; Ma, Z.; Zhou, Z.; Cheng, Y.; et al. PDGF-D Promotes Cell Growth, Aggressiveness, Angiogenesis and EMT Transformation of Colorectal Cancer by Activation of Notch1/Twist1 Pathway. Oncotarget 2016, 8, 9961–9973. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Lennartsson, J. Structural and Functional Properties of Platelet-Derived Growth Factor and Stem Cell Factor Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009100. [Google Scholar] [CrossRef]
- Gacche, R.N.; Assaraf, Y.G. Redundant Angiogenic Signaling and Tumor Drug Resistance. Drug Resist. Updates 2018, 36, 47–76. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, E.; Wu, Q.; Guryanova, O.; Hitomi, M.; Lathia, J.D.; Serwanski, D.; Sloan, A.E.; Weil, R.J.; Lee, J.; et al. Platelet-Derived Growth Factor Receptors Differentially Inform Intertumoral and Intratumoral Heterogeneity. Genes Dev. 2012, 26, 1247–1262. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Lennartsson, J.; Westermark, B. Involvement of Platelet-Derived Growth Factor Ligands and Receptors in Tumorigenesis. J. Intern. Med. 2018, 283, 16–44. [Google Scholar] [CrossRef]
- Guo, P.; Hu, B.; Gu, W.; Xu, L.; Wang, D.; Huang, H.-J.S.; Cavenee, W.K.; Cheng, S.-Y. Platelet-Derived Growth Factor-B Enhances Glioma Angiogenesis by Stimulating Vascular Endothelial Growth Factor Expression in Tumor Endothelia and by Promoting Pericyte Recruitment. Am. J. Pathol. 2003, 162, 1083–1093. [Google Scholar] [CrossRef]
- Hermansson, M.; Nistér, M.; Betsholtz, C.; Heldin, C.H.; Westermark, B.; Funa, K. Endothelial Cell Hyperplasia in Human Glioblastoma: Coexpression of mRNA for Platelet-Derived Growth Factor (PDGF) B Chain and PDGF Receptor Suggests Autocrine Growth Stimulation. Proc. Natl. Acad. Sci. USA 1988, 85, 7748–7752. [Google Scholar] [CrossRef]
- Cenciarelli, C.; Marei, H.E.; Zonfrillo, M.; Pierimarchi, P.; Paldino, E.; Casalbore, P.; Felsani, A.; Vescovi, A.L.; Maira, G.; Mangiola, A. PDGF Receptor Alpha Inhibition Induces Apoptosis in Glioblastoma Cancer Stem Cells Refractory to Anti-Notch and Anti-EGFR Treatment. Mol. Cancer 2014, 13, 247. [Google Scholar] [CrossRef]
- Lane, R.; Cilibrasi, C.; Chen, J.; Shah, K.; Messuti, E.; Mazarakis, N.K.; Stebbing, J.; Critchley, G.; Song, E.; Simon, T.; et al. PDGF-R Inhibition Induces Glioblastoma Cell Differentiation via DUSP1/p38MAPK Signalling. Oncogene 2022, 41, 2749–2763. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; Ryan, T.E.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of Angiopoietin-1, a Ligand for the TIE2 Receptor, by Secretion-Trap Expression Cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, D.M.; Griffiths, J.A.; Rojas, J.; Aldrich, T.H.; Jones, P.F.; Zhou, H.; McClain, J.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; et al. Angiopoietins 3 and 4: Diverging Gene Counterparts in Mice and Humans. Proc. Natl. Acad. Sci. USA 1999, 96, 1904–1909. [Google Scholar] [CrossRef] [PubMed]
- Barton, W.A.; Tzvetkova, D.; Nikolov, D.B. Structure of the Angiopoietin-2 Receptor Binding Domain and Identification of Surfaces Involved in Tie2 Recognition. Structure 2005, 13, 825–832. [Google Scholar] [CrossRef]
- Kontos, C.D.; Stauffer, T.P.; Yang, W.-P.; York, J.D.; Huang, L.; Blanar, M.A.; Meyer, T.; Peters, K.G. Tyrosine 1101 of Tie2 Is the Major Site of Association of P85 and Is Required for Activation of Phosphatidylinositol 3-Kinase and Akt. Mol. Cell. Biol. 1998, 18, 4131–4140. [Google Scholar] [CrossRef]
- Reusch, P.; Barleon, B.; Weindel, K.; Martiny-Baron, G.; Gödde, A.; Siemeister, G.; Marmé, D. Identification of a Soluble Form of the Angiopoietin Receptor TIE-2 Released from Endothelial Cells and Present in Human Blood. Angiogenesis 2001, 4, 123–131. [Google Scholar] [CrossRef]
- Schnürch, H.; Risau, W. Expression of Tie-2, a Member of a Novel Family of Receptor Tyrosine Kinases, in the Endothelial Cell Lineage. Development 1993, 119, 957–968. [Google Scholar] [CrossRef]
- La Porta, S.; Roth, L.; Singhal, M.; Mogler, C.; Spegg, C.; Schieb, B.; Qu, X.; Adams, R.H.; Baldwin, H.S.; Savant, S.; et al. Endothelial Tie1–Mediated Angiogenesis and Vascular Abnormalization Promote Tumor Progression and Metastasis. J. Clin. Investig. 2018, 128, 834–845. [Google Scholar] [CrossRef]
- Savant, S.; Porta, S.L.; Budnik, A.; Busch, K.; Hu, J.; Tisch, N.; Korn, C.; Valls, A.F.; Benest, A.V.; Terhardt, D.; et al. The Orphan Receptor Tie1 Controls Angiogenesis and Vascular Remodeling by Differentially Regulating Tie2 in Tip and Stalk Cells. Cell Rep. 2015, 12, 1761–1773. [Google Scholar] [CrossRef]
- Jeansson, M.; Gawlik, A.; Anderson, G.; Li, C.; Kerjaschki, D.; Henkelman, M.; Quaggin, S.E. Angiopoietin-1 Is Essential in Mouse Vasculature during Development and in Response to Injury. J. Clin. Investig. 2011, 121, 2278–2289. [Google Scholar] [CrossRef]
- Mueller, S.B.; Kontos, C.D. Tie1: An Orphan Receptor Provides Context for Angiopoietin-2/Tie2 Signaling. J. Clin. Investig. 2016, 126, 3188–3191. [Google Scholar] [CrossRef] [PubMed]
- Brunckhorst, M.K.; Xu, Y.; Lu, R.; Yu, Q. Angiopoietins Promote Ovarian Cancer Progression by Establishing a Procancer Microenvironment. Am. J. Pathol. 2014, 184, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Martoglio, A.-M.; Tom, B.D.M.; Starkey, M.; Corps, A.N.; Charnock-Jones, D.S.; Smith, S.K. Changes in Tumorigenesis- and Angiogenesis-Related Gene Transcript Abundance Profiles in Ovarian Cancer Detected by Tailored High Density cDNA Arrays. Mol. Med. 2000, 6, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jarzynka, M.J.; Guo, P.; Imanishi, Y.; Schlaepfer, D.D.; Cheng, S.-Y. Angiopoietin 2 Induces Glioma Cell Invasion by Stimulating Matrix Metalloprotease 2 Expression through the Avβ1 Integrin and Focal Adhesion Kinase Signaling Pathway. Cancer Res. 2006, 66, 775–783. [Google Scholar] [CrossRef]
- Dong, Z.; Chen, J.; Yang, X.; Zheng, W.; Wang, L.; Fang, M.; Wu, M.; Yao, M.; Yao, D. Ang-2 Promotes Lung Cancer Metastasis by Increasing Epithelial-Mesenchymal Transition. Oncotarget 2018, 9, 12705–12717. [Google Scholar] [CrossRef]
- Machein, M.R.; Knedla, A.; Knoth, R.; Wagner, S.; Neuschl, E.; Plate, K.H. Angiopoietin-1 Promotes Tumor Angiogenesis in a Rat Glioma Model. Am. J. Pathol. 2004, 165, 1557–1570. [Google Scholar] [CrossRef]
- Stratmann, A.; Risau, W.; Plate, K.H. Cell Type-Specific Expression of Angiopoietin-1 and Angiopoietin-2 Suggests a Role in Glioblastoma Angiogenesis. Am. J. Pathol. 1998, 153, 1459–1466. [Google Scholar] [CrossRef]
- Brunckhorst, M.K.; Wang, H.; Lu, R.; Yu, Q. Angiopoietin-4 Promotes Glioblastoma Progression by Enhancing Tumor Cell Viability and Angiogenesis. Cancer Res. 2010, 70, 7283–7293. [Google Scholar] [CrossRef]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual Inhibition of Ang-2 and VEGF Receptors Normalizes Tumor Vasculature and Prolongs Survival in Glioblastoma by Altering Macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef]
- Choi, Y.C.; Dalakas, M.C. Expression of Matrix Metalloproteinases in the Muscle of Patients with Inflammatory Myopathies. Neurology 2000, 54, 65–71. [Google Scholar] [CrossRef]
- Jaoude, J.; Koh, Y. Matrix Metalloproteinases in Exercise and Obesity. Vasc. Health Risk Manag. 2016, 12, 287–295. [Google Scholar] [PubMed]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [PubMed]
- Gobin, E.; Bagwell, K.; Wagner, J.; Mysona, D.; Sandirasegarane, S.; Smith, N.; Bai, S.; Sharma, A.; Schleifer, R.; She, J.-X. A Pan-Cancer Perspective of Matrix Metalloproteases (MMP) Gene Expression Profile and Their Diagnostic/Prognostic Potential. BMC Cancer 2019, 19, 581. [Google Scholar] [CrossRef]
- Rundhaug, J.E. Matrix Metalloproteinases and Angiogenesis. J. Cell Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Stojic, J.; Hagemann, C.; Haas, S.; Herbold, C.; Kühnel, S.; Gerngras, S.; Roggendorf, W.; Roosen, K.; Vince, G.H. Expression of Matrix Metalloproteinases MMP-1, MMP-11 and MMP-19 Is Correlated with the WHO-Grading of Human Malignant Gliomas. Neurosci. Res. 2008, 60, 40–49. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, Y.; Chen, M.; Zha, Y. Matrix Metalloproteinase 9 Expression and Glioblastoma Survival Prediction Using Machine Learning on Digital Pathological Images. Sci. Rep. 2024, 14, 15065. [Google Scholar] [CrossRef]
- Xue, Q.; Cao, L.; Chen, X.-Y.; Zhao, J.; Gao, L.; Li, S.-Z.; Fei, Z. High Expression of MMP9 in Glioma Affects Cell Proliferation and Is Associated with Patient Survival Rates. Oncol. Lett. 2017, 13, 1325–1330. [Google Scholar] [CrossRef]
- Jiguet-Jiglaire, C.; Boissonneau, S.; Denicolai, E.; Hein, V.; Lasseur, R.; Garcia, J.; Romain, S.; Appay, R.; Graillon, T.; Mason, W.; et al. Plasmatic MMP9 Released from Tumor-Infiltrating Neutrophils Is Predictive for Bevacizumab Efficacy in Glioblastoma Patients: An AVAglio Ancillary Study. Acta Neuropathol. Commun. 2022, 10, 1. [Google Scholar] [CrossRef]
- Gabelloni, P.; Da Pozzo, E.; Bendinelli, S.; Costa, B.; Nuti, E.; Casalini, F.; Orlandini, E.; Da Settimo, F.; Rossello, A.; Martini, C. Inhibition of Metalloproteinases Derived from Tumours: New Insights in the Treatment of Human Glioblastoma. Neuroscience 2010, 168, 514–522. [Google Scholar] [CrossRef]
- Goldie, L.C.; Nix, M.K.; Hirschi, K.K. Embryonic Vasculogenesis and Hematopoietic Specification. Organogenesis 2008, 4, 257–263. [Google Scholar] [CrossRef]
- Drake, C.J. Embryonic and Adult Vasculogenesis. Birth Defects Res. C Embryo Today 2003, 69, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kässmeyer, S.; Plendl, J.; Custodis, P.; Bahramsoltani, M. New Insights in Vascular Development: Vasculogenesis and Endothelial Progenitor Cells. Anat. Histol. Embryol. 2009, 38, 1–11. [Google Scholar] [CrossRef]
- Tepper, O.M.; Capla, J.M.; Galiano, R.D.; Ceradini, D.J.; Callaghan, M.J.; Kleinman, M.E.; Gurtner, G.C. Adult Vasculogenesis Occurs through in Situ Recruitment, Proliferation, and Tubulization of Circulating Bone Marrow-Derived Cells. Blood 2005, 105, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Chopra, H.; Hung, M.K.; Kwong, D.L.; Zhang, C.F.; Pow, E.H.N. Insights into Endothelial Progenitor Cells: Origin, Classification, Potentials, and Prospects. Stem Cells Int. 2018, 2018, 9847015. [Google Scholar] [CrossRef]
- Blatchley, M.R.; Hall, F.; Wang, S.; Pruitt, H.C.; Gerecht, S. Hypoxia and Matrix Viscoelasticity Sequentially Regulate Endothelial Progenitor Cluster-Based Vasculogenesis. Sci. Adv. 2019, 5, eaau7518. [Google Scholar] [CrossRef]
- Huizer, K.; Sacchetti, A.; Dik, W.A.; Mustafa, D.A.; Kros, J.M. Circulating Proangiogenic Cells and Proteins in Patients with Glioma and Acute Myocardial Infarction: Differences in Neovascularization between Neoplasia and Tissue Regeneration. J. Oncol. 2019, 2019, 3560830. [Google Scholar] [CrossRef]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha Induces the Recruitment of Bone Marrow-Derived Vascular Modulatory Cells to Regulate Tumor Angiogenesis and Invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation Activates HIF-1 to Regulate Vascular Radiosensitivity in Tumors: Role of Reoxygenation, Free Radicals, and Stress Granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef]
- Jin, D.K.; Shido, K.; Kopp, H.-G.; Petit, I.; Shmelkov, S.V.; Young, L.M.; Hooper, A.T.; Amano, H.; Avecilla, S.T.; Heissig, B.; et al. Cytokine-Mediated Deployment of SDF-1 Induces Revascularization through Recruitment of CXCR4+ Hemangiocytes. Nat. Med. 2006, 12, 557–567. [Google Scholar] [CrossRef]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma Tumor Stem-like Cells Promote Tumor Angiogenesis and Vasculogenesis via Vascular Endothelial Growth Factor and Stromal-Derived Factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar] [CrossRef]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of Vasculogenesis, but Not Angiogenesis, Prevents the Recurrence of Glioblastoma after Irradiation in Mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Pezzella, F. Vascular Co-Option and Other Alternative Modalities of Growth of Tumor Vasculature in Glioblastoma. Front. Oncol. 2022, 12, 874554. [Google Scholar] [CrossRef] [PubMed]
- Pichol-Thievend, C.; Anezo, O.; Pettiwala, A.M.; Bourmeau, G.; Montagne, R.; Lyne, A.-M.; Guichet, P.-O.; Deshors, P.; Ballestín, A.; Blanchard, B.; et al. VC-Resist Glioblastoma Cell State: Vessel Co-Option as a Key Driver of Chemoradiation Resistance. Nat. Commun. 2024, 15, 3602. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Jain, R.K. Vessel Co-Option in Glioblastoma: Emerging Insights and Opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef]
- Bae, J.; Kim, M.-H.; Han, S.; Park, S. Development of Tumor-Vasculature Interaction on Chip Mimicking Vessel Co-Option of Glioblastoma. BioChip J. 2023, 17, 77–84. [Google Scholar] [CrossRef]
- Nico, B.; Crivellato, E.; Guidolin, D.; Annese, T.; Longo, V.; Finato, N.; Vacca, A.; Ribatti, D. Intussusceptive Microvascular Growth in Human Glioma. Clin. Exp. Med. 2010, 10, 93–98. [Google Scholar] [CrossRef]
- Hlushchuk, R.; Riesterer, O.; Baum, O.; Wood, J.; Gruber, G.; Pruschy, M.; Djonov, V. Tumor Recovery by Angiogenic Switch from Sprouting to Intussusceptive Angiogenesis after Treatment with PTK787/ZK222584 or Ionizing Radiation. Am. J. Pathol. 2008, 173, 1173–1185. [Google Scholar] [CrossRef]
- Hlushchuk, R.; Makanya, A.N.; Djonov, V. Escape Mechanisms after Antiangiogenic Treatment, or Why Are the Tumors Growing Again? Int. J. Dev. Biol. 2011, 55, 563–567. [Google Scholar] [CrossRef]
- Kirschmann, D.A.; Seftor, E.A.; Hardy, K.M.; Seftor, R.E.B.; Hendrix, M.J.C. Molecular Pathways: Vasculogenic Mimicry in Tumor Cells: Diagnostic and Therapeutic Implications. Clin. Cancer Res. 2012, 18, 2726–2732. [Google Scholar] [CrossRef]
- Angara, K.; Borin, T.F.; Arbab, A.S. Vascular Mimicry: A Novel Neovascularization Mechanism Driving Anti-Angiogenic Therapy (AAT) Resistance in Glioblastoma. Transl. Oncol. 2017, 10, 650–660. [Google Scholar] [CrossRef]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.-L.; Eichmann, A.; et al. A New Alternative Mechanism in Glioblastoma Vascularization: Tubular Vasculogenic Mimicry. Brain 2010, 133, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.-Y.; Chen, Z.-P. Does Vasculogenic Mimicry Exist in Astrocytoma? J. Histochem. Cytochem. 2005, 53, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ke, Y.; Lu, G.; Song, Z.; Yu, L.; Xiao, S.; Sun, X.; Jiang, X.; Yang, Z.; Hu, C. Vasculogenic Mimicry Is a Prognostic Factor for Postoperative Survival in Patients with Glioblastoma. J. Neurooncol. 2013, 112, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Chen, Y.-S.; Zhang, Q.-P.; Chen, F.-R.; Xi, S.-Y.; Long, Y.-K.; Zhang, J.; Cai, H.-P.; Ke, C.; Wang, J.; et al. Association between Glioblastoma Cell-Derived Vessels and Poor Prognosis of the Patients. Cancer Commun. 2020, 40, 211–221. [Google Scholar] [CrossRef]
- Angara, K.; Rashid, M.H.; Shankar, A.; Ara, R.; Iskander, A.; Borin, T.F.; Jain, M.; Achyut, B.R.; Arbab, A.S. Vascular Mimicry in Glioblastoma Following Anti-Angiogenic and Anti-20-HETE Therapies. Histol. Histopathol. 2017, 32, 917–928. [Google Scholar]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of Glioblastoma Cells into Vascular Endothelial Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef]
- Niechi, I.; Erices, J.I.; Carrillo-Beltrán, D.; Uribe-Ojeda, A.; Torres, Á.; Rocha, J.D.; Uribe, D.; Toro, M.A.; Villalobos-Nova, K.; Gaete-Ramírez, B.; et al. Cancer Stem Cell and Aggressiveness Traits Are Promoted by Stable Endothelin-Converting Enzyme-1c in Glioblastoma Cells. Cells 2023, 12, 506. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Xu, S.; Liu, Z.; Cheng, Q. The Adaptive Transition of Glioblastoma Stem Cells and Its Implications on Treatments. Signal Transduct. Target. Ther. 2021, 6, 124. [Google Scholar] [CrossRef]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef]
- Scully, S.; Francescone, R.; Faibish, M.; Bentley, B.; Taylor, S.L.; Oh, D.; Schapiro, R.; Moral, L.; Yan, W.; Shao, R. Transdifferentiation of Glioblastoma Stem-Like Cells into Mural Cells Drives Vasculogenic Mimicry in Glioblastomas. J. Neurosci. 2012, 32, 12950–12960. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour Vascularization via Endothelial Differentiation of Glioblastoma Stem-like Cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, T.; Liu, A.Y.; Ouyang, G. Differentiation and Transdifferentiation Potentials of Cancer Stem Cells. Oncotarget 2015, 6, 39550–39563. [Google Scholar] [CrossRef] [PubMed]
- De Pascalis, I.; Morgante, L.; Pacioni, S.; D’Alessandris, Q.G.; Giannetti, S.; Martini, M.; Ricci-Vitiani, L.; Malinverno, M.; Dejana, E.; Larocca, L.M.; et al. Endothelial Trans-Differentiation in Glioblastoma Recurring after Radiotherapy. Mod. Pathol. 2018, 31, 1361–1366. [Google Scholar] [CrossRef]
- Buccarelli, M.; D’Alessandris, Q.G.; Matarrese, P.; Mollinari, C.; Signore, M.; Cappannini, A.; Martini, M.; D’Aliberti, P.; De Luca, G.; Pedini, F.; et al. Elesclomol-Induced Increase of Mitochondrial Reactive Oxygen Species Impairs Glioblastoma Stem-like Cell Survival and Tumor Growth. J. Exp. Clin. Cancer Res. 2021, 40, 228. [Google Scholar] [CrossRef]
- Cai, Q.; Fan, H.; Li, X.; Giannotta, M.; Bachoo, R.; Qin, Z. Optical Modulation of the Blood-Brain Barrier for Glioblastoma Treatment. Bio Protoc. 2024, 14, e4920. [Google Scholar] [CrossRef]
- Gerritsen, J.K.W.; Broekman, M.L.D.; De Vleeschouwer, S.; Schucht, P.; Nahed, B.V.; Berger, M.S.; Vincent, A.J.P.E. Safe Surgery for Glioblastoma: Recent Advances and Modern Challenges. Neurooncol. Pract. 2022, 9, 364–379. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of Temozolomide Resistance in Glioblastoma—A Comprehensive Review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Fan, J.; Liu, J.; Zhang, B.; Wang, X.; Wang, X.; Liang, J.; Li, Y.; Zhang, Y.; Zhang, C.; Yu, S.; et al. GPR65 Contributes to Constructing Immunosuppressive Microenvironment in Glioma. Neurosurg. Rev. 2024, 47, 417. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B.; Tataranu, L.G.; Kamel, A.; Turliuc, S.; Rizea, R.E.; Dricu, A. Glioblastoma and Immune Checkpoint Inhibitors: A Glance at Available Treatment Options and Future Directions. Int. J. Mol. Sci. 2024, 25, 10765. [Google Scholar] [CrossRef]
- Rosińska, S.; Gavard, J. Tumor Vessels Fuel the Fire in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6514. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.J.; Gleeson, J.P.; O’Reilly, S.; Bambury, R.M. Management of Newly Diagnosed Glioblastoma Multiforme: Current State of the Art and Emerging Therapeutic Approaches. Med. Oncol. 2022, 39, 129. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Moen, M.D. Bevacizumab: In Previously Treated Glioblastoma. Drugs 2010, 70, 181–189. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic Signaling Pathways and Anti-Angiogenic Therapy for Cancer. Signal Transduct. Target. Ther. 2023, 8, 1–39. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Reardon, D.A.; de Groot, J.F.; Wick, W.; Weller, M. Antiangiogenic Therapy for Glioblastoma: Current Status and Future Prospects. Clin. Cancer Res. 2014, 20, 5612–5619. [Google Scholar] [CrossRef]
- Ghiaseddin, A.; Peters, K.B. Use of Bevacizumab in Recurrent Glioblastoma. CNS Oncol. 2015, 4, 157–169. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.A.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination with Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II Trial of Single-Agent Bevacizumab Followed by Bevacizumab Plus Irinotecan at Tumor Progression in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Ren, X.; Ai, D.; Li, T.; Xia, L.; Sun, L. Effectiveness of Lomustine Combined With Bevacizumab in Glioblastoma: A Meta-Analysis. Front. Neurol. 2021, 11, 603947. [Google Scholar] [CrossRef] [PubMed]
- Tsien, C.I.; Pugh, S.L.; Dicker, A.P.; Raizer, J.J.; Matuszak, M.M.; Lallana, E.C.; Huang, J.; Algan, O.; Deb, N.; Portelance, L.; et al. NRG Oncology/RTOG1205: A Randomized Phase II Trial of Concurrent Bevacizumab and Reirradiation Versus Bevacizumab Alone as Treatment for Recurrent Glioblastoma. J. Clin. Oncol. 2023, 41, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.E.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.J.; Buter, J.; Honkoop, A.H.; Boerman, D.; de Vos, F.Y.F.; et al. Single-Agent Bevacizumab or Lomustine versus a Combination of Bevacizumab plus Lomustine in Patients with Recurrent Glioblastoma (BELOB Trial): A Randomised Controlled Phase 2 Trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- She, L.; Su, L.; Liu, C. Bevacizumab Combined with Re-Irradiation in Recurrent Glioblastoma. Front. Oncol. 2022, 12, 961014. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef]
- Sandmann, T.; Bourgon, R.; Garcia, J.; Li, C.; Cloughesy, T.; Chinot, O.L.; Wick, W.; Nishikawa, R.; Mason, W.; Henriksson, R.; et al. Patients With Proneural Glioblastoma May Derive Overall Survival Benefit From the Addition of Bevacizumab to First-Line Radiotherapy and Temozolomide: Retrospective Analysis of the AVAglio Trial. J. Clin. Oncol. 2015, 33, 2735–2744. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Rogers, L.R.; Chaudhary, R.; Newton, H.; Ozair, A.; Khosla, A.A.; Nixon, A.B.; Adams, B.J.; Seon, B.K.; Peereboom, D.M.; et al. Endoglin Inhibitor TRC105 with or without Bevacizumab for Bevacizumab-Refractory Glioblastoma (ENDOT): A Multicenter Phase II Trial. Commun. Med. 2023, 3, 120. [Google Scholar] [CrossRef]
- Lombardi, G.; Caccese, M.; Padovan, M.; Cerretti, G.; Pintacuda, G.; Manara, R.; Di Sarra, F.; Zagonel, V. Regorafenib in Recurrent Glioblastoma Patients: A Large and Monocentric Real-Life Study. Cancers 2021, 13, 4731. [Google Scholar] [CrossRef]
- Caccese, M.; Desideri, I.; Villani, V.; Simonelli, M.; Buglione, M.; Chiesa, S.; Franceschi, E.; Gaviani, P.; Stasi, I.; Caserta, C.; et al. REGOMA-OSS: A Large, Italian, Multicenter, Prospective, Observational Study Evaluating the Efficacy and Safety of Regorafenib in Patients with Recurrent Glioblastoma. ESMO Open 2024, 9, 102943. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Ben Aissa, A.; Espeli, V.; Squiban, D.; Dunkel, N.; Vargas, M.I.; Hundsberger, T.; Mach, N.; Schaller, K.; Weber, D.C.; et al. Phase I Study of Sorafenib Combined with Radiation Therapy and Temozolomide as First-Line Treatment of High-Grade Glioma. Br. J. Cancer 2014, 110, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, S.; Ferrari, V.D.; Buglione, M.; Agazzi, G.M.; Liserre, R.; Poliani, L.; Buttolo, L.; Gipponi, S.; Pedersini, R.; Consoli, F.; et al. Second Line Treatment of Recurrent Glioblastoma with Sunitinib: Results of a Phase II Study and Systematic Review of Literature. J. Neurosurg. Sci. 2019, 63, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Vredenburgh, J.J.; Coan, A.; Desjardins, A.; Peters, K.B.; Gururangan, S.; Sathornsumetee, S.; Rich, J.N.; Herndon, J.E.; Friedman, H.S. Phase I Study of Sunitinib and Irinotecan for Patients with Recurrent Malignant Glioma. J. Neurooncol. 2011, 105, 621–627. [Google Scholar] [CrossRef]
- de Groot, J.F.; Lamborn, K.R.; Chang, S.M.; Gilbert, M.R.; Cloughesy, T.F.; Aldape, K.; Yao, J.; Jackson, E.F.; Lieberman, F.; Robins, H.I.; et al. Phase II Study of Aflibercept in Recurrent Malignant Glioma: A North American Brain Tumor Consortium Study. J. Clin. Oncol. 2011, 29, 2689–2695. [Google Scholar] [CrossRef]
- Nayak, L.; de Groot, J.; Wefel, J.S.; Cloughesy, T.F.; Lieberman, F.; Chang, S.M.; Omuro, A.; Drappatz, J.; Batchelor, T.T.; DeAngelis, L.M.; et al. Phase I Trial of Aflibercept (VEGF Trap) with Radiation Therapy and Concomitant and Adjuvant Temozolomide in Patients with High-Grade Gliomas. J. Neurooncol. 2017, 132, 181–188. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib Compared with Lomustine in Patients with Relapsed Glioblastoma (REGOMA): A Multicentre, Open-Label, Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Janssen, J.B.E.; Brahm, C.G.; Driessen, C.M.L.; Nuver, J.; Labots, M.; Kouwenhoven, M.C.M.; Sanchez Aliaga, E.; Enting, R.H.; de Groot, J.C.; Walenkamp, A.M.E.; et al. The STELLAR Trial: A Phase II/III Randomized Trial of High-Dose, Intermittent Sunitinib in Patients with Recurrent Glioblastoma. Brain Commun. 2024, 6, fcae241. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III Randomized Trial Comparing the Efficacy of Cediranib as Monotherapy, and in Combination with Lomustine, versus Lomustine Alone in Patients with Recurrent Glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef]
- Xiong, W.; Li, C.; Kong, G.; Wan, B.; Wang, S.; Fan, J. Glioblastoma: Two Immune Subtypes under the Surface of the Cold Tumor. Aging 2022, 14, 4357–4375. [Google Scholar] [CrossRef]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic Cancer Vaccine and Combinations With Antiangiogenic Therapies and Immune Checkpoint Blockade. Front. Immunol. 2019, 10, 467. [Google Scholar] [CrossRef]
- Weathers, S.-P.; Han, X.; Liu, D.D.; Conrad, C.A.; Gilbert, M.R.; Loghin, M.E.; O’Brien, B.J.; Penas-Prado, M.; Puduvalli, V.K.; Tremont-Lukats, I.; et al. A Randomized Phase II Trial of Standard Dose Bevacizumab versus Low Dose Bevacizumab plus Lomustine (CCNU) in Adults with Recurrent Glioblastoma. J. Neurooncol. 2016, 129, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Rodà, F.; Caraffi, R.; Picciolini, S.; Tosi, G.; Vandelli, M.A.; Ruozi, B.; Bedoni, M.; Ottonelli, I.; Duskey, J.T. Recent Advances on Surface-Modified GBM Targeted Nanoparticles: Targeting Strategies and Surface Characterization. Int. J. Mol. Sci. 2023, 24, 2496. [Google Scholar] [CrossRef] [PubMed]
- Lo Dico, A.; Martelli, C.; Diceglie, C.; Lucignani, G.; Ottobrini, L. Hypoxia-Inducible Factor-1α Activity as a Switch for Glioblastoma Responsiveness to Temozolomide. Front. Oncol. 2018, 8, 249. [Google Scholar] [CrossRef] [PubMed]
- Hassn Mesrati, M.; Behrooz, A.B.; Abuhamad, A.Y.; Syahir, A. Understanding Glioblastoma Biomarkers: Knocking a Mountain with a Hammer. Cells 2020, 9, 1236. [Google Scholar] [CrossRef]
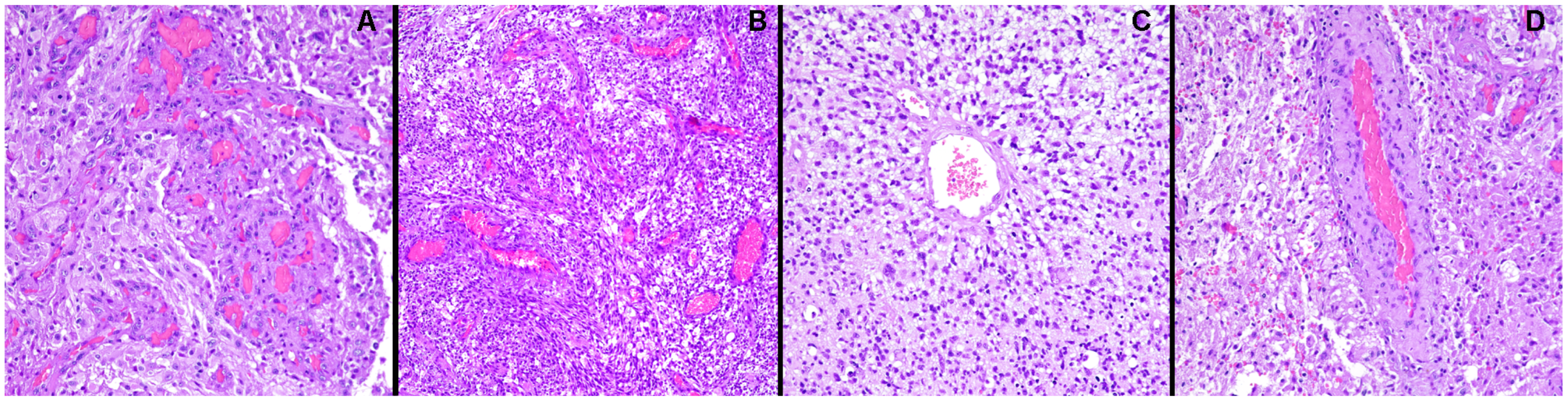
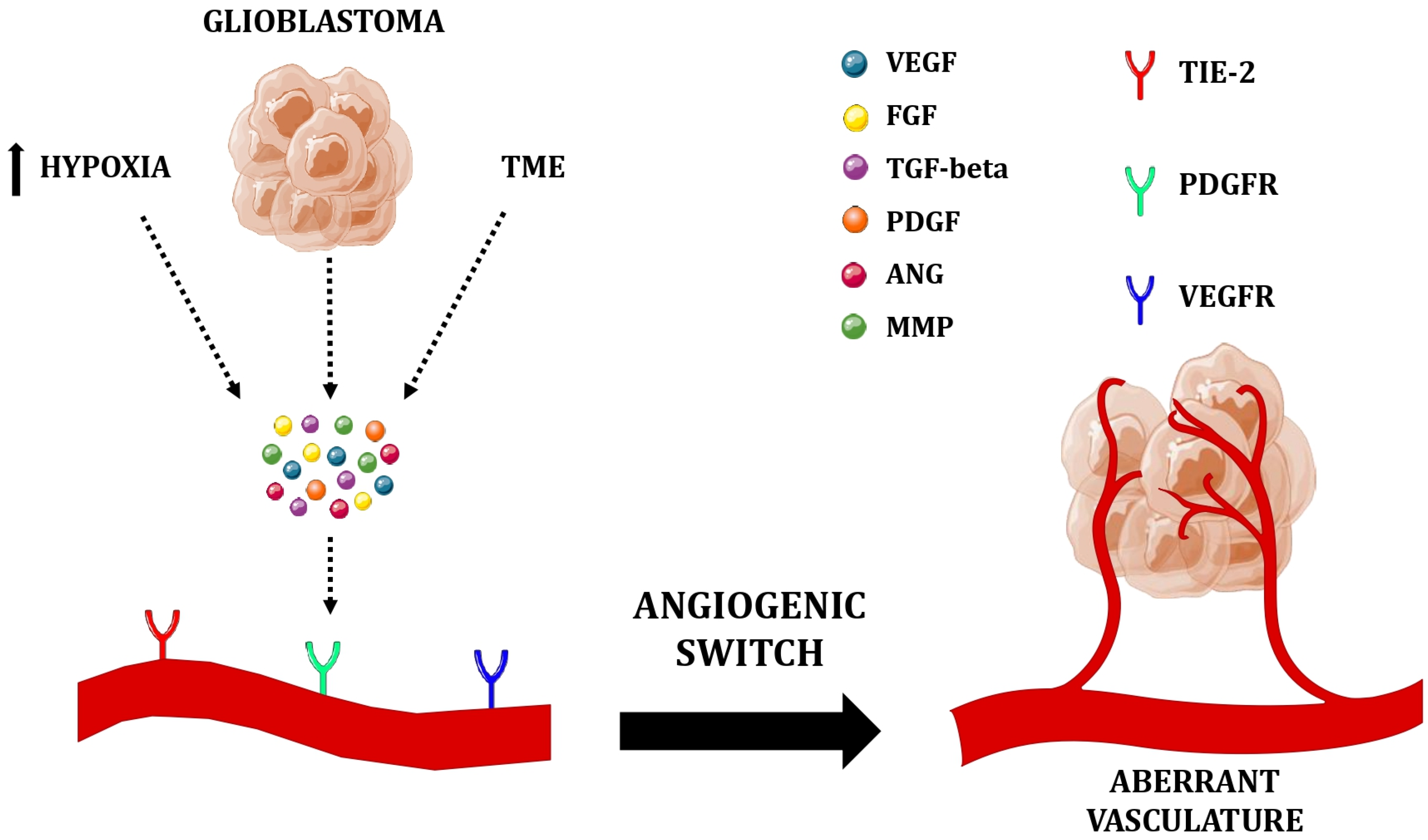

{kind=link}
{kind=link}
| Trial Name/ Author | Treatment | Phase | No. of Patients | Dose | 6-Months PFS | Median OS | Adverse Effects |
|---|---|---|---|---|---|---|---|
| BRAIN [268] | BEV alone/ BEV+ Irinotecan | II | 167 patients with recurrent GBM | 10 mg/kg every 2 weeks of BEV alone or 10 mg/kg of BEV + 340 mg/m2 or 125 mg/m2 of Irinotecan (if taking or not taking enzyme-inducing antiepileptic drugs, respectively) every 2 weeks | 42.6% BEV alone 50.3% BEV+ Irinotecan | 9.2 months BEV alone 8.7 months BEV+ Irinotecan | Hypertension, fatigue, headache, convulsion, diarrhea, nausea, and neutropenia |
| NCI 06-C-0064E [269] | BEV | II | 48 patients with recurrent GBM | 10 mg/kg every 2 weeks | 29% | 31 weeks | Thromboembolic events, hypertension, hypophosphatemia, and thrombocytopenia |
| BELOB [273] | BEV/ Lomustine alone or BEV + Lomustine | II | 153 patients with recurrent GBM | 110 mg/m2 every 6 weeks of Lomustine alone or 10 mg/kg every 2 weeks of BEV alone or 90/110 mg/m2 every 6 weeks of Lomustine combined with 10 mg/kg every 2 weeks of BEV | 13% Lomustine alone 16% BEV alone 42% BEV + Lomustine | 8 months Lomustine alone 8 months BEV alone 12 months BEV+ Lomustine | Hypertension, fatigue, and infections |
| EORTC 26101 [275] | BEV + Lomustine or Lomustine alone | III | 437 patients with progressive GBM | 10 mg/kg every 2 weeks of BEV + 90 mg/m2 every 6 weeks of Lomustine or 110 mg/m2 every 6 weeks of Lomustine alone | Not available | 9.1 months BEV + Lomustine 8.6 months Lomustine alone | Pulmonary embolism, arterial hypertension, and hematologic toxic effects |
| RTOG 0825 [263] | BEV/placebo + TMZ + radiotherapy | III | 637 patients with newly diagnosed GBM | 10 mg/kg every 2 weeks of BEV + TMZ + radiotherapy | Not available | 15.7 months | Lymphopenia, neutropenia, fatigue, and thrombocytopenia |
| AVAglio [276] | BEV/placebo + TMZ + radiotherapy | III | 921 patients with newly diagnosed GBM | 10 mg/kg every 2 weeks of BEV + TMZ + radiotherapy | Not available | 16.8 months BEV 16.7 months placebo | Hypertension, proteinuria, thromboembolia, and wound healing complications |
| ENDOT [278] | Carotuximab (TRC105) alone or TRC105 + BEV following radiation, TMZ, and BEV therapy | II | 22 patients with GBM that had progressed after chemoradiation | 10 mg/kg weekly TRC105 alone/ 10 mg/kg split into two doses with 3 mg/kg administered on cycle 1 day 8 and 7 mg/kg administered on cycle 1 day 11 with TRC105 + BEV | 13.3% (calculated for the 15 evaluable patients treated with BEV + TRC105) | 5.7 months (calculated for the 15 evaluable patients treated with BEV + TRC105) | Headache, epistaxis, fatigue, TIA, lower leg edema, pulmonary embolism, and sinusitis |
| REGOMA [286] | Regorafenib or Lomustine | II | 119 patients with recurrent GBM | 160 mg once daily for the first 3 weeks of each 4-week cycle of Regorafenib or 110 mg/m2 every 6 weeks of Lomustine | Not available | 7.4 months in the Regorafenib group/ 5.6 months in the Lomustine group | Hand–foot skin reaction, increased lipase, increased blood bilirubin, decreased platelet/ lymphocyte count, and neutropenia |
| Hottinger, A.F [281] | Sorafenib + TMZ + radiotherapy | I | 17 patients with newly diagnosed GBM | 400 mg 2 times daily | 86.7% | 17.8 months | Thrombocytopenia, neutropenia, alopecia, nausea, vomiting, hypophosphatemia, and fatigue |
| STELLAR [287] | Sunitinib or Lomustine (in the control arm) | I | 32 patients with recurrent GBM in part I 37 patients with recurrent GBM in part II | Part I 300 mg Q1W of Sunitinib or 110 mg/m2 of Lomustine once every six weeks Part II 700 mg Q2W of Sunitinib or 110 mg/m2 of Lomustine once every six weeks | 8% in part I with Sunitinib vs. 29% with Lomustine 14% in part II with Sunitinib vs. 15% with Lomustine | 6.5 months in part I with Sunitinib vs. 4.7 months with Lomustine 4.7 months in part II with Sunitinib vs. 7 months with Lomustine | Thrombocytopenia, fatigue, leukopenia, diarrhea, nausea, and vomiting |
| REGAL [288] | Cediranib alone/in combination with Lomustine vs. Lomustine plus placebo | III | 325 patients with recurrent GBM | Cediranib alone (30 mg), Cediranib (20 mg) + Lomustine (110 mg/m2), or Lomustine (110 mg/m2) + placebo | 16% Cediranib alone 35% Cediranib + Lomustine 25% Lomustine alone | 8.0 months Cediranib alone, 9.4 months Cediranib + Lomustine 9.8 months Lomustine alone | Diarrhea, thrombocytopenia, neutropenia, and hypertension |
| De Groot, J.F [284] | Aflibercept | II | 42 patients with GBM and 16 patients with anaplastic glioma | 4 mg/kg on day 1 of every 2-week cycle. | 7.7% for GBM | 39 weeks | Fatigue, thromboembolia, wound healing, and CNS ischemia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballato, M.; Germanà, E.; Ricciardi, G.; Giordano, W.G.; Tralongo, P.; Buccarelli, M.; Castellani, G.; Ricci-Vitiani, L.; D’Alessandris, Q.G.; Giuffrè, G.; et al. Understanding Neovascularization in Glioblastoma: Insights from the Current Literature. Int. J. Mol. Sci. 2025, 26, 2763. https://doi.org/10.3390/ijms26062763
Ballato M, Germanà E, Ricciardi G, Giordano WG, Tralongo P, Buccarelli M, Castellani G, Ricci-Vitiani L, D’Alessandris QG, Giuffrè G, et al. Understanding Neovascularization in Glioblastoma: Insights from the Current Literature. International Journal of Molecular Sciences. 2025; 26(6):2763. https://doi.org/10.3390/ijms26062763
Chicago/Turabian StyleBallato, Mariagiovanna, Emanuela Germanà, Gabriele Ricciardi, Walter Giuseppe Giordano, Pietro Tralongo, Mariachiara Buccarelli, Giorgia Castellani, Lucia Ricci-Vitiani, Quintino Giorgio D’Alessandris, Giuseppe Giuffrè, and et al. 2025. "Understanding Neovascularization in Glioblastoma: Insights from the Current Literature" International Journal of Molecular Sciences 26, no. 6: 2763. https://doi.org/10.3390/ijms26062763
APA StyleBallato, M., Germanà, E., Ricciardi, G., Giordano, W. G., Tralongo, P., Buccarelli, M., Castellani, G., Ricci-Vitiani, L., D’Alessandris, Q. G., Giuffrè, G., Pizzimenti, C., Fiorentino, V., Zuccalà, V., Ieni, A., Caffo, M., Fadda, G., & Martini, M. (2025). Understanding Neovascularization in Glioblastoma: Insights from the Current Literature. International Journal of Molecular Sciences, 26(6), 2763. https://doi.org/10.3390/ijms26062763