
Cultured Macrophage Models for the Investigation of Lysosomal Glucocerebrosidase and Gaucher Disease

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
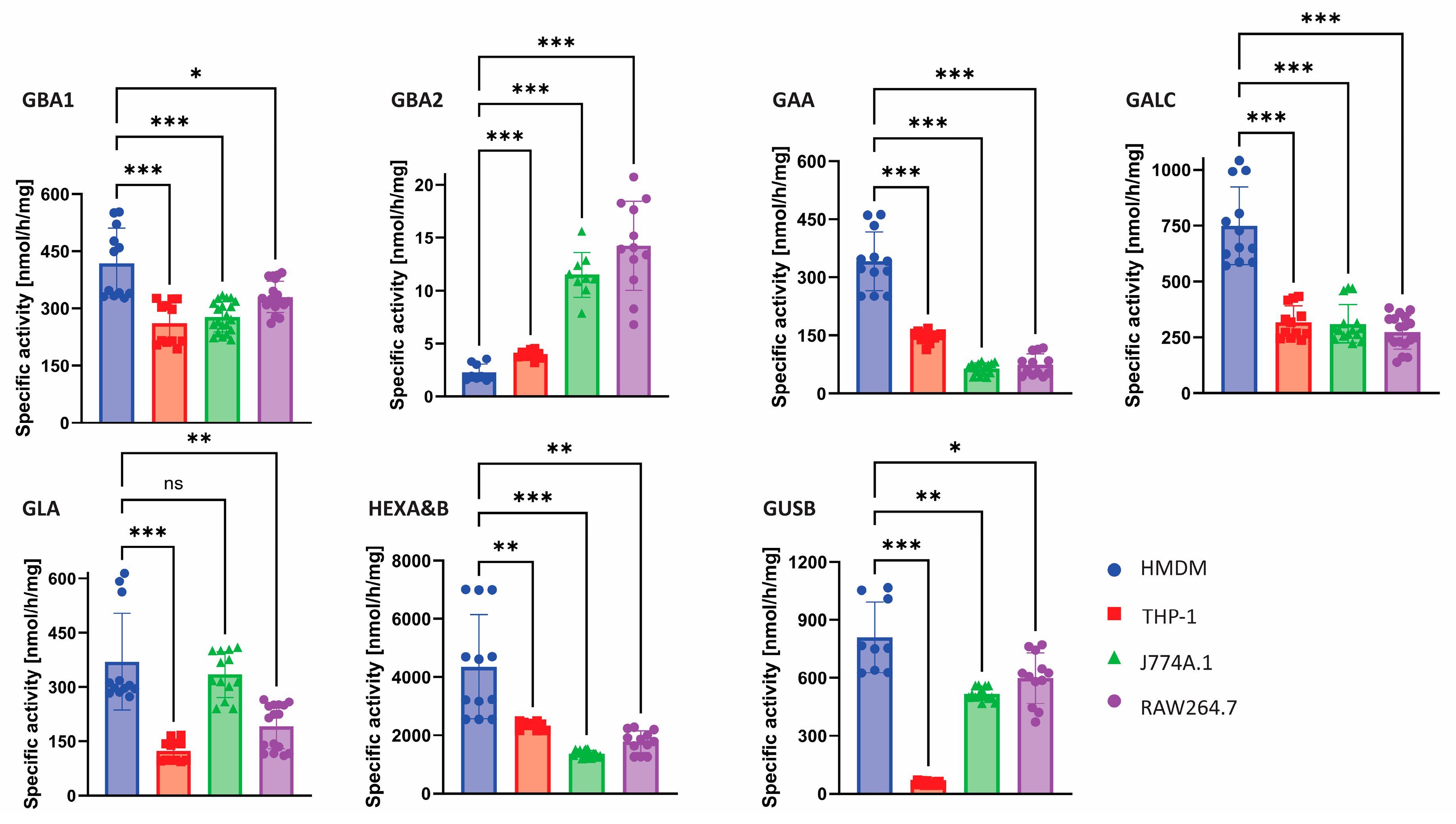
2.1. Glycosidase Activity Measurements
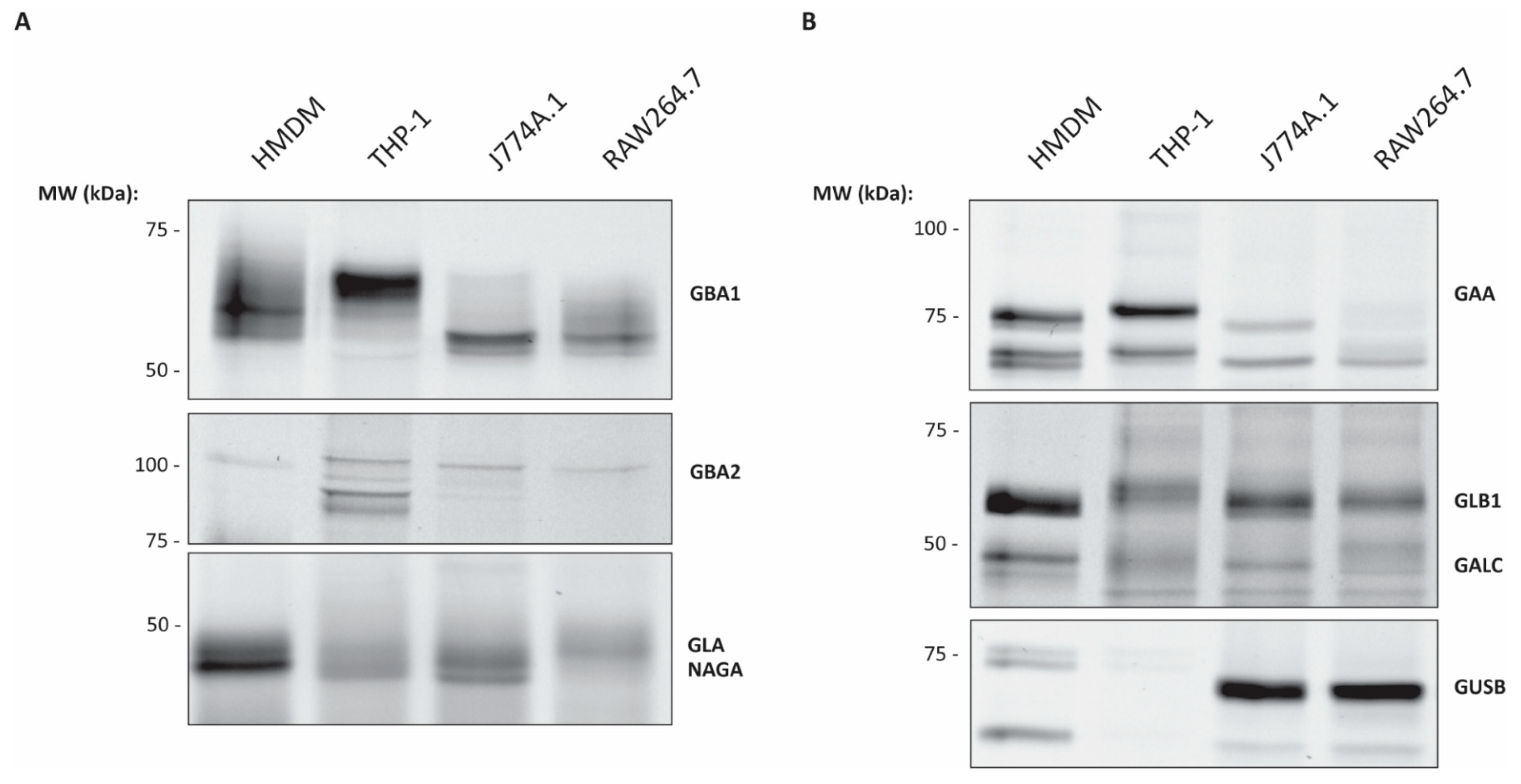
2.2. ABP Mediated Detection of Glycosidases in Various Macrophages
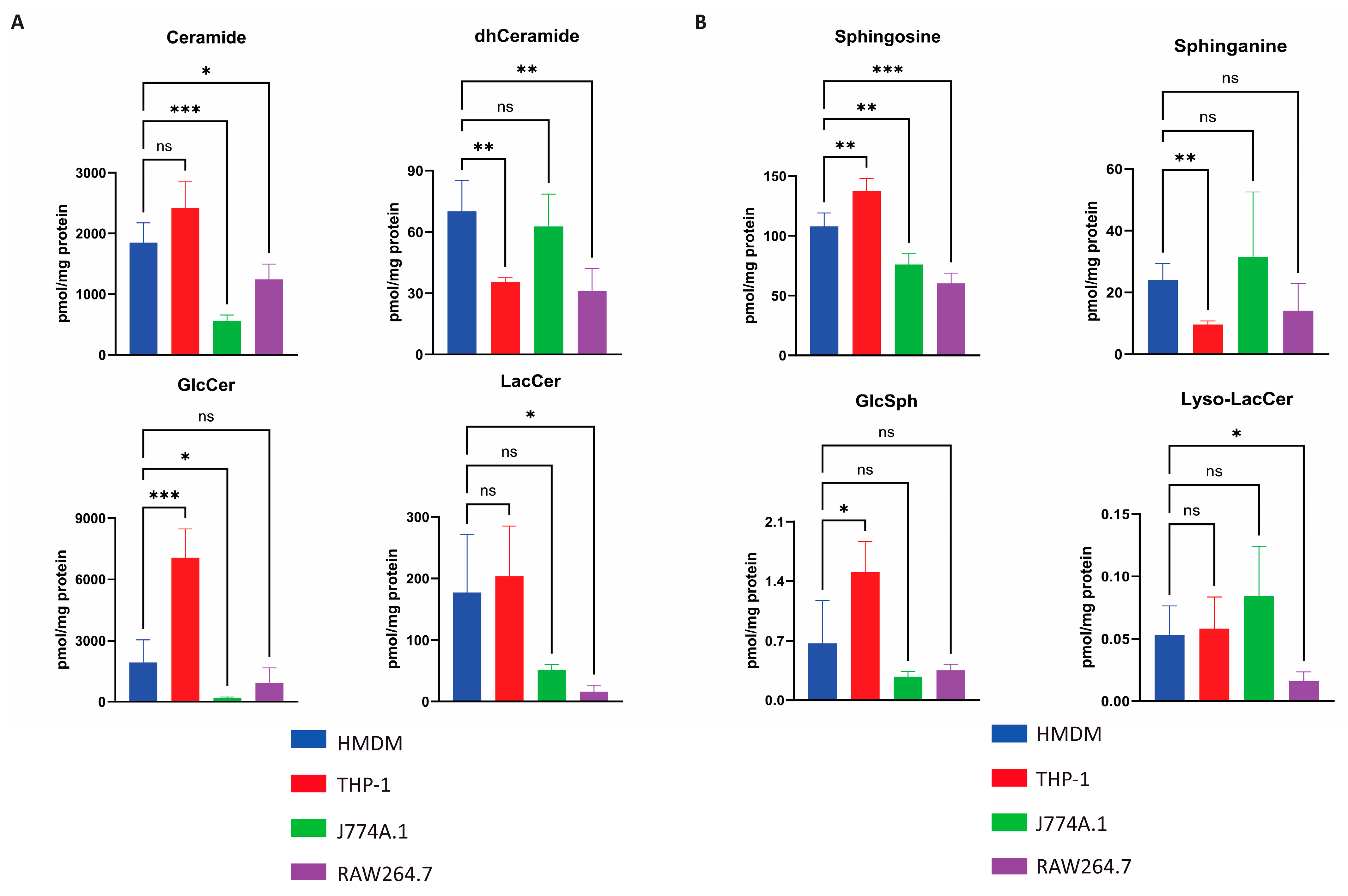
2.3. Quantification of Selected Glycosphingolipids in Various Macrophages
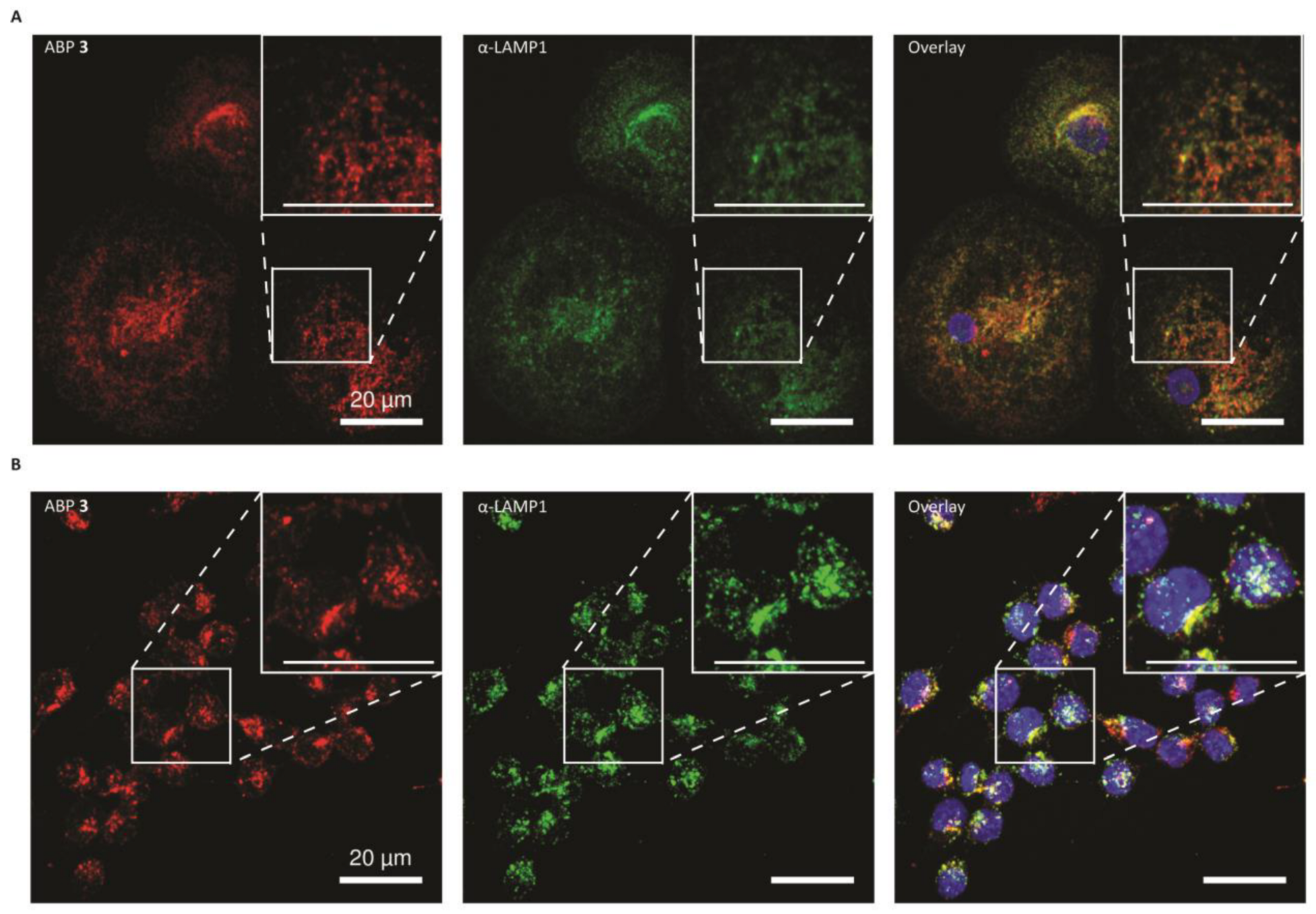
2.4. Glucocerebrosidase Distribution in HMDMs and RAW264.7 Cells
2.5. HMDMs with Inactivated Glucocerebrosidase as Model for Gaucher Disease
2.6. Recombinant Glucocerebrosidase Uptake in HMDMs
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Lysis
4.4. Primary Monocyte Isolation
4.5. Activity-Based Probe Analysis
4.6. Western Blotting
4.7. Recombinant Human Glucocerebrosidase Uptake
4.8. Enzyme Activity Assay
4.9. Confocal Microscopy
4.10. Glycosphingolipid Analysis
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LSDs | Lysosomal storage disorders |
| GD | Gaucher disease |
| GD1 | Non-neuropathic Gaucher disease type 1 |
| ERT | Enzyme replacement therapy |
| rhGBA1 | Recombinant human glucocerebrosidase |
| GlcCer | Glucosylceramide |
| GlcSph | Glucosylsphingosine |
| ABP | Activity-based probe |
| PMA | Phorbol-12-myristate-13-acetate |
| HMDMs | Human monocyte-derived macrophages |
| GAA | α-glucosidase |
| GBA1 | Glucocerebrosidase |
| GBA2 | Non-lysosomal β-glucosidase |
| GLA | α-galactosidase |
| GALC | β-galactosylceramidase |
| GUSB | β-glucuronidase |
| HEXA&B | β-hexosaminidases |
| GLB1 | Acid β-galactosidase |
| NAGA | α-N-acetylgalactosaminidase |
| GANAB | ER α-glucosidase II |
| MW | Molecular weight |
| LAMP1 | Lysosomal associated membrane protein 1 |
| Cer | Ceramide |
| dhCer | Dihydroceramide |
| LacCer | Lactosylceramide |
| Lyso-LacCer | Lyso-Lactocylceramide |
| HexSph | Hexosylsphingosine |
| HexCer | Hexosylceramide |
| Sph | Sphingosine |
| Spa | Sphinganine |
| GlcChol | Glucosylated cholesterol |
| HexChol | Hexosylcholesterol |
| MR | Mannose receptor |
References
- de Duve, C. The Lysosome Turns Fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as Dynamic Regulators of Cell and Organismal Homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M.; Cachón-González, M.B. The Cellular Pathology of Lysosomal Diseases. J. Pathol. 2012, 226, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Gaucher Disease and Other Storage Disorders. Hematology Am. Soc. Hematol. Educ. Program. 2012, 2012, 13–18. [Google Scholar] [CrossRef]
- Aerts, J.M.F.G.; Kuo, C.-L.; Lelieveld, L.T.; Boer, D.E.C.; van der Lienden, M.J.C.; Overkleeft, H.S.; Artola, M. Glycosphingolipids and Lysosomal Storage Disorders as Illustrated by Gaucher Disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. Lysosome: Regulator of Lipid Degradation Pathways. Trends Cell Biol. 2014, 24, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H. Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef]
- Poorthuis, B.J.H.M.; Wevers, R.A.; Kleijer, W.J.; Groener, J.E.M.; De Jong, J.G.N.; Van Weely, S.; Niezen-Koning, K.E.; Van Diggelen, O.P. The Frequency of Lysosomal Storage Diseases in The Netherlands. Hum. Genet. 1999, 105, 151–156. [Google Scholar] [CrossRef]
- Pinto, R.; Caseiro, C.; Lemos, M.; Lopes, L.; Fontes, A.; Ribeiro, H.; Pinto, E.; Silva, E.; Rocha, S.; Marcão, A.; et al. Prevalence of Lysosomal Storage Diseases in Portugal. Eur. J. Human. Genet. 2004, 12, 87–92. [Google Scholar] [CrossRef]
- Chin, S.J.; Fuller, M. Prevalence of Lysosomal Storage Disorders in Australia from 2009 to 2020. Lancet Reg. Health West Pac. 2022, 19, 100344. [Google Scholar] [CrossRef]
- Cox, T.M.; Schofield, J.P. Gaucher’s Disease: Clinical Features and Natural History. Baillieres Clin. Haematol. 1997, 10, 657–689. [Google Scholar] [CrossRef]
- Wenstrup, R.J.; Roca-Espiau, M.; Weinreb, N.J.; Bembi, B. Skeletal Aspects of Gaucher Disease: A Review. Br. J. Radiol. 2002, 75, A2–A12. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Horowitz, M.; Abel, R.B.; Currie, J.N.; Yu, K.-T.; Kaneski, C.; Higgins, J.J.; O’Neill, R.R.; Fedio, P.; Pikus, A.; et al. Isolated Horizontal Supranuclear Gaze Palsy as a Marker of Severe Systemic Involvement in Gaucher’s Disease. Neurology 1993, 43, 1993. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O. Gaucher’s Disease: Past, Present and Future. Baillieres Clin. Haematol. 1997, 10, 621–634. [Google Scholar] [CrossRef]
- Gupta, N.; Oppenheim, I.M.; Kauvar, E.F.; Tayebi, N.; Sidransky, E. Type 2 Gaucher Disease: Phenotypic Variation and Genotypic Heterogeneity. Blood Cells Mol. Dis. 2011, 46, 75–84. [Google Scholar] [CrossRef]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement Therapy for Inherited Enzyme Deficiency—Macrophage-Targeted Glucocerebrosidase for Gaucher’s Disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef]
- van Eijk, M.; Ferraz, M.J.; Boot, R.G.; Aerts, J.M.F.G. Lyso-Glycosphingolipids: Presence and Consequences. Essays Biochem. 2020, 64, 565–578. [Google Scholar] [CrossRef]
- Koshland, D.E. Sterochemistry and the Mechanism of Enzymatic Reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Artola, M.; Kuo, C.L.; Lelieveld, L.T.; Rowland, R.J.; Van Der Marel, G.A.; Codée, J.D.C.; Boot, R.G.; Davies, G.J.; Aerts, J.M.F.G.; Overkleeft, H.S. Functionalized Cyclophellitols Are Selective Glucocerebrosidase Inhibitors and Induce a Bona Fide Neuropathic Gaucher Model in Zebrafish. J. Am. Chem. Soc. 2019, 141, 4214–4218. [Google Scholar] [CrossRef]
- Kuo, C.-L.; Kallemeijn, W.W.; Lelieveld, L.T.; Mirzaian, M.; Zoutendijk, I.; Vardi, A.; Futerman, A.H.; Meijer, A.H.; Spaink, H.P.; Overkleeft, H.S.; et al. In Vivo Inactivation of Glycosidases by Conduritol B Epoxide and Cyclophellitol as Revealed by Activity-Based Protein Profiling. FEBS J. 2019, 286, 584–600. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.D.; Kallemeijn, W.W.; Aten, J.; Li, K.-Y.; Strijland, A.; Donker-Koopman, W.E.; van den Nieuwendijk, A.M.C.H.; Bleijlevens, B.; Kramer, G.; Florea, B.I.; et al. Ultrasensitive in Situ Visualization of Active Glucocerebrosidase Molecules. Nat. Chem. Biol. 2010, 6, 907–913. [Google Scholar] [CrossRef]
- Wu, L.; Armstrong, Z.; Schröder, S.P.; de Boer, C.; Artola, M.; Aerts, J.M.; Overkleeft, H.S.; Davies, G.J. An Overview of Activity-Based Probes for Glycosidases. Curr. Opin. Chem. Biol. 2019, 53, 25–36. [Google Scholar] [CrossRef]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.B.; Dockrell, D.H. The Identification of Markers of Macrophage Differentiation in PMA-Stimulated THP-1 Cells and Monocyte-Derived Macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef]
- Starr, T.; Bauler, T.J.; Malik-Kale, P.; Steele-Mortimer, O. The Phorbol 12-Myristate-13-Acetate Differentiation Protocol Is Critical to the Interaction of THP-1 Macrophages with Salmonella Typhimurium. PLoS ONE 2018, 13, e193601. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, M.A.; Kopytova, A.E.; Baidakova, G.V.; Emel’yanov, A.K.; Salogub, G.N.; Senkevich, K.A.; Usenko, T.S.; Gorchakova, M.V.; Koval’chuk, Y.P.; Berkovich, O.A.; et al. Human Peripheral Blood Macrophages As a Model for Studying Glucocerebrosidase Dysfunction. Cell Tissue Biol. 2019, 13, 100–106. [Google Scholar] [CrossRef]
- Boot, R.G.; Verhoek, M.; Donker-Koopman, W.; Strijland, A.; van Marle, J.; Overkleeft, H.S.; Wennekes, T.; Aerts, J.M.F.G. Identification of the Non-Lysosomal Glucosylceramidase as β-Glucosidase 2. J. Biol. Chem. 2007, 282, 1305–1312. [Google Scholar] [CrossRef]
- Erickson, A.H.; Ginns, E.I.; Barranger, J.A. Biosynthesis of the Lysosomal Enzyme Glucocerebrosidase. J. Biol. Chem. 1985, 260, 14319–14324. [Google Scholar] [CrossRef]
- Van Weely, S.; Aerts, J.M.; Van Leeuwen, M.B.; Heikoop, J.C.; Donker-Koopman, W.E.; Barranger, J.A.; Tager, J.M.; Schram, A.W. Function of Oligosaccharide Modification in Glucocerebrosidase, a Membrane-Associated Lysosomal Hydrolase. Eur. J. Biochem. 1990, 191, 669–677. [Google Scholar] [CrossRef]
- Matern, H.; Boermans, H.; Matern, S.; Lottspeich, F. Molecular Cloning and Expression of Human Bile Acid β-Glucosidase. J. Biol. Chem. 2001, 276, 37929–37933. [Google Scholar] [CrossRef]
- Kallemeijn, W.W.; Li, K.; Witte, M.D.; Marques, A.R.A.; Aten, J.; Scheij, S.; Jiang, J.; Willems, L.I.; Voorn-Brouwer, T.M.; van Roomen, C.P.A.A.; et al. Novel Activity-Based Probes for Broad-Spectrum Profiling of Retaining Β-Exoglucosidases In Situ and In Vivo. Angew. Chem. Int. Ed. 2012, 51, 12529–12533. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-L.; Su, Q.; van den Nieuwendijk, A.M.C.H.; Beenakker, T.J.M.; Offen, W.A.; Willems, L.I.; Boot, R.G.; Sarris, A.J.; Marques, A.R.A.; Codée, J.D.C.; et al. The Development of a Broad-Spectrum Retaining β-Exo-Galactosidase Activity-Based Probe. Org. Biomol. Chem. 2023, 21, 7813–7820. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Grubb, J.H.; Sly, W.S. C-Terminal Processing of Human Beta-Glucuronidase. The Propeptide Is Required for Full Expression of Catalytic Activity, Intracellular Retention, and Proper Phosphorylation. J. Biol. Chem. 1993, 268, 22627–22633. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked Elevation of Plasma Chitotriosidase Activity. A Novel Hallmark of Gaucher Disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Mirzaian, M.; Akiyama, H.; Wisse, P.; Ferraz, M.J.; Gaspar, P.; Ghauharali-van der Vlugt, K.; Meijer, R.; Giraldo, P.; Alfonso, P.; et al. Glucosylated Cholesterol in Mammalian Cells and Tissues: Formation and Degradation by Multiple Cellular β-Glucosidases. J. Lipid Res. 2016, 57, 451–463. [Google Scholar] [CrossRef]
- Doebber, T.W.; Wu, M.S.; Bugianesi, R.L.; Ponpipom, M.M.; Furbish, F.S.; Barranger, J.A.; Brady, R.O.; Shen, T.Y. Enhanced Macrophage Uptake of Synthetically Glycosylated Human Placental Beta-Glucocerebrosidase. J. Biol. Chem. 1982, 257, 2193–2199. [Google Scholar] [CrossRef]
- Friedman, B.; Vaddi, K.; Preston, C.; Mahon, E.; Cataldo, J.R.; McPherson, J.M. A Comparison of the Pharmacological Properties of Carbohydrate Remodeled Recombinant and Placental-Derived β-Glucocerebrosidase: Implications for Clinical Efficacy in Treatment of Gaucher Disease. Blood 1999, 93, 2807–2816. [Google Scholar] [CrossRef]
- Taciak, B.; Białasek, M.; Braniewska, A.; Sas, Z.; Sawicka, P.; Kiraga, Ł.; Rygiel, T.; Król, M. Evaluation of Phenotypic and Functional Stability of RAW 264.7 Cell Line through Serial Passages. PLoS ONE 2018, 13, e0198943. [Google Scholar] [CrossRef]
- Ralph, P.; Prichard, J.; Cohn, M. Reticulum Cell Sarcoma: An Effector Cell in Antibody-Dependent Cell-Mediated Immunity. J. Immunol. 1975, 114, 898–905. [Google Scholar] [CrossRef]
- Raschke, W.C.; Baird, S.; Ralph, P.; Nakoinz, I. Functional Macrophage Cell Lines Transformed by Abelson Leukemia Virus. Cell 1978, 15, 261–267. [Google Scholar] [CrossRef]
- Brady, R.O.; Murray, G.J.; Barton, N.W. Modifying Exogenous Glucocerebrosidase for Effective Replacement Therapy in Gaucher Disease. J. Inherit. Metab. Dis. 1994, 17, 510–519. [Google Scholar] [CrossRef] [PubMed]
- ATCC, TIB-71. Available online: https://www.atcc.org/products/tib-71 (accessed on 13 March 2025).
- ATCC, TIB-67. Available online: https://www.atcc.org/products/tib-67 (accessed on 13 March 2025).
- ATCC, TIB-202. Available online: https://www.atcc.org/products/tib-202 (accessed on 13 March 2025).
- van Eijk, M.; van Roomen, C.P.A.A.; Renkema, G.H.; Bussink, A.P.; Andrews, L.; Blommaart, E.F.C.; Sugar, A.; Verhoeven, A.J.; Boot, R.G.; Aerts, J.M.F.G. Characterization of Human Phagocyte-Derived Chitotriosidase, a Component of Innate Immunity. Int. Immunol. 2005, 17, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.F.G.; Donker-Koopman, W.E.; Murray, G.J.; Barranger, J.A.; Tager, J.M.; Schram, A.W. A Procedure for the Rapid Purification in High Yield of Human Glucocerebrosidase Using Immunoaffinity Chromatography with Monoclonal Antibodies. Anal. Biochem. 1986, 154, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Martino, S.; Tiribuzi, R.; Tortori, A.; Conti, D.; Visigalli, I.; Lattanzi, A.; Biffi, A.; Gritti, A.; Orlacchio, A. Specific Determination of β-Galactocerebrosidase Activity via Competitive Inhibition of β-Galactosidase. Clin. Chem. 2009, 55, 541–548. [Google Scholar] [CrossRef]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Marques, A.R.A.; Gaspar, P.; Oussoren, S.V.; Kytidou, K.; Codée, J.D.C.; van der Marel, G.; Overkleeft, H.S.; et al. Simultaneous Quantitation of Sphingoid Bases by UPLC-ESI-MS/MS with Identical 13 C-Encoded Internal Standards. Clin. Chim. Acta 2017, 466, 178–184. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louwerse, M.; Bila, K.O.; van der Lienden, M.J.C.; de Beaufort, A.J.M.; Boot, R.G.; Artola, M.; van Eijk, M.; Aerts, J.M.F.G. Cultured Macrophage Models for the Investigation of Lysosomal Glucocerebrosidase and Gaucher Disease. Int. J. Mol. Sci. 2025, 26, 2726. https://doi.org/10.3390/ijms26062726
Louwerse M, Bila KO, van der Lienden MJC, de Beaufort AJM, Boot RG, Artola M, van Eijk M, Aerts JMFG. Cultured Macrophage Models for the Investigation of Lysosomal Glucocerebrosidase and Gaucher Disease. International Journal of Molecular Sciences. 2025; 26(6):2726. https://doi.org/10.3390/ijms26062726
Chicago/Turabian StyleLouwerse, Max, Kateryna O. Bila, Martijn J. C. van der Lienden, Arnout Jan M. de Beaufort, Rolf G. Boot, Marta Artola, Marco van Eijk, and Johannes M. F. G. Aerts. 2025. "Cultured Macrophage Models for the Investigation of Lysosomal Glucocerebrosidase and Gaucher Disease" International Journal of Molecular Sciences 26, no. 6: 2726. https://doi.org/10.3390/ijms26062726
APA StyleLouwerse, M., Bila, K. O., van der Lienden, M. J. C., de Beaufort, A. J. M., Boot, R. G., Artola, M., van Eijk, M., & Aerts, J. M. F. G. (2025). Cultured Macrophage Models for the Investigation of Lysosomal Glucocerebrosidase and Gaucher Disease. International Journal of Molecular Sciences, 26(6), 2726. https://doi.org/10.3390/ijms26062726