
Deciphering the Genetic Basis of Degenerative and Developmental Eye Disorders in 50 Pakistani Consanguineous Families Using Whole-Exome Sequencing

 , and
, and
Abstract
1. Introduction
2. Results
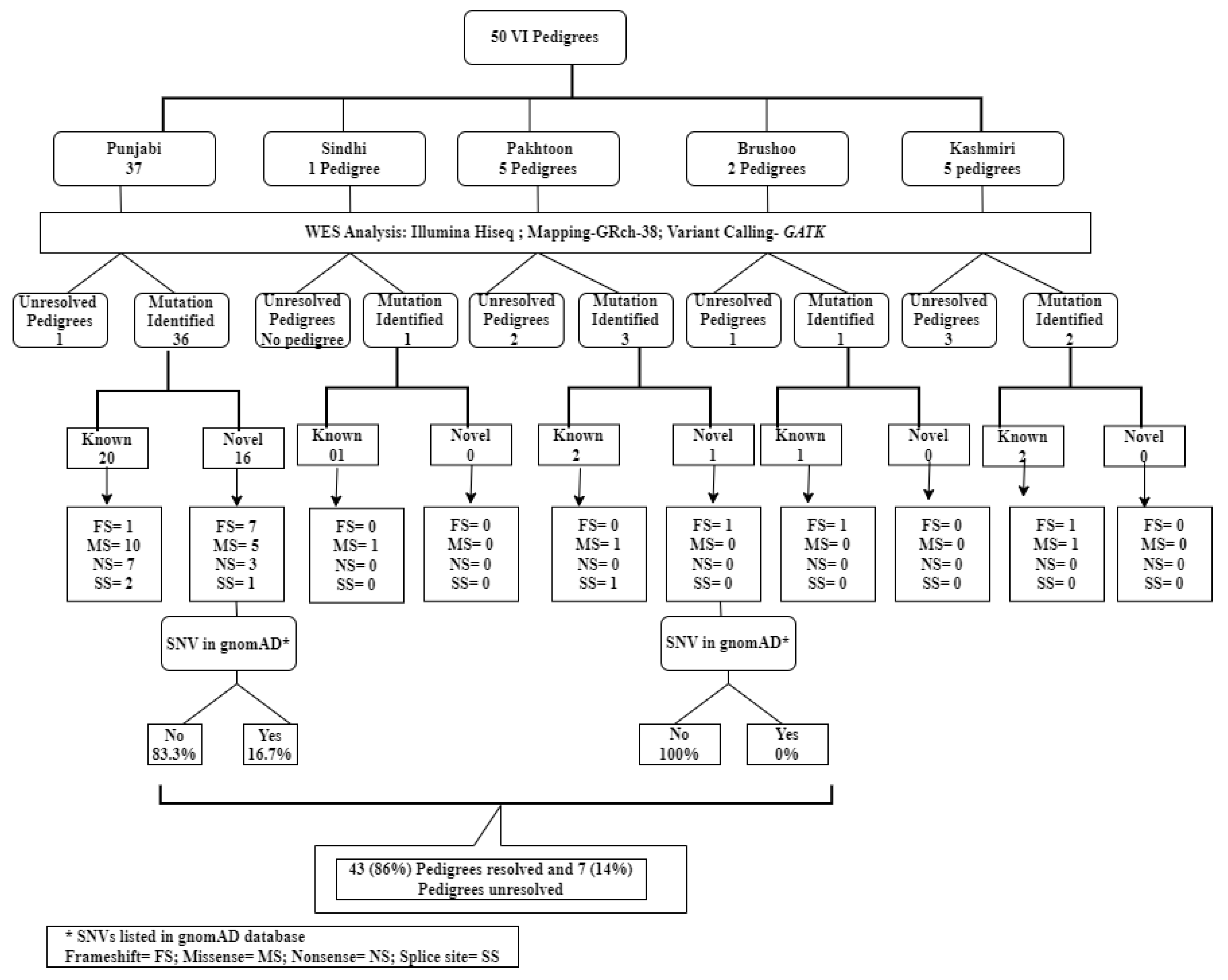
2.1. Pedigrees Analyzed
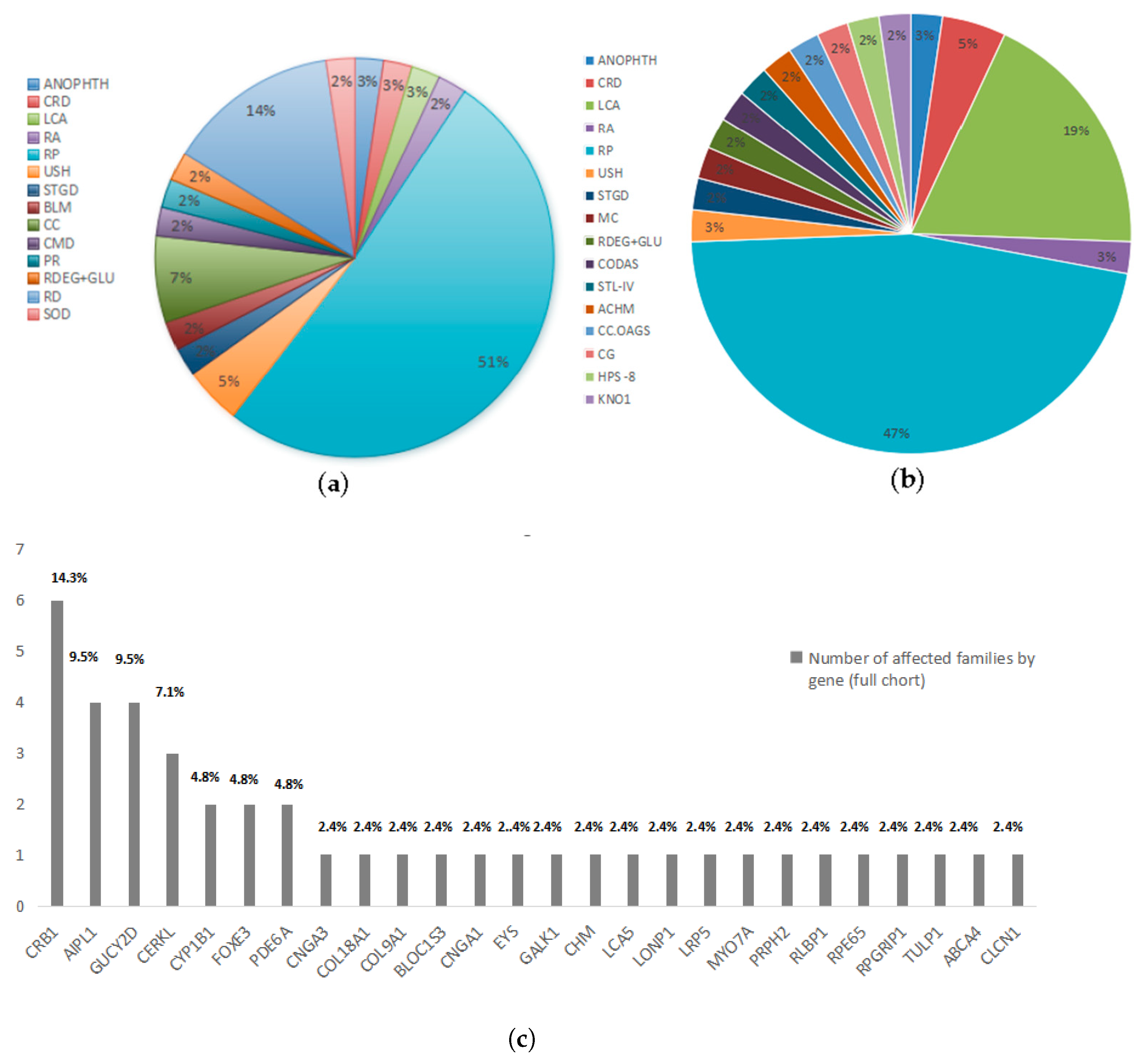
2.2. Reclassification of Presenting Clinical Phenotypes Based on WES Analysis Results
2.3. Previously Reported Variants Identified in Known IRD Genes
2.4. Novel Potentially Causative Variants Detected in Known IRD Genes
2.4.1. Non-Sense Mutations Observed in Unrelated IRD Pedigrees
2.4.2. Novel Frameshift Variants Detected in Known IRD Genes
- A 20 bps homozygous frameshift deletion in COL18A1 (NM_030582.4):c.3559_3577del p.(Ser1187AlafsTer18) was detected in a large Punjabi consanguineous pedigree Ref. MA0468 Figure S6. This homozygous deletion included 20 bps of exon 36. The deletion of the 20 bps sequence in exon 36 of COL18A1 might produce a truncated and consequently non-functional protein owing to the frameshift of the coding region. PCR amplification and Sanger sequencing of the deleted region using primers located in the flanking sequence identified these specific boundaries of the frame shift deletion and its further segregation with Knobloch syndrome in the RF.MA0468 pedigree. The first affected individual presented with microphthalmia, early childhood photophobia, congenital cataract, severe visual impairment with light perception, bilateral retinal detachment, and chorioretinal atrophy. The second affected individual exhibited congenital strabismus, early childhood photophobia, corneal opacity, congenital cataract, unilateral retinal detachment, and chorioretinal atrophy. Notably, night blindness was absent in both individuals. While some of these features (e.g., retinal detachment, chorioretinal atrophy) are consistent with Knobloch syndrome, others are more unusual (e.g., microphthalmia, lack of nyctalopia) but may represent the phenotypic heterogeneity of this condition. MetaDome analysis revealed that the truncation is in a mildly intolerant region 20 residues before the endostatin domain. This deletion likely leads to the loss of the endostatin domain in collagen type XVIII alpha chain, which is associated with Knobloch syndrome. As endostatin binds zinc near its N-terminus, the mutation may impact the structure rather than the function [15].
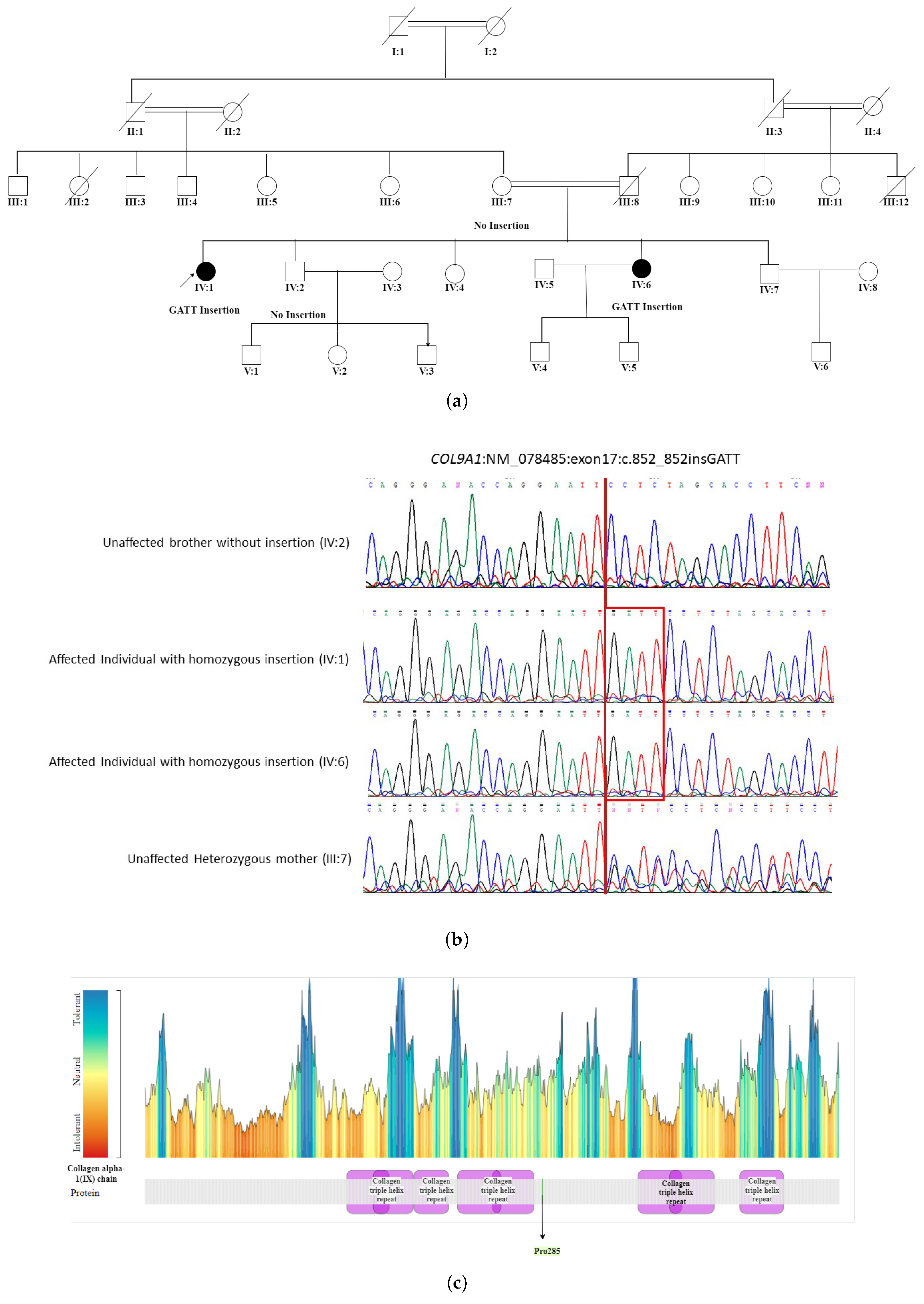
- The WES in patient IV:1 (proband) of a five-generation pedigree RF. MA0537 Figure 5 identified a novel homozygous nonsense mutation c.851_852insCAAT, p. Pro285AsnfsTer20 in the COL9A1 gene. This novel sequence change led to a premature stop signal p. Pro285AsnfsTer20 in the COL9A1 gene. This resulted in an expected disrupted or absent protein product. The family was initially suspected to have Usher syndrome due to hearing loss in both affected individuals. However, based on WES findings, it was reclassified as STL-IV. The affected members included two female patients, IV:1 (49 years) and IV:6 (35 years), who were born to first-degree relatives. Patient (IV:1) experienced severe visual impairment, including night and day blindness, deafness, and speech impairment, progressing to total blindness by age 40. In contrast, patient (IV:6) had milder symptoms with progressive vision loss, night blindness, cataracts, and hearing loss while retaining color and light perception. Neither patient exhibited facial dysmorphism. The reclassification to Stickler syndrome type 4 (STL-IV) aligns with key phenotypic features, including progressive vision loss, hearing impairment, and cataracts. In patient IV:1, night blindness occurred secondary to retinal detachment, which is a known complication of Stickler syndrome. In contrast, patient IV:6 exhibited night blindness while retaining light and color perception, suggesting milder retinal involvement or a distinct disease mechanism. Additionally, the absence of facial dysmorphism in both patients is consistent with STL-IV, which is typically associated with a non-syndromic presentation compared to other Stickler subtypes.
2.4.3. Novel Missense Variants in Known IRD Genes
- A novel CNGA3 (NM_001298) variant, c.1774C>G, p. Pro592Ala, seggregated with the achromatopsia phenotype in the proband (IV:2) of a Punjabi family RF. MA0425, as seen in Figure S5. Both affected males were initially diagnosed with RP based on clinical evaluation. Their phenotype, characterized by photophobia, congenital nystagmus, and moderate visual impairment, was consistent with CNGA3-related achromatopsia.HOPE analysis showed that the wild-type residue proline is located in the cyclic nucleotide-binding domain. There are six invariant amino acids in this domain, three of which are glycine residues that are thought to be vital for maintaining the structural integrity of the beta barrel. The c.1774C>G substitution may affect the local structure by disrupting proline-induced special backbone conformation and impair binding site properties.
- In the RF.MA0449 Punjabi pedigree (Figure S7) which included two affected members, WES identified a pathogenic novel homozygous variant c.1171T>C, p.(Cys391Arg) in exon 4 of the GUCY2D (NM_000180) gene, which was initially associated with bilateral maculopathy. However, due to the presence of congenital nystagmus, early-onset blindness, bilateral maculopathy, and photophobia in both affected individuals, the condition was reclassified as LCA. Bioinformatics analysis revealed a difference in charge between cysteine (neutral) and arginine (positive) residues. Cysteine is located in the receptor–ligand binding region, and the c.1171T>C variation introduces a charge that may generate the repulsion of ligands or other nearby residues bearing the same charge.
- Analysis of a Punjabi pedigree RF. MA0454 Figure S8 with three affected members with the proband (IV:3) available for the WES study detected a homozygous potentially pathogenic novel variant c.626T>G, p. (Val209Gly) in the PRPH2 gene linked with the autosomal recessive LCA phenotype. The clinical changes in the affected individuals including nystagmus, mottled fovea, macular atrophy, and congenital photophobia are consistent with the phenotype of autosomal recessive LCA. Bioinformatics analysis revealed that the variation is located within a stretch of residues, namely lumenal repeats, that are repeated in the peripherin/rom-1 protein. The substitution replaces valine with glycine at position 209. Glycine’s high flexibility could disrupt the rigidity of the specific repeat in the protein structure. Since valine is located near a highly conserved position, this substitution with glycine can lead to a loss of function in the protein.
3. Discussion
4. Materials and Methods
4.1. Identification of Consanguineous Families Manifesting Various Eye Disorders
4.2. Inclusion Criteria
4.3. Phenotyping and Collection of Saliva
4.4. Approval by Ethical Committee
4.5. Whole Exome Sequencing
4.6. Sanger Sequencing of Samples from Selected VI Families
4.7. Bioinformatics Analysis for Protein Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kolawole, O.; Huang, A.; Gregory-Evans, C.; Shunmugam, M.; Weaver, T.; Gregory-Evans, K. Molecular genetic diagnostics for inherited retinal dystrophies in the clinical setting. Can. J. Ophthalmol. 2024, 59, e575–e581. [Google Scholar] [CrossRef] [PubMed]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef]
- Tavares, J.; Lorenz, B.; Born, I.; Marques, J.; Stingl, K.; Pilotto, E.; Issa, P.; Leroux, D.; Dollfus, H.; Scholl, H. Current management of Inherited Retinal Degenerations (IRD) patients in Europe. Results of a 2 years follow-up multinational survey by EVICR. net and ERN-EYE. Investig. Ophthalmol. Vis. Sci. 2022, 63, 4481-F0268. [Google Scholar]
- Fleckenstein, M.; Schmitz-Valckenberg, S.; Chakravarthy, U. Age-related macular degeneration: A review. Jama 2024, 331, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.; Reid, J.; Bainbridge, M.; Willis, A.; Ward, P.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef]
- Rashid, M.; Qasim, M.; Ishaq, R.; Bukhari, S.; Sajid, Z.; Ashfaq, U.; Haque, A.; Ahmed, Z. Pathogenic variants of AIPL1, MERTK, GUCY2D, and FOXE3 in Pakistani families with clinically heterogeneous eye diseases. PLoS ONE 2020, 15, e0239748. [Google Scholar] [CrossRef] [PubMed]
- Tehreem, R.; Chen, I.; Shah, M.; Li, Y.; Khan, M.; Afshan, K.; Chen, R.; Firasat, S. Exome sequencing identified molecular determinants of retinal dystrophies in nine consanguineous Pakistani families. Genes 2022, 13, 1630. [Google Scholar] [CrossRef]
- Reiners, J.; Nagel-Wolfrum, K.; Jürgens, K.; Märker, T.; Wolfrum, U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006, 83, 97–119. [Google Scholar] [CrossRef]
- Periyandavan, J.; Stephen, M. A brief review on microphthalmia and anophthalmia. Kerala J. Ophthalmol. 2024, 36, 224–228. [Google Scholar] [CrossRef]
- Şekeroğlu, H.; Utine, G. Congenital cataract and its genetics: The era of next-generation sequencing. Turk. J. Ophthalmol. 2021, 51, 107–113. [Google Scholar] [CrossRef]
- Kemmanu, V.; Giliyar, S.; Rao, H.; Shetty, B.; Kumaramanickavel, G.; McCarty, C. Consanguinity and its association with visual impairment in southern India: The Pavagada Pediatric Eye Disease Study 2. J. Community Genet. 2019, 10, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Hamamy, H. Consanguineous marriages: Preconception consultation in primary health care settings. J. Community Genet. 2012, 3, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Khan, I.; Khan, M.; Latif, M.; Siddiqui, M.; Khan, S.; Htar, T.; Wahid, G.; Ullah, I.; Bibi, F.; et al. Whole exome sequencing identifies a novel compound heterozygous GFM1 variant underlying developmental delay, dystonia, polymicrogyria, and severe intellectual disability in a Pakhtun family. Am. J. Med. Genet. Part A 2022, 188, 2693–2700. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, Y.; Jiao, X.; Jin, C.; Jiang, D.; Tanwar, M.; Ma, Z.; Huang, L.; Ma, X.; Sun, W.; et al. Homozygosity mapping and genetic analysis of autosomal recessive retinal dystrophies in 144 consanguineous Pakistani families. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2218–2238. [Google Scholar] [CrossRef]
- Sasaki, T.; Larsson, H.; Tisi, D.; Claesson-Welsh, L.; Hohenester, E.; Timpl, R. Endostatins derived from collagens XV and XVIII differ in structural and binding properties, tissue distribution and anti-angiogenic activity. J. Mol. Biol. 2000, 301, 1179–1190. [Google Scholar] [CrossRef]
- Hoppe, B.; Danpure, C.; Rumsby, G.; Fryer, P.; Jennings, P.; Blau, N.; Schubiger, G.; Neuhaus, T.; Leumann, E. A vertical (pseudodominant) pattern of inheritance in the autosomal recessive disease primary hyperoxaluria type 1: Lack of relationship between genotype, enzymic phenotype, and disease severity. Am. J. Kidney Dis. 1997, 29, 36–44. [Google Scholar] [CrossRef]
- Darr, A.; Small, N.; Ahmad, W.; Atkin, K.; Corry, P.; Modell, B. Addressing key issues in the consanguinity-related risk of autosomal recessive disorders in consanguineous communities: Lessons from a qualitative study of British Pakistanis. J. Community Genet. 2016, 7, 65–79. [Google Scholar] [CrossRef]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 3000 families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef]
- Munir, A.; Afsar, S.; Rehman, A. A systematic review of inherited retinal dystrophies in Pakistan: Updates from 1999 to April 2023. BMC Ophthalmol. 2024, 24, 55. [Google Scholar] [CrossRef]
- Ur Rehman, A.; Peter, V.; Quinodoz, M.; Rashid, A.; Khan, S.; Superti-Furga, A.; Rivolta, C. Exploring the genetic landscape of retinal diseases in North-Western Pakistan reveals a high degree of autozygosity and a prevalent founder mutation in ABCA4. Genes 2019, 11, 12. [Google Scholar] [CrossRef]
- Michaelides, M.; Laich, Y.; Wong, S.; Oluonye, N.; Zaman, S.; Kumaran, N.; Kalitzeos, A.; Petrushkin, H.; Georgiou, M.; Tailor, V.; et al. Gene therapy in children with AIPL1-associated severe retinal dystrophy: An open-label, first-in-human interventional study. Lancet 2025, 405, 648–657. [Google Scholar] [CrossRef]
- Alapati, A.; Goetz, K.; Suk, J.; Navani, M.; Al-Tarouti, A.; Jayasundera, T.; Tumminia, S.; Lee, P.; Ayyagari, R. Molecular diagnostic testing by eyeGENE: Analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5510–5521. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.; Wheaton, D.; Bowne, S.; Sullivan, L.; Birch, D.; Chen, R.; Daiger, S. Next-generation sequencing to solve complex inherited retinal dystrophy: A case series of multiple genes contributing to disease in extended families. Mol. Vis. 2017, 23, 470–481. [Google Scholar]
- Daiger, S.; Sullivan, L.; Bowne, S. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef]
- Bruijn, S.; Fiorentino, A.; Ottaviani, D.; Fanucchi, S.; Melo, U.; Corral-Serrano, J.; Mulders, T.; Georgiou, M.; Rivolta, C.; Pontikos, N.; et al. Structural variants create new topological-associated domains and ectopic retinal enhancer-gene contact in dominant retinitis pigmentosa. Am. J. Hum. Genet. 2020, 107, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Shami, S.; Schmitt, L.; Bittles, A. Consanguinity related prenatal and postnatal mortality of the populations of seven Pakistani Punjab cities. J. Med. Genet. 1989, 26, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Hussain The impact of consanguinity and inbreeding on perinatal mortality in Karachi, Pakistan. Paediatr. Perinat. Epidemiol. 1998, 12, 370–382. [CrossRef]
- Biswas, P.; Villanueva, A.; Soto-Hermida, A.; Duncan, J.; Matsui, H.; Borooah, S.; Kurmanov, B.; Richard, G.; Khan, S.; Branham, K.; et al. Deciphering the genetic architecture and ethnographic distribution of IRD in three ethnic populations by whole genome sequence analysis. PLoS Genet. 2021, 17, e1009848. [Google Scholar] [CrossRef]
- Maranhao, B.; Biswas, P.; Gottsch, A.; Navani, M.; Naeem, M.; Suk, J.; Chu, J.; Khan, S.; Poleman, R.; Akram, J.; et al. Investigating the molecular basis of retinal degeneration in a familial cohort of Pakistani decent by exome sequencing. PLoS ONE 2015, 10, e0136561. [Google Scholar] [CrossRef]
- Zhang, Q. Retinitis pigmentosa: Progress and perspective. Asia-Pac. J. Ophthalmol. 2016, 5, 265–271. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, W.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Zhang, Q. Clinical and genetic analysis of 63 families demonstrating early and advanced characteristic fundus as the signature of CRB1 mutations. Am. J. Ophthalmol. 2021, 223, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Jin, C.; Jiao, X.; Li, L.; Bushra, T.; Naeem, M.; Butt, N.; Husnain, T.; Sieving, P.; Riazuddin, S.; et al. AIPL1 implicated in the pathogenesis of two cases of autosomal recessive retinal degeneration. Mol. Vis. 2014, 20, 1–14. [Google Scholar]
- Maranhao, B.; Biswas, P.; Duncan, J.; Branham, K.; Silva, G.; Naeem, M.; Khan, S.; Riazuddin, S.; Hejtmancik, J.; Heckenlively, J.; et al. exomeSuite: Whole exome sequence variant filtering tool for rapid identification of putative disease causing SNVs/indels. Genomics 2014, 103, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.; Khan, M.; Micheal, S.; Ahmed, W.; Shah, A.; Venselaar, H.; Bokhari, H.; Azam, A.; Waheed, N.; Collin, R.; et al. Identification of recurrent and novel mutations in TULP1 in Pakistani families with early-onset retinitis pigmentosa. Mol. Vis. 2012, 18, 1226–1237. [Google Scholar]
- Azam, M.; Khan, M.; Gal, A.; Hussain, A.; Shah, S.; Khan, M.; Sadeque, A.; Bokhari, H.; Collin, R.; Orth, U.; et al. A homozygous p. Glu150Lys mutation in the opsin gene of two Pakistani families with autosomal recessive retinitis pigmentosa. Mol. Vis. 2009, 15, 2526–2534. [Google Scholar]
- Shenk, M.; Naz, S.; Chaudhry, T. Intensive kinship, development, and demography: Why Pakistan has the highest rates of cousin marriage in the world. Popul. Dev. Rev. 2024, 50, 1045–1090. [Google Scholar] [CrossRef]
- Ansar, M.; Chung, H.; Waryah, Y.; Makrythanasis, P.; Falconnet, E.; Rao, A.; Guipponi, M.; Narsani, A.; Fingerhut, R.; Santoni, F.; et al. Visual impairment and progressive phthisis bulbi caused by recessive pathogenic variant in MARK3. Hum. Mol. Genet. 2018, 27, 2703–2711. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- DePristo, M.; Banks, E.; Poplin, R.; Garimella, K.; Maguire, J.; Hartl, C.; Philippakis, A.; Del Angel, G.; Rivas, M.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Karczewski, K.; Francioli, L.; Tiao, G.; Cummings, B.; Alföldi, J.; Wang, Q.; Collins, R.; Laricchia, K.; Ganna, A.; Birnbaum, D.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.; Jain, P.; O’roak, B.; Cooper, G.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Cooper, G.; Stone, E.; Asimenos, G.; Green, E.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, H.; Waryah, A.; Narsani, A.; Iqbal, M.; Shahzad, M.; Waryah, Y.; Shaikh, N.; Mahmood, A. Genetic testing of non-familial deaf patients for CIB2 and GJB2 mutations: Phenotype and genetic counseling. Biochem. Genet. 2017, 55, 410–420. [Google Scholar] [CrossRef]
- Ansar, M.; Riazuddin, S.; Sarwar, M.; Makrythanasis, P.; Paracha, S.; Iqbal, Z.; Khan, J.; Assir, M.; Hussain, M.; Razzaq, A.; et al. Biallelic variants in LINGO1 are associated with autosomal recessive intellectual disability, microcephaly, speech and motor delay. Genet. Med. 2018, 20, 778–784. [Google Scholar] [CrossRef]
- Wiel, L.; Baakman, C.; Gilissen, D.; Veltman, J.; Vriend, G.; Gilissen, C. MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat. 2019, 40, 1030–1038. [Google Scholar] [CrossRef]
- Wang, X.; Zein, W.; D’Souza, L.; Roberson, C.; Wetherby, K.; He, H.; Villarta, A.; Turriff, A.; Johnson, K.; Fann, Y. Applying next generation sequencing with microdroplet PCR to determine the disease-causing mutations in retinal dystrophies. BMC Ophthalmol. 2017, 17, 157. [Google Scholar] [CrossRef]
- Carss, K.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Zampaglione, E.; Maher, M.; Place, E.; Wagner, N.; DiTroia, S.; Chao, K.; England, E.; Broad, C.; Catomeris, A.; Nassiri, S.; et al. The importance of automation in genetic diagnosis: Lessons from analyzing an inherited retinal degeneration cohort with the Mendelian Analysis Toolkit (MATK). Genet. Med. 2022, 24, 332–343. [Google Scholar] [CrossRef]
- Sohocki, M.; Perrault, I.; Leroy, B.; Payne, A.; Dharmaraj, S.; Bhattacharya, S.; Kaplan, J.; Maumenee, I.; Koenekoop, R.; Meire, F.; et al. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol. Genet. Metab. 2000, 70, 142–150. [Google Scholar] [CrossRef]
- Suetterlin, K.; Matthews, E.; Sud, R.; McCall, S.; Fialho, D.; Burge, J.; Jayaseelan, D.; Haworth, A.; Sweeney, M.; Kullmann, D.; et al. Translating genetic and functional data into clinical practice: A series of 223 families with myotonia. Brain 2022, 145, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Bianco, L.; Antropoli, A.; Arrigo, A.; Saladino, A.; Berni, A.; Bandello, F.; Mansour, A.; Parodi, M. RPGRIP1 variant associated with pigmented paravenous chorioretinal atrophy. Eur. J. Ophthalmol. 2023, 33, NP6–NP9. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, D.; Heon, E.; Cideciyan, A.; Ratnapriya, R.; Lu, M.; Sumaroka, A.; Roman, A.; Batmanabane, V.; Garafalo, A.; Stone, E.; et al. EYS mutations causing autosomal recessive retinitis pigmentosa: Changes of retinal structure and function with disease progression. Genes 2017, 8, 178. [Google Scholar] [CrossRef]
- Schulz, H.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Huchzermeyer, C.; Baier, M.; Weber, B.; et al. Mutation spectrum of the ABCA4 gene in 335 Stargardt disease patients from a multicenter German cohort—Impact of selected deep intronic variants and common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef]
- Reis, L.; Sorokina, E.; Dudakova, L.; Moravikova, J.; Skalicka, P.; Malinka, F.; Seese, S.; Thompson, S.; Bardakjian, T.; Capasso, J.; et al. Comprehensive phenotypic and functional analysis of dominant and recessive FOXE3 alleles in ocular developmental disorders. Hum. Mol. Genet. 2021, 30, 1591–1606. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.; Reece, R. Functional analysis of disease-causing mutations in human galactokinase. Eur. J. Biochem. 2003, 270, 1767–1774. [Google Scholar] [CrossRef]
- Rashid, M.; Yousaf, S.; Sheikh, S.; Sajid, Z.; Shabbir, A.; Kausar, T.; Tariq, N.; Usman, M.; Shaikh, R.; Ali, M.; et al. Identities and frequencies of variants in CYP1B1 causing primary congenital glaucoma in Pakistan. Mol. Vis. 2019, 25, 144–154. [Google Scholar]
- Aparisi, M.; Aller, E.; Fuster-García, C.; García-García, G.; Rodrigo, R.; Vázquez-Manrique, R.; Blanco-Kelly, F.; Ayuso, C.; Roux, A.; Jaijo, T.; et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J. Rare Dis. 2014, 9, 168. [Google Scholar] [CrossRef]
- Bouzia, Z.; Georgiou, M.; Hull, S.; Robson, A.; Fujinami, K.; Rotsos, T.; Pontikos, N.; Arno, G.; Webster, A.; Hardcastle, A.; et al. GUCY2D-associated Leber congenital amaurosis: A retrospective natural history study in preparation for trials of novel therapies. Am. J. Ophthalmol. 2020, 210, 59–70. [Google Scholar] [CrossRef]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.; Cho, T.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.; et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. Part A 2015, 167, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Panigrahi, A.; Mahalingam, K.; Singh, A.; Somarajan, B.; Gupta, S. Expanding the phenotypic spectrum of CYP1B1 associated primary congenital glaucoma. Clin. Exp. Ophthalmol. 2022, 50, 1112. [Google Scholar] [CrossRef] [PubMed]
- McKibbin, M.; Ali, M.; Mohamed, M.; Booth, A.; Bishop, F.; Pal, B.; Springell, K.; Raashid, Y.; Jafri, H.; Inglehearn, C. Genotype-phenotype correlation for leber congenital amaurosis in Northern Pakistan. Arch. Ophthalmol. 2010, 128, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Lotery, A.; Malik, A.; Shami, S.; Sindhi, M.; Chohan, B.; Maqbool, C.; Moore, P.; Denton, M.; Stone, E. CRB1 mutations may result in retinitis pigmentosa without para- arteriolar RPE preservation. Ophthalmic Genet. 2001, 22, 163–169. [Google Scholar] [CrossRef]
- McLaren, T.; De Roach, J.; Thompson, J.; Chen, F.; Mackey, D.; Hoffmann, L.; Urwin, I.; Lamey, T. Expanding the genetic spectrum of choroideremia in an Australian cohort: Report of five novel CHM variants. Hum. Genome Var. 2020, 7, 35. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pedigree ID | Initial Phenotype | Re-Assessed Phenotype | Gene | Variant |
|---|---|---|---|---|
| MA0315 | Retinal dystrophy | Leber congenital amaurosis | LCA5 | c.1151del |
| (NM_001122769) | p.(Pro384GlnfsTer18) | |||
| MA0383 | Retinitis pigmentosa | Myotonia congenita | CLCN1 | c.2647C>A |
| (NM_000083) | p.(Pro883Thr) | |||
| MA0392 | Septo-optic dysplasia | Leber congenital amaurosis | AIPL1 | c.645G>A |
| (NM_001033054.3) | p.(Trp215Ter) | |||
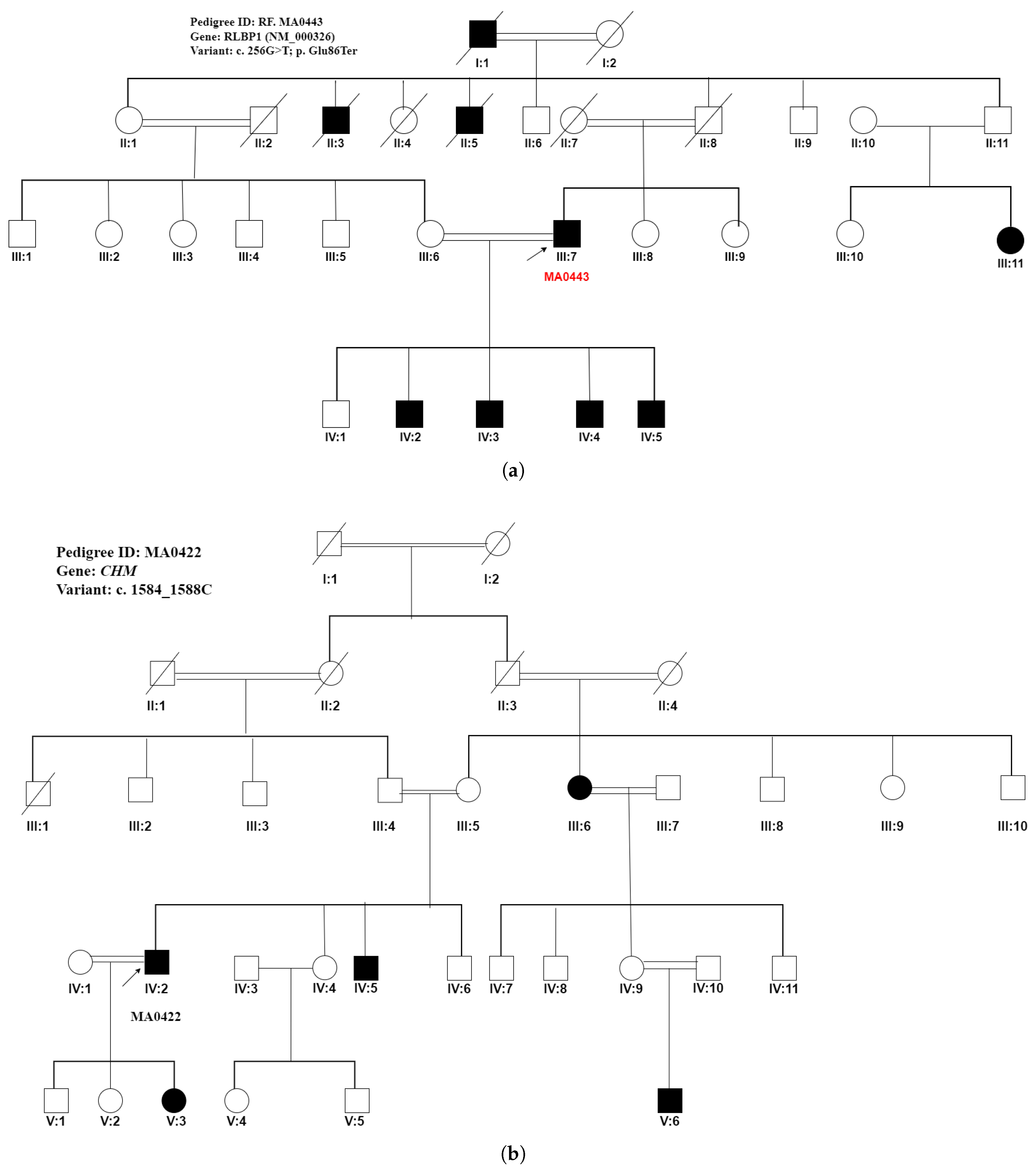
| MA0422 | Pigmentary | Retinitis pigmentosa | CHM | c.1584_1587del |
| retinopathy | (NM_000390) | p.(Val529HisfsTer7) | ||
| MA0425 | Retinitis pigmentosa | Achromatopsia | CNGA3 | c.1720C>G |
| (NM_001079878) | p.Pro574Ala | |||
| MA0435 | Stargardt disease | Leber congenital amaurosis | AIPL1 | c.834G>A |
| (NM_014336) | p.Trp278Ter | |||
| MA0449 | Bilateral maculopathy | Leber congenital amaurosis | GUCY2D | c.1171T>C |
| (NM_000180) | p.Cys391Arg | |||
| MA0454 | Congenital macular | Leber congenital amaurosis | PRPH2 | c.626T>G |
| (NM_000322) | p.Val209Gly | |||
| MA0468 | Retinal dystrophy | Knobloch syndrome | COL18A1 | c.3559_3577del |
| (NM_030582.4) | p.(Ser1187AlafsTer18) | |||
| MA0477 | Retinal dystrophy | Hermansky–Pudlak | BLOC1S3 | c.499C>T |
| syndrome-8 | (NM_212550) | p.(Leu167Phe) | ||
| MA0489 | Retinal dystrophy | Congenital syndrome | FOXE3 | c.720C>A |
| cataract–glaucoma | (NM_012186) | p.(Cys240Ter) | ||
| MA0503 | Retinal dystrophy | Leber congenital amaurosis | CRB1 | c.3007_3016del |
| (NM_001193640.2) | p.(Gly1003IlefsTer23) | |||
| MA0507 | Retinal dystrophy | Leber congenital amaurosis | AIPL1 | c.664T>C |
| (NM_014336) | p.(Trp222Arg) | |||
| MA0510 | Congenital cataract | Cone–rod dystrophy | GUCY2D | c.2965dup |
| (NM_000180) | p.(Val989GlyfsTer83) | |||
| MA0515 | Congenital cataract | Congenital glaucoma | CYP1B1 | c.1063C>T |
| (NM_000104) | p.(Arg355Ter) | |||
| MA0524 | Congenital cataract | CODAS syndrome | LONP1 | c.1448G>A |
| (NM_001276480) | p. (Arg483His) | |||
| MA0537 | Usher syndrome | Stickler syndrome type-IV | COL9A1 | c.851_852insCAAT |
| (NM_078485.4) | p.(Pro285AsnfsTer20) |
| Family ID | Patient ID/Gender | Age at Enrollment (Years) | Initial Phenotype | Phenotype After WES | Initial Complaint with Age of Onset (Years) | No. of Affected Individuals | Clinical Symptoms | Identified Variant |
|---|---|---|---|---|---|---|---|---|
| MA0320 | IV:2/Female (Proband) | 27 | ar RP | ar RP | By birth night blindness | 02 | Night blindness, photophobia, blindness since 13 years of age | TULP1(NM_001289395.2):c.417del p.(Lys140ArgfsTer2) Homozygous |
| IV:3/Female | 23 | ar RP | ar RP | By birth night blindness | 02 | Night blindness, photophobia, blindness since 13 years of age | TULP1(NM_001289395.2):c.417del p.(Lys140ArgfsTer2) Homozygous | |
| MA0324 | IV:6/Male | 31 | ar RP | ar RP | Low vision and color blindness | 02 | Night blindness, photophobia, nystagmus, complete blindness at 14 years of age | CRB1(NM_001193640.2):c.107C>G p.(Ser36Ter) Homozygous |
| IV:7/Female | ar RP | ar RP | Low vision | 02 | Night blindness, photophobia, nystagmus, complete blindness at 12 years of age | CRB1(NM_001193640.2):c.107C>G p.(Ser36Ter) Homozygous | ||
| MA0356 | III:4/Male (Proband) | 35 | ar RP | ar RP | By birth night blindness | 05 | Night blindness, photophobia, complete blindness at 25 years of age | CRB1(NM_001193640.2):c.3626G>C p.(Cys1209Ser) Homozygous |
| III:7/Male | 44 | ar RP | ar RP | By birth night blindness | 05 | Night blindness, photophobia, complete blindness at 25 years of age, corneal opacities | CRB1(NM_001193640.2):c.3626G>C p.(Cys1209Ser) Homozygous | |
| MA0406 | IV:1/Male | 18 | ar RP | ar RP | Low vision at 10 years | 02 | Night blindness, photophobia, low vision, slow progression of RP | CRB1(NM_001193640.2):c.1123T>C p.(Ser375Pro) Homozygous |
| IV:2/Female (Proband) | 32 | ar RP | ar RP | Night blindness plus photophobia at 26 years | 02 | Night blindness, photophobia, low vision, slow progression of RP | CRB1(NM_001193640.2):c.1123T>C p.(Ser375Pro) Homozygous | |
| MA0425 | IV:2/Male (Proband) | 06 | arRP | ar achromatopsia | Photophobia, nystagmus, low vision | 02 | Night blindness, photophobia, nystagmus, low vision, microphthalmia | CNGA3(NM_001079878):c.1720C>G p.Pro574Ala Homozygous |
| IV:1/Male | 02 | arRP | ar achromatopsia | Myopia since 4 months of age | 02 | Night blindness, photophobia, nystagmus, low vision | CNGA3(NM_001079878):c.1720C>G p.Pro574Ala Homozygous | |
| MA0510 | IV:8/Male (Proband) | 36 | Congenital cataract | Cone–rod dystrophy | By birth day blindness | 02 | Day blindness, night blindness, photophobia, nystagmus, complete blindness since 23 years of age | GUCY2D(NM_000180):c.2965dup p.(Val989GlyfsTer83) Homozygous |
| IV:9/Male | 27 | Congenital cataract | Cone–rod dystrophy | By birth day blindness | 02 | Day blindness, photophobia, nystagmus, complete blindness since 23 years of age, bilateral cataracts | GUCY2D(NM_000180):c.2965dup p.(Val989GlyfsTer83) Homozygous | |
| MA0337 | IV:1/Male | 36 | arRP | arRP | Low vision since childhood | 03 | Night blindness, nystagmus, complete blindness at 25 years of age | CERKL(NM_001160277.2):c.715C>T, p.(Arg239Ter) Homozygous |
| IV:3/Male (Proband) | 40 | arRP | arRP | Low vision since childhood | 03 | Night blindness, nystagmus, partial blindness | CERKL(NM_001160277.2):c.715C>T, p.(Arg239Ter) Homozygous | |
| MA0371 | IV:2/Female | 27 | arRP | arRP | Night blindness since 11 years | 02 | Night blindness, photophobia, complete blindness since 24 years of age, microphthalmia | CERKL(NM_001160277.2):c.715C>T, p.(Arg239Ter) Homozygous |
| IV:4/Male (Proband) | 34 | arRP | arRP | Night blindness since 18 years | 02 | Night blindness, photophobia, complete blindness since 27 years of age | CERKL(NM_001160277.2):c.715C>T, p.(Arg239Ter) Homozygous | |
| MA0380 | III:1/Male (Proband) | 34 | arRP | arRP | Night blindness since 10 years | 03 | Night blindness, photophobia, severe visual impairment blindness since 29 years of age | CRB1(NM_001193640.2):c.601T>C, p. Cys210Arg Homozygous |
| III:7/Male | 37 | arRP | arRP | By birth low vision | 03 | Night blindness, photophobia, nystagmus, severe visual impairment since 33 years of age | CRB1(NM_001193640.2):c.601T>C, p. Cys210Arg Homozygous | |
| MA0400 | IV:3/Male | 23 | arRP | arRP | Low vision since 7 years of age | 02 | Night blindness, photophobia, nystagmus, complete blindness in one eye since 19 years of age and cataracts | LRP5(NM_001291902.2):c. 430G>A, p.(Val144Ile) Homozygous |
| IV:4/Male | 27 | arRP | arRP | Low vision since 7 years of age | 02 | Night blindness, photophobia, nystagmus, Complete blindness in one eye since 19 years of age and cataracts | LRP5(NM_001291902.2):c. 430G>A, p.(Val144Ile) Homozygous | |
| MA0413 | III:4/Female (Proband) | 46 | arRP | arRP | Low vision since 14 years of age | 02 | Night blindness, complete blindness since 17 years of age | PDE6A(NM_000440): c.650_651dup, p.(Ala218LeufsTer4) Homozygous |
| III:7/Male | 36 | arRP | arRP | Night blindness since 4 years of age | 02 | Night blindness, photophobia, blindness since 4 years of age and cataracts | PDE6A(NM_000440): c.650_651dup, p.(Ala218LeufsTer4) Homozygous | |
| MA0430 | IV:4/Female (Proband) | 12 | arRP | arRP | Low vision since 3.5 years of age | 02 | Night blindness, blindness since 3.5 years of age and squint | PDE6A(NM_000440): c.650_651dup, p.(Ala218LeufsTer4) Homozygous |
| IV:6/Male | 06 | arRP | arRP | Low vision since 2.5 years of age | 02 | Night blindness, blindness since 2.5 years of age and squint | PDE6A(NM_000440): c.650_651dup, p.(Ala218LeufsTer4) Homozygous | |
| MA0341 | III:1/Male | 20 | arRP | arRP | By birth low vision | 02 | By birth night blindness and photophobia, nystagmus, complete blindness at the age of 15 years, corneal opacities | CRB1(NM_001193640.2): c.3007_3016del, p.(Gly1003IlefsTer23) Homozygous |
| III:4/Male (Proband) | 22 | arRP | arRP | Low vision at the age of 2 years | 02 | Night blindness and photophobia since childhood, nystagmus, blindness at the age of 15 years, corneal opacities | CRB1(NM_001193640.2): c.3007_3016del, p.(Gly1003IlefsTer23) Homozygous | |
| MA0503 | IV:1/Male (Proband) | 43 | arRD | arLCA | By birth blindness | 02 | By birth night blindness, by birth photophobia, complete blindness at 30 years of age, cataracts | CRB1(NM_001193640.2):c.3007_3016del, p.(Gly1003IlefsTer23) Homozygous |
| IV:11/Male | 46 | arRD | arLCA | Very low vision since two years of age | 02 | By birth night blindness, by birth photophobia, complete blindness at 2 years of age | CRB1(NM_001193640.2):c.3007_3016del, p.(Gly1003IlefsTer23) Homozygous | |
| MA0315 | IV:3/Male | 22 | arRD | arLCA | By birth nystagmus and reduced vision | 02 | Night blindness, photophobia, nystagmus, congenital blindness | LCA5(NM_001122769): c.1151del, p.(Pro384GlnfsTer18) Homozygous |
| IV:4/Female (Proband) | 25 | arRD | arLCA | By birth nystagmus and reduced vision | 02 | Night blindness, photophobia, nystagmus, congenital blindness | LCA5(NM_001122769): c.1151del, p.(Pro384GlnfsTer18) Homozygous | |
| MA0461 | IV:2/Female (Proband) | 22 | arLCA | arLCA | By birth blindness, nystagmus | 05 | Complete blindness since 2 years of age | GUCY2D(NM_000180):c.3056A>C, p.His1019Pro Homozygous |
| IV:3/Female | 18 | arLCA | arLCA | By birth blindness | 05 | Complete blindness since 2 years of age | GUCY2D(NM_000180):c.3056A>C, p.His1019Pro Homozygous | |
| MA0422 | III:2/Male (Proband) | 45 | Pigmentary retinopathy | arRP | Low vision since 7 years of age | 02 | Night blindness at 9 years of age, complete blindness at 12 years of age | CHM(NM_000390):c.1584_1587del, p.(Val529HisfsTer7) Homozygous |
| III:5/Male | Pigmentary retinopathy | arRP | Low vision since 6 years of age | 02 | Night blindness at 9 years of age, complete blindness at 7 years of age | CHM(NM_000390):c.1584_1587del, p.(Val529HisfsTer7) Homozygous | ||
| MA0534 | IV:14/Male | 22 | Usher syndrome | Usher syndrome | Sensorineural hearing loss and low vision | 02 | RP and Hearing problem | MY07A (NM_001369365.1):c.55962A>G Homozygous |
| IV:15 | 23 | Usher syndrome | Usher syndrome | By birth night blindness, RP | 02 | Sensorineural hearing loss, low vision and bilateral cataracts | MY07A (NM_001369365.1):c.55962A>G Homozygous |
| Pakistani Population | |
|---|---|
| Summary of genetic findings and population distribution of variants identified in 43 pedigrees | 43 |
| Mutation identification rate | 86% |
| Novel IRD detected mutations | 16 |
| Novel SNVs not reported in gnomAD | 06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafar, A.; Baig, R.M.; Arshad, A.; Rashid, A.; Oreshkov, S.; Frederiksen, H.N.; Ansar, M. Deciphering the Genetic Basis of Degenerative and Developmental Eye Disorders in 50 Pakistani Consanguineous Families Using Whole-Exome Sequencing. Int. J. Mol. Sci. 2025, 26, 2715. https://doi.org/10.3390/ijms26062715
Zafar A, Baig RM, Arshad A, Rashid A, Oreshkov S, Frederiksen HN, Ansar M. Deciphering the Genetic Basis of Degenerative and Developmental Eye Disorders in 50 Pakistani Consanguineous Families Using Whole-Exome Sequencing. International Journal of Molecular Sciences. 2025; 26(6):2715. https://doi.org/10.3390/ijms26062715
Chicago/Turabian StyleZafar, Ainee, Ruqia Mehmood Baig, Abida Arshad, Abdur Rashid, Sergey Oreshkov, Helen Nabiryo Frederiksen, and Muhammad Ansar. 2025. "Deciphering the Genetic Basis of Degenerative and Developmental Eye Disorders in 50 Pakistani Consanguineous Families Using Whole-Exome Sequencing" International Journal of Molecular Sciences 26, no. 6: 2715. https://doi.org/10.3390/ijms26062715
APA StyleZafar, A., Baig, R. M., Arshad, A., Rashid, A., Oreshkov, S., Frederiksen, H. N., & Ansar, M. (2025). Deciphering the Genetic Basis of Degenerative and Developmental Eye Disorders in 50 Pakistani Consanguineous Families Using Whole-Exome Sequencing. International Journal of Molecular Sciences, 26(6), 2715. https://doi.org/10.3390/ijms26062715